
Could Lower Testosterone in Older Men Explain Higher COVID-19 Morbidity and Mortalities?

 , , , , ,
, , , , ,  , and
, and 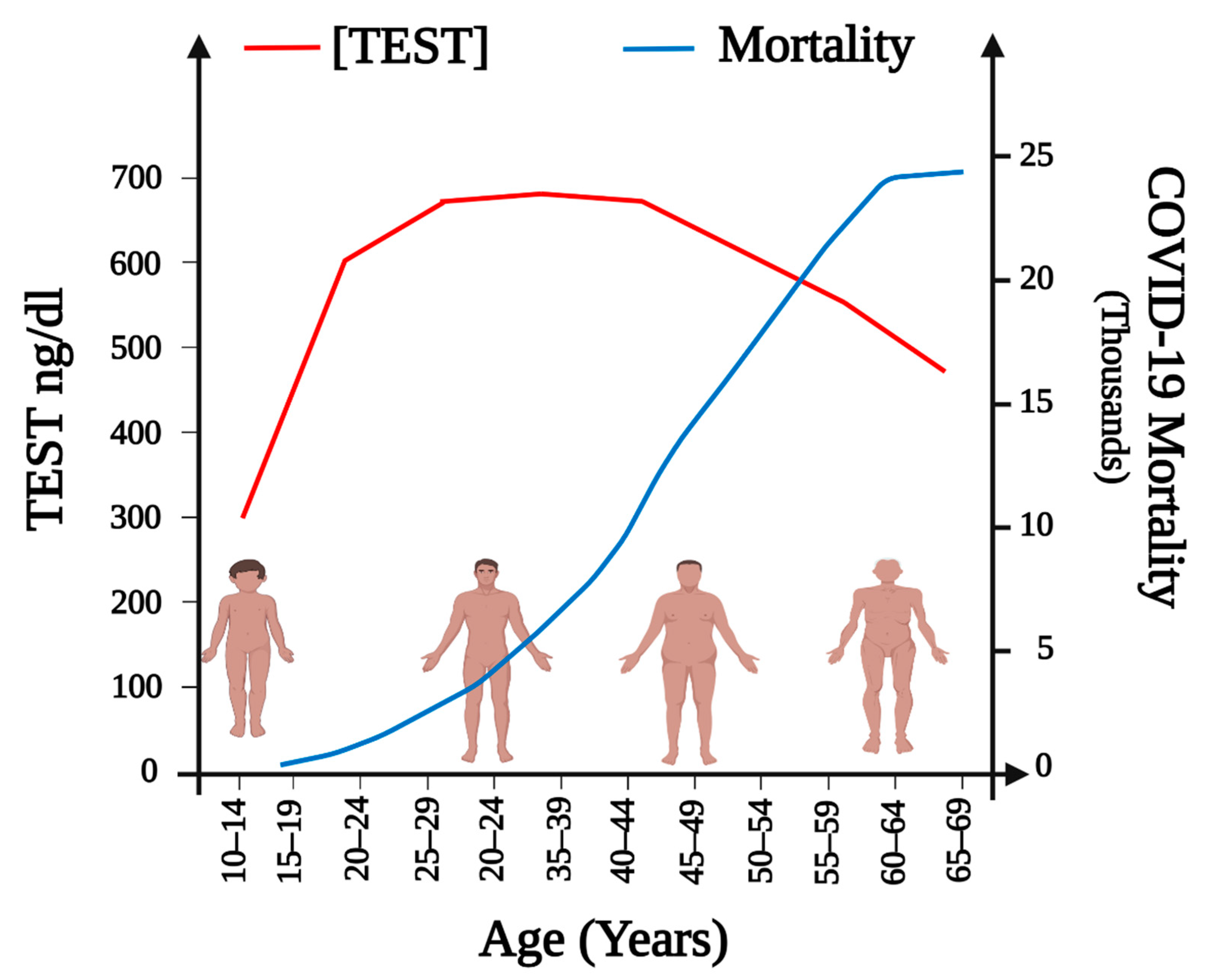{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Calcium Signaling
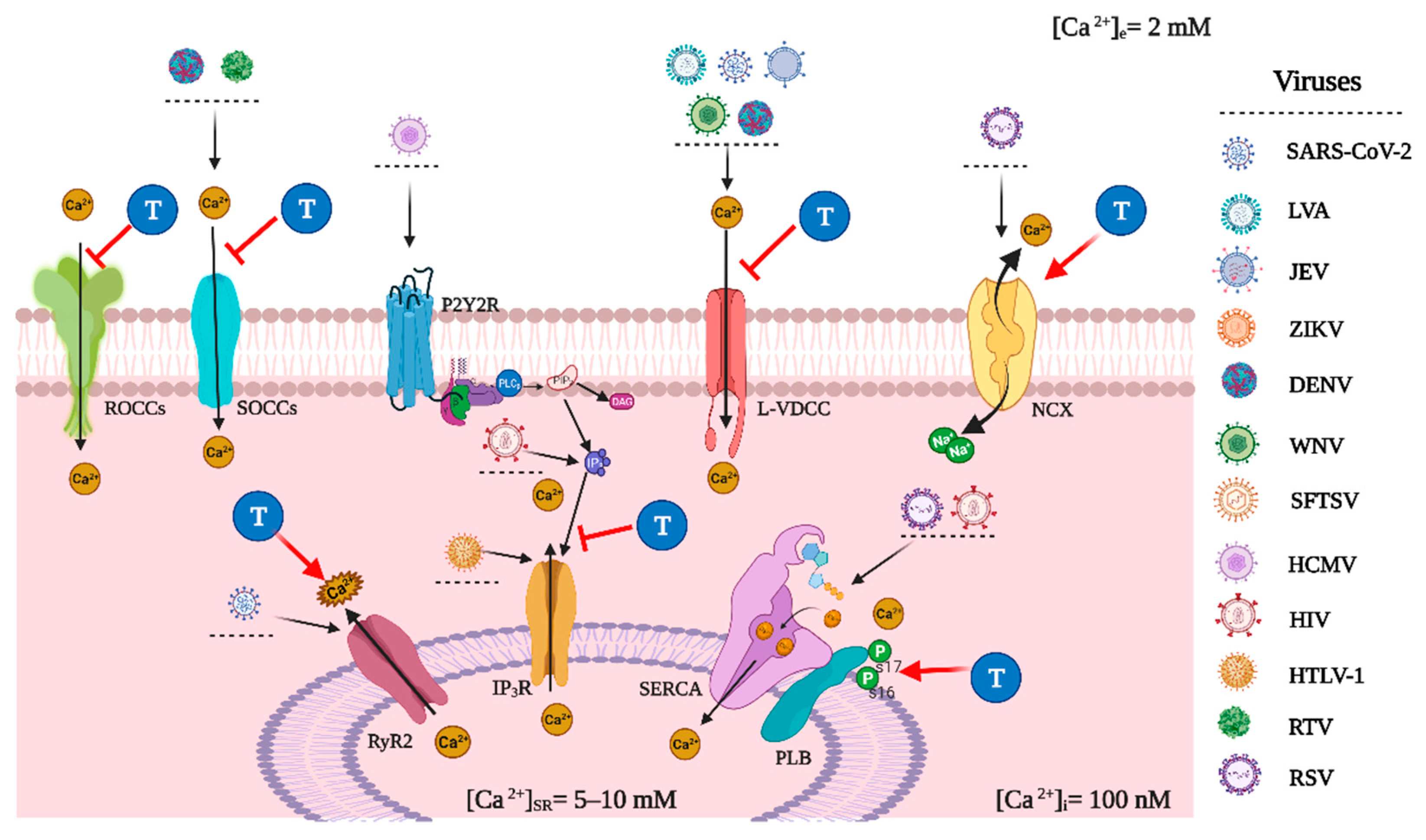
3. Viral Modifications of Host Cell Calcium Homeostasis
4. Role of COVID-19 in Testosterone Production
5. Testosterone’s Modes of Action at the Cellular Level
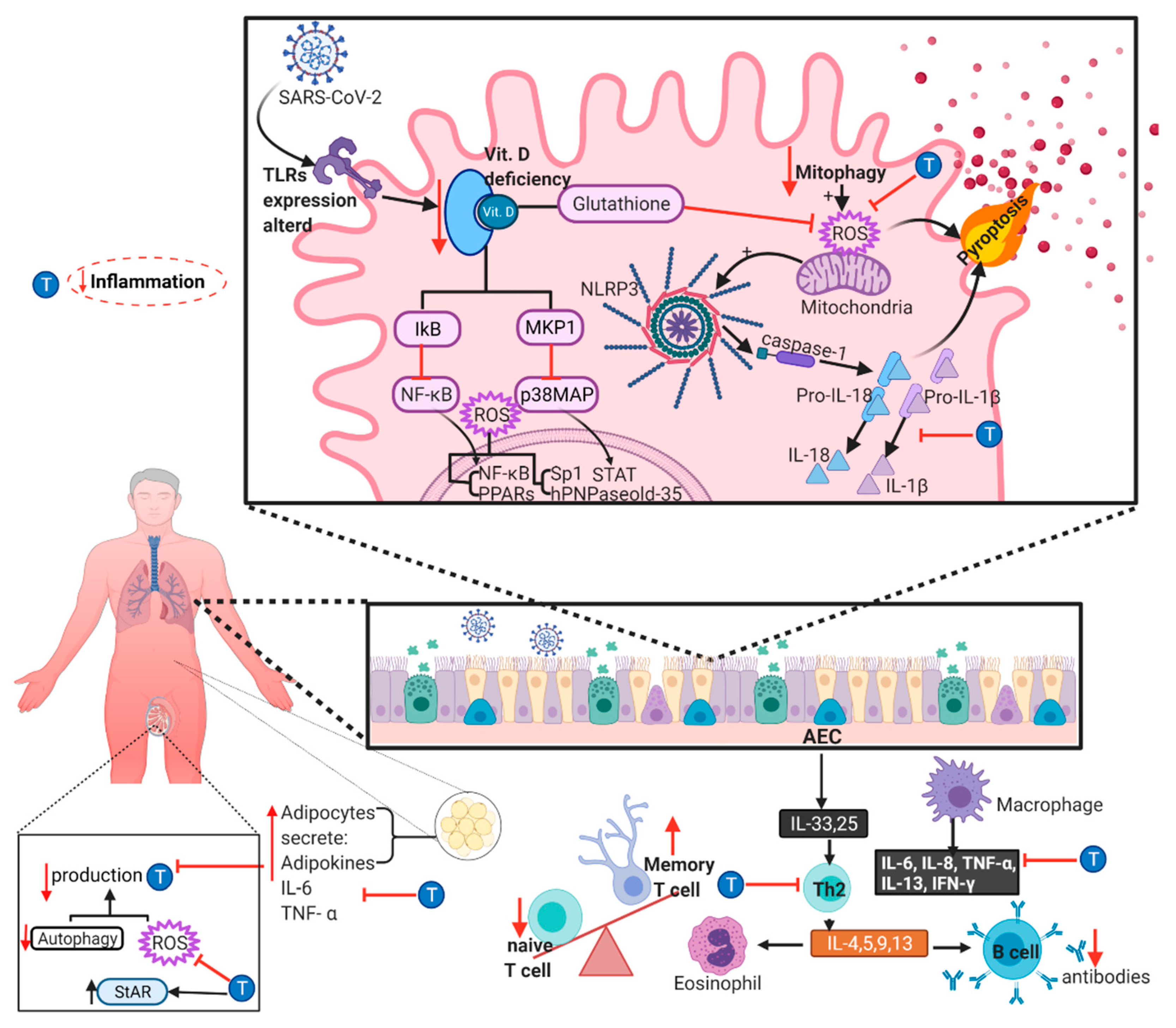
6. Role of Inflammaging in the Pathogenesis of COVID
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Bordea, I.R.; Candrea, S.; Sălăgean, T.; Pop, I.D.; Lucaciu, O.; Ilea, A.; Manole, M.; Băbțan, A.M.; Sirbu, A.; Hanna, R. Impact of COVID-19 Pandemic on Healthcare Professionals and Oral Care Operational Services: A Systemic Review. Risk Manag. Healthc. Policy 2021, 14, 453–463. [Google Scholar] [CrossRef]
- The Sex, Gender and Covid-19 Project. Available online: https://globalhealth5050.org/the-sex-gender-and-covid-19-project/about-us/ (accessed on 4 November 2021).
- World Health Organization (WHO). Weekly Epidemiological Update on COVID-19—26 October 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---26-october-2021 (accessed on 9 November 2021).
- México, G.D. Covid-19 México. Available online: Datos.covid-19.conacyt.mx (accessed on 9 November 2021).
- Wu, Z.; Mcgoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China. JAMA 2020, 323, 1239. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; Mcginn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, J. Do Men Have a Higher Case Fatality Rate of Severe Acute Respiratory Syndrome than Women Do? Am. J. Epidemiol. 2004, 159, 229–231. [Google Scholar] [CrossRef] [Green Version]
- Gomez, C.R.; Nomellini, V.; Kovacs, E.J. Sex Hormones and Immunosenescence. In Handbook of Immunosenescence; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Vermeulen, A.; Verdonck, L.; Kaufman, J.M. A Critical Evaluation of Simple Methods for the Estimation of Free Testosterone in Serum. J. Clin. Endocrinol. Metab. 1999, 84, 3666–3672. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, M.; Zhang, X.; Liu, T.; Libby, P.; Shi, G.P. COVID-19, the Pandemic of the Century and Its Impact on Cardiovascular Diseases. Cardiol. Discov. 2021, 1, 233–258. [Google Scholar] [CrossRef]
- Foo, Y.Z.; Nakagawa, S.; Rhodes, G.; Simmons, L.W. The effects of sex hormones on immune function: A meta-analysis. Biol. Rev. 2017, 92, 551–571. [Google Scholar] [CrossRef] [Green Version]
- Patil, A.; Tripathy, J.P.; Deshmukh, V.; Sontakke, B.; Tripathi, S.C. SeXX and COVID-19: Tussle between the two. Monaldi Arch. Chest Dis. 2020, 90, 2020060159. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Mantovani, A.; Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 2020, 12, 10087–10098. [Google Scholar] [CrossRef]
- Bordea, I.R.; Xhajanka, E.; Candrea, S.; Bran, S.; Onișor, F.; Inchingolo, A.D.; Malcangi, G.; Pham, V.H.; Inchingolo, A.M.; Scarano, A.; et al. Coronavirus (SARS-CoV-2) Pandemic: Future Challenges for Dental Practitioners. Microorganisms 2020, 8, 1704. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Dalpiaz, P.L.M.; Lamas, A.Z.; Caliman, I.F.; Ribeiro, R.F.; Abreu, G.R.; Moyses, M.R.; Andrade, T.U.; Gouvea, S.A.; Alves, M.F.; Carmona, A.K.; et al. Sex Hormones Promote Opposite Effects on ACE and ACE2 Activity, Hypertrophy and Cardiac Contractility in Spontaneously Hypertensive Rats. PLoS ONE 2015, 10, e0127515. [Google Scholar] [CrossRef]
- Li, D.; Jin, M.; Bao, P.; Zhao, W.; Zhang, S. Clinical Characteristics and Results of Semen Tests Among Men With Coronavirus Disease 2019. JAMA Netw. Open 2020, 3, e208292. [Google Scholar] [CrossRef]
- Temiz, M.Z.; Dincer, M.M.; Hacibey, I.; Yazar, R.O.; Celik, C.; Kucuk, S.H.; Alkurt, G.; Doganay, L.; Yuruk, E.; Muslumanoglu, A.Y. Investigation of SARS-CoV-2 in semen samples and the effects of COVID-19 on male sexual health by using semen analysis and serum male hormone profile: A cross-sectional, pilot study. Andrologia 2021, 53, e13912. [Google Scholar] [CrossRef]
- Holtmann, N.; Edimiris, P.; Andree, M.; Doehmen, C.; Baston-Buest, D.; Adams, O.; Kruessel, J.-S.; Bielfeld, A.P. Assessment of SARS-CoV-2 in human semen—A cohort study. Fertil. Steril. 2020, 114, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, S.; Huang, B.; Zhong, J.-M.; Su, H.; Chen, Y.-J.; Cao, Q.; Ma, L.; He, J.; Li, X.-F.; et al. Pathological Findings in the Testes of COVID-19 Patients: Clinical Implications. Eur. Urol. Focus 2020, 6, 1124–1129. [Google Scholar] [CrossRef]
- Chen, X.; Cao, R.; Zhong, W. Host Calcium Channels and Pumps in Viral Infections. Cells 2019, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Liu, Y.; Guo, J.; Wang, P.; Zhang, L.; Xiao, G.; Wang, W. Screening of FDA-Approved Drugs for Inhibitors of Japanese Encephalitis Virus Infection. J. Virol. 2017, 91, e01055-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saurav, S.; Tanwar, J.; Ahuja, K.; Motiani, R.K. Dysregulation of host cell calcium signaling during viral infections: Emerging paradigm with high clinical relevance. Mol. Asp. Med. 2021, 81, 101004. [Google Scholar] [CrossRef]
- Jiang, B.; Liang, S.; Liang, G.; Wei, H. Could dantrolene be explored as a repurposed drug to treat COVID-19 patients by restoring intracellular calcium homeostasis? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10228–10238. [Google Scholar] [CrossRef]
- Straus, M.R.; Bidon, M.K.; Tang, T.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Inhibitors of L-Type Calcium Channels Show Therapeutic Potential for Treating SARS-CoV-2 Infections by Preventing Virus Entry and Spread. ACS Infect. Dis. 2021, 7, 2807–2815. [Google Scholar] [CrossRef]
- Montaño, L.M.; Flores-Soto, E.; Sommer, B.; Solís-Chagoyán, H.; Perusquía, M. Androgens are effective bronchodilators with anti-inflammatory properties: A potential alternative for asthma therapy. Steroids 2020, 153, 108509. [Google Scholar] [CrossRef]
- Flores-Soto, E.; Reyes-García, J.; Carbajal-García, A.; Campuzano-González, E.; Perusquía, M.; Sommer, B.; Montaño, L.M. Sex steroids effects on guinea pig airway smooth muscle tone and intracellular Ca2+ basal levels. Mol. Cell. Endocrinol. 2017, 439, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Perusquía, M.; Flores-Soto, E.; Sommer, B.; Campuzano-González, E.; Martínez-Villa, I.; Martínez-Banderas, A.I.; Montaño, L.M. Testosterone-induced relaxation involves L-type and store-operated Ca2+ channels blockade, and PGE2 in guinea pig airway smooth muscle. Pflügers Arch.-Eur. J. Physiol. 2015, 467, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Montaño, L.M.; Flores-Soto, E.; Reyes-García, J.; Díaz-Hernández, V.; Carbajal-García, A.; Campuzano-González, E.; Ramírez-Salinas, G.L.; Velasco-Velázquez, M.A.; Sommer, B. Testosterone induces hyporesponsiveness by interfering with IP3 receptors in guinea pig airway smooth muscle. Mol. Cell. Endocrinol. 2018, 473, 17–30. [Google Scholar] [CrossRef]
- Flores-Soto, E.; Reyes-García, J.; Sommer, B.; Montaño, L.M. Sarcoplasmic reticulum Ca2+ refilling is determined by L-type Ca2+ and store operated Ca2+ channels in guinea pig airway smooth muscle. Eur. J. Pharmacol. 2013, 721, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Bootman, M.D.; Lipp, P.; Berridge, M.J. The organisation and functions of local Ca2+ signals. J. Cell Sci. 2001, 114, 2213–2222. [Google Scholar] [CrossRef]
- Reyes-García, J.; Flores-Soto, E.; Carbajal-García, A.; Sommer, B.; Montaño, L.M. Maintenance of intracellular Ca2+ basal concentration in airway smooth muscle (Review). Int. J. Mol. Med. 2018, 42, 2998–3008. [Google Scholar] [CrossRef] [Green Version]
- Bazán-Perkins, B.; Flores-Soto, E.; Barajas-López, C.; Montaño, L.M. Role of sarcoplasmic reticulum Ca2+ content in Ca2+ entry of bovine airway smooth muscle cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 368, 277–283. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Janssen, L.J.; Walters, D.K.; Wattie, J. Regulation of [Ca2+]i in canine airway smooth muscle by Ca2+-ATPase and Na+/Ca2+ exchange mechanisms. Am. J. Physiol. 1997, 273, L322–L330. [Google Scholar] [CrossRef]
- Floyd, R.; Wray, S. Calcium transporters and signalling in smooth muscles. Cell Calcium 2007, 42, 467–476. [Google Scholar] [CrossRef]
- Philipson, K.D.; Nicoll, D.A. Sodium-Calcium Exchange: A Molecular Perspective. Annu. Rev. Physiol. 2000, 62, 111–133. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.K.; Eisenstein, M.E. Targeting Host Store-Operated Ca2+ Release to Attenuate Viral Infections. Curr. Top. Med. Chem. 2013, 13, 1916–1932. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Tsuda, M.; Nanbo, A.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. A Ca2+-dependent signalling circuit regulates influenza A virus internalization and infection. Nat. Commun. 2013, 4, 2763. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, L.-K.; Li, S.-F.; Zhang, S.-F.; Wan, W.-W.; Zhang, Y.-L.; Xin, Q.-L.; Dai, K.; Hu, Y.-Y.; Wang, Z.-B.; et al. Calcium channel blockers reduce severe fever with thrombocytopenia syndrome virus (SFTSV) related fatality. Cell Res. 2019, 29, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Lavanya, M.; Cuevas, C.D.; Thomas, M.; Cherry, S.; Ross, S.R. siRNA Screen for Genes That Affect Junín Virus Entry Uncovers Voltage-Gated Calcium Channels as a Therapeutic Target. Sci. Transl. Med. 2013, 5, 204ra131–204ra201. [Google Scholar] [CrossRef] [Green Version]
- Tammineni, E.R.; Carrillo, E.D.; Soto-Acosta, R.; Angel-Ambrocio, A.H.; García, M.C.; Bautista-Carbajal, P.; Del Angel, R.M.; Sánchez, J.A. The β4 subunit of Cav1.2 channels is required for an optimal interferon response in cardiac muscle cells. Sci. Signal. 2018, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Danta, C.C. Calcium Channel Blockers: A Possible Potential Therapeutic Strategy for the Treatment of Alzheimer’s Dementia Patients with SARS-CoV-2 Infection. ACS Chem. Neurosci. 2020, 11, 2145–2148. [Google Scholar] [CrossRef]
- Han, Z.; Madara, J.J.; Herbert, A.; Prugar, L.I.; Ruthel, G.; Lu, J.; Liu, Y.; Liu, W.; Liu, X.; Wrobel, J.E.; et al. Calcium Regulation of Hemorrhagic Fever Virus Budding: Mechanistic Implications for Host-Oriented Therapeutic Intervention. PLoS Pathog. 2015, 11, e1005220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solaimanzadeh, I. Nifedipine and Amlodipine Are Associated With Improved Mortality and Decreased Risk for Intubation and Mechanical Ventilation in Elderly Patients Hospitalized for COVID-19. Cureus 2020, 12, e8069. [Google Scholar] [CrossRef] [PubMed]
- Reiken, S.; Dridi, H.; Sittenfeld, L.; Liu, X.; Marks, A.R. Alzheimer’s-like remodeling of neuronal ryanodine receptor in COVID-19. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wei, H.; Liang, G.; Vera, R.M. Dantrolene repurposed to treat sepsis or septic shock and COVID-19 patients. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 3136–3144. [Google Scholar]
- Chen, S.; Shenk, T.; Nogalski, M.T. P2Y2 purinergic receptor modulates virus yield, calcium homeostasis, and cell motility in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2019, 116, 18971–18982. [Google Scholar] [CrossRef] [Green Version]
- Keay, S.; Baldwin, B.R.; Smith, M.W.; Wasserman, S.S.; Goldman, W.F. Increases in [Ca2+]i mediated by the 92.5-kDa putative cell membrane receptor for HCMV gp86. Am. J. Physiol. 1995, 269, C11–C21. [Google Scholar] [CrossRef]
- Ehrlich, L.S.; Medina, G.N.; Photiadis, S.; Whittredge, P.B.; Watanabe, S.; Taraska, J.W.; Carter, C.A. Tsg101 regulates PI(4,5)P2/Ca(2+) signaling for HIV-1 Gag assembly. Front. Microbiol. 2014, 5, 234. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Albrecht, B.R.; Kelley, R.E.; Muthusamy, N.; Kim, S.-J.; Altschuld, R.A.; Lairmore, M.D. Human T-Cell Lymphotropic Virus Type 1 p12 I Expression Increases Cytoplasmic Calcium To Enhance the Activation of Nuclear Factor of Activated T Cells. J. Virol. 2002, 76, 10374–10382. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Harty, R.N. Influence of calcium/calmodulin on budding of Ebola VLPs: Implications for the involvement of the Ras/Raf/MEK/ERK pathway. Virus Genes 2007, 35, 511–520. [Google Scholar] [CrossRef]
- Dionicio, C.L.; Peña, F.; Constantino-Jonapa, L.A.; Vazquez, C.; Yocupicio-Monroy, M.; Rosales, R.; Zambrano, J.L.; Ruiz, M.C.; Del Angel, R.M.; Ludert, J.E. Dengue virus induced changes in Ca2+ homeostasis in human hepatic cells that favor the viral replicative cycle. Virus Res. 2018, 245, 17–28. [Google Scholar] [CrossRef]
- Michelangeli, F.; Ruiz, M.C.; del Castillo, J.R.; Ludert, J.E.; Liprandi, F. Effect of rotavirus infection on intracellular calcium homeostasis in cultured cells. Virology 1991, 181, 520–527. [Google Scholar] [CrossRef]
- Pham, T.; Perry, J.L.; Dosey, T.L.; Delcour, A.H.; Hyser, J.M. The Rotavirus NSP4 Viroporin Domain is a Calcium-conducting Ion Channel. Sci. Rep. 2017, 7, 43487. [Google Scholar] [CrossRef]
- Panda, S.; Behera, S.; Alam, M.F.; Syed, G.H. Endoplasmic reticulum & mitochondrial calcium homeostasis: The interplay with viruses. Mitochondrion 2021, 58, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Wang, Y.; Wang, L.; Li, G.; Lan, K.; Altmeyer, R.; Zou, G. Cyclopiazonic acid, an inhibitor of calcium-dependent ATPases with antiviral activity against human respiratory syncytial virus. Antivir. Res. 2016, 132, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Grupp, I.L.; Grupp, G.; Hoit, B.; Morris, R.; Samarel, A.M.; Bruggeman, L.; Klotman, P. Cardiac Dysfunction Occurs in the HIV-1 Transgenic Mouse Treated with Zidovudine. Lab. Investig. 2000, 80, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz, Y.; Peña, F.; Aristimuño, O.C.; Matteo, L.; De Agrela, M.; Chemello, M.E.; Michelangeli, F.; Ruiz, M.C. Dissecting the Ca2+ entry pathways induced by rotavirus infection and NSP4-EGFP expression in Cos-7 cells. Virus Res. 2012, 167, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Subramaniam, M.D.; Chakraborty, R.; Myakala, H.; Iyer, M.; Bharathi, G.; Siva, K.; Vellingiri, B.; Valsala Gopalakrishnan, A. The role of Interleukin-4 in COVID-19 associated male infertility—A hypothesis. J. Reprod. Immunol. 2020, 142, 103213. [Google Scholar] [CrossRef]
- Fan, C.; Lu, W.; Li, K.; Ding, Y.; Wang, J. ACE2 Expression in Kidney and Testis May Cause Kidney and Testis Infection in COVID-19 Patients. Front. Med. 2021, 7, 1045. [Google Scholar] [CrossRef]
- Shen, Q.; Xiao, X.; Aierken, A.; Yue, W.; Wu, X.; Liao, M.; Hua, J. The ACE2 expression in Sertoli cells and germ cells may cause male reproductive disorder after SARS-CoV-2 infection. J. Cell. Mol. Med. 2020, 24, 9472–9477. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, X. scRNA-seq Profiling of Human Testes Reveals the Presence of the ACE2 Receptor, A Target for SARS-CoV-2 Infection in Spermatogonia, Leydig and Sertoli Cells. Cells 2020, 9, 920. [Google Scholar] [CrossRef] [Green Version]
- Haghpanah, A.; Masjedi, F.; Alborzi, S.; Hosseinpour, A.; Dehghani, A.; Malekmakan, L.; Roozbeh, J. Potential mechanisms of SARS-CoV-2 action on male gonadal function and fertility: Current status and future prospects. Andrologia 2021, 53, e13883. [Google Scholar] [CrossRef] [PubMed]
- Saylam, B.; Uguz, M.; Yarpuzlu, M.; Efesoy, O.; Akbay, E.; Çayan, S. The presence of SARS-CoV-2 virus in semen samples of patients with COVID-19 pneumonia. Andrologia 2021, 53, e14145. [Google Scholar] [CrossRef]
- Song, C.; Wang, Y.; Li, W.; Hu, B.; Chen, G.; Xia, P.; Wang, W.; Li, C.; Diao, F.; Hu, Z.; et al. Absence of 2019 novel coronavirus in semen and testes of COVID-19 patients. Biol. Reprod. 2020, 103, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Banihani, S.A. Human semen quality as affected by SARS-CoV-2 infection: An up-to-date review. Andrologia 2021, e14295. [Google Scholar] [CrossRef] [PubMed]
- Paoli, D.; Pallotti, F.; Colangelo, S.; Basilico, F.; Mazzuti, L.; Turriziani, O.; Antonelli, G.; Lenzi, A.; Lombardo, F. Study of SARS-CoV-2 in semen and urine samples of a volunteer with positive naso-pharyngeal swab. J. Endocrinol. Investig. 2020, 43, 1819–1822. [Google Scholar] [CrossRef]
- Ruan, Y.; Hu, B.; Liu, Z.; Liu, K.; Jiang, H.; Li, H.; Li, R.; Luan, Y.; Liu, X.; Yu, G.; et al. No detection of SARS-CoV-2 from urine, expressed prostatic secretions, and semen in 74 recovered COVID-19 male patients: A perspective and urogenital evaluation. Andrology 2021, 9, 99–106. [Google Scholar] [CrossRef]
- Guo, L.; Zhao, S.; Li, W.; Wang, Y.; Li, L.; Jiang, S.; Ren, W.; Yuan, Q.; Zhang, F.; Kong, F.; et al. Absence of SARS-CoV-2 in semen of a COVID-19 patient cohort. Andrology 2021, 9, 42–47. [Google Scholar] [CrossRef]
- Pan, F.; Xiao, X.; Guo, J.; Song, Y.; Li, H.; Patel, D.P.; Spivak, A.M.; Alukal, J.P.; Zhang, X.; Xiong, C.; et al. No evidence of severe acute respiratory syndrome–Coronavirus 2 in semen of males recovering from coronavirus disease 2019. Fertil. Steril. 2020, 113, 1135–1139. [Google Scholar] [CrossRef]
- Loveland, K.L.; Klein, B.; Pueschl, D.; Indumathy, S.; Bergmann, M.; Loveland, B.E.; Hedger, M.P.; Schuppe, H.C. Cytokines in Male Fertility and Reproductive Pathologies: Immunoregulation and Beyond. Front. Endocrinol. 2017, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Schuppe, H.-C.; Pilatz, A.; Hossain, H.; Diemer, T.; Wagenlehner, F.; Weidner, W. Urogenital Infection as a Risk Factor for Male Infertility. Dtsch. Ärzteblatt Int. 2017, 114, 339. [Google Scholar] [CrossRef] [Green Version]
- Hedger, M.P.; Meinhardt, A. Cytokines and the immune-testicular axis. J. Reprod. Immunol. 2003, 58, 1–26. [Google Scholar] [CrossRef]
- Li, H.; Xiao, X.; Zhang, J.; Zafar, M.I.; Wu, C.; Long, Y.; Lu, W.; Pan, F.; Meng, T.; Zhao, K.; et al. Impaired spermatogenesis in COVID-19 patients. EClinicalMedicine 2020, 28, 100604. [Google Scholar] [CrossRef]
- Carlsen, E. History of febrile illness and variation in semen quality. Hum. Reprod. 2003, 18, 2089–2092. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Schuppe, H.-C.; Schill, W.-B. Fieber als Ursache einer temporären Fertilitätseinschränkung des Mannes. Hautarzt 2001, 52, 1090–1093. [Google Scholar] [CrossRef]
- Salonia, A.; Pontillo, M.; Capogrosso, P.; Gregori, S.; Carenzi, C.; Ferrara, A.M.; Rowe, I.; Boeri, L.; Larcher, A.; Ramirez, G.A.; et al. Testosterone in males with COVID-19: A 7-month cohort study. Andrology 2021, 10, 34–41. [Google Scholar] [CrossRef]
- Kadihasanoglu, M.; Aktas, S.; Yardimci, E.; Aral, H.; Kadioglu, A. SARS-CoV-2 Pneumonia Affects Male Reproductive Hormone Levels: A Prospective, Cohort Study. J. Sex. Med. 2021, 18, 256–264. [Google Scholar] [CrossRef]
- Higgins, V.; Sohaei, D.; Diamandis, E.P.; Prassas, I. COVID-19: From an acute to chronic disease? Potential long-term health consequences. Crit. Rev. Clin. Lab. Sci. 2021, 58, 297–310. [Google Scholar] [CrossRef]
- Callard, F.; Perego, E. How and why patients made Long Covid. Soc. Sci. Med. 2021, 268, 113426. [Google Scholar] [CrossRef]
- Mendelson, M.; Nel, J.; Blumberg, L.; Madhi, S.A.; Dryden, M.; Stevens, W.; Venter, F.W.D. Long-COVID: An evolving problem with an extensive impact. S. Afr. Med. J. 2020, 111, 10. [Google Scholar] [CrossRef]
- Bermejo-Martin, J.F.; González-Rivera, M.; Almansa, R.; Micheloud, D.; Tedim, A.P.; Domínguez-Gil, M.; Resino, S.; Martín-Fernández, M.; Ryan Murua, P.; Pérez-García, F.; et al. Viral RNA load in plasma is associated with critical illness and a dysregulated host response in COVID-19. Crit. Care 2020, 24, 691. [Google Scholar] [CrossRef]
- Bermejo-Martin, J.F.; Almansa, R.; Tedim, A.P.; De La Fuente, A.; Eiros, J.M.; Torres, A.; Kelvin, D.J. Mounting evidence of impaired viral control in severe COVID-19. Lancet Microbe 2021, 2, e228–e229. [Google Scholar] [CrossRef]
- Rastrelli, G.; Di Stasi, V.; Inglese, F.; Beccaria, M.; Garuti, M.; Di Costanzo, D.; Spreafico, F.; Greco, G.F.; Cervi, G.; Pecoriello, A.; et al. Low testosterone levels predict clinical adverse outcomes in SARS-CoV-2 pneumonia patients. Andrology 2021, 9, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Herald, A.K.; Alves-Lopes, R.; Montezano, A.C.; Ahmed, S.F.; Touyz, R.M. Genomic and non-genomic effects of androgens in the cardiovascular system: Clinical implications. Clin. Sci. 2017, 131, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Markle, J.G.; Fish, E.N. SeXX matters in immunity. Trends Immunol. 2014, 35, 97–104. [Google Scholar] [CrossRef]
- Kjellman, B.; Gustafsson, P.M. Asthma from childhood to adulthood: Asthma severity, allergies, sensitization, living conditions, gender influence and social consequences. Respir. Med. 2000, 94, 454–465. [Google Scholar] [CrossRef] [Green Version]
- De Marco, R.; Locatelli, F.; Sunyer, J.; Burney, P. Differences in Incidence of Reported Asthma Related to Age in Men and Women. Am. J. Respir. Crit. Care Med. 2000, 162, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.; Montaño, L.M.; Perusquía, M. Nongenomic bronchodilating action elicited by dehydroepiandrosterone (DHEA) in a guinea pig asthma model. J. Steroid Biochem. Mol. Biol. 2013, 138, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Carbajal-García, A.; Reyes-García, J.; Casas-Hernández, M.F.; Flores-Soto, E.; Díaz-Hernández, V.; Solís-Chagoyán, H.; Sommer, B.; Montaño, L.M. Testosterone augments β2 adrenergic receptor genomic transcription increasing salbutamol relaxation in airway smooth muscle. Mol. Cell. Endocrinol. 2020, 510, 110801. [Google Scholar] [CrossRef]
- Laffont, S.; Blanquart, E.; Guéry, J.C. Sex Differences in Asthma: A Key Role of Androgen-Signaling in Group 2 Innate Lymphoid Cells. Front. Immunol. 2017, 8, 1069. [Google Scholar] [CrossRef]
- Fuseini, H.; Yung, J.A.; Cephus, J.Y.; Zhang, J.; Goleniewska, K.; Polosukhin, V.V.; Peebles, R.S.; Newcomb, D.C. Testosterone Decreases House Dust Mite–Induced Type 2 and IL-17A–Mediated Airway Inflammation. J. Immunol. 2018, 201, 1843–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalidhindi, R.; Katragadda, R.; Beauchamp, K.L.; Pabelick, C.M.; Prakash, Y.S.; Sathish, V. Androgen Receptor-Mediated Regulation of Intracellular Calcium in Human Airway Smooth Muscle Cells. Cell. Physiol. Biochem. 2019, 53, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Kalidhindi, R.S.R.; Borkar, N.A.; Ambhore, N.S.; Pabelick, C.M.; Prakash, Y.S.; Sathish, V. Sex steroids skew ACE2 expression in human airway: A contributing factor to sex differences in COVID-19? Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 319, L843–L847. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; de Marañon, A.M.; Diaz-Morales, N.; Jover, A.; Garzon, S.; Rocha, M.; Victor, V.M.; Hernandez-Mijares, A. Low testosterone levels are related to oxidative stress, mitochondrial dysfunction and altered subclinical atherosclerotic markers in type 2 diabetic male patients. Free Radic. Biol. Med. 2017, 108, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Grandys, M.; Majerczak, J.; Zapart-Bukowska, J.; Duda, K.; Kulpa, J.K.; Zoladz, J.A. Lowered Serum Testosterone Concentration Is Associated With Enhanced Inflammation and Worsened Lipid Profile in Men. Front. Endocrinol. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Golden, K.L.; Marsh, J.D.; Jiang, Y. Castration Reduces mRNA Levels for Calcium Regulatory Proteins in Rat Heart. Endocrine 2002, 19, 339–344. [Google Scholar] [CrossRef]
- Golden, K.L.; Marsh, J.D.; Jiang, Y.; Brown, T.; Moulden, J. Gonadectomy of adult male rats reduces contractility of isolated cardiac myocytes. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E449–E453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayaz, O.; Howlett, S.E. Testosterone modulates cardiac contraction and calcium homeostasis: Cellular and molecular mechanisms. Biol. Sex Differ. 2015, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Er, F.; Gassanov, N.; Brandt, M.C.; Madershahian, N.; Hoppe, U.C. Impact of dihydrotestosterone on L-type calcium channels in human ventricular cardiomyocytes. Endocr. Res. 2009, 34, 59–67. [Google Scholar] [CrossRef]
- Callies, F.; Strömer, H.; Schwinger, R.H.; Bölck, B.; Hu, K.; Frantz, S.; Leupold, A.; Beer, S.; Allolio, B.; Bonz, A.W. Administration of testosterone is associated with a reduced susceptibility to myocardial ischemia. Endocrinology 2003, 144, 4478–4483. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.; Wong, S.S.; Wu, S.; Kravtsov, G.M.; Wong, T.M. Testosterone-augmented contractile responses to alpha1- and beta1-adrenoceptor stimulation are associated with increased activities of RyR, SERCA, and NCX in the heart. Am. J. Physiol. Cell Physiol. 2009, 296, C766–C782. [Google Scholar] [CrossRef]
- Weerateerangkul, P.; Shinlapawittayatorn, K.; Palee, S.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Early testosterone replacement attenuates intracellular calcium dyshomeostasis in the heart of testosterone-deprived male rats. Cell Calcium 2017, 67, 22–30. [Google Scholar] [CrossRef]
- Murphy, J.G.; Marsh, J.D.; Smith, T.W. The role of calcium in ischemic myocardial injury. Circulation 1987, 75, V15–V24. [Google Scholar] [PubMed]
- Vittone, L.; Mundiña-Weilenmann, C.; Said, M.; Ferrero, P.; Mattiazzi, A. Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts. Dissociation of phospholamban phosphorylation pathways. J. Mol. Cell. Cardiol. 2002, 34, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Sebag, I.A.; Gillis, M.A.; Calderone, A.; Kasneci, A.; Meilleur, M.; Haddad, R.; Noiles, W.; Patel, B.; Chalifour, L.E. Sex hormone control of left ventricular structure/function: Mechanistic insights using echocardiography, expression, and DNA methylation analyses in adult mice. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1706–H1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witayavanitkul, N.; Woranush, W.; Bupha-Intr, T.; Wattanapermpool, J. Testosterone regulates cardiac contractile activation by modulating SERCA but not NCX activity. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H465–H472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.A.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Z.; Tse, G.; Zhang, L.; Wan, E.Y.; Guo, Y.; Lip, G.Y.H.; Li, G.; Lu, Z.; Liu, T. Cardiac arrhythmias in patients with COVID-19. J. Arrhythmia 2020, 36, 827–836. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Lake, M.A. What we know so far: COVID-19 current clinical knowledge and research. Clin. Med. 2020, 20, 124–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, S.; Jiang, F.; Su, W.; Chen, C.; Chen, J.; Mei, W.; Zhan, L.-Y.; Jia, Y.; Zhang, L.; Liu, D.; et al. Clinical characteristics and outcomes of patients undergoing surgeries during the incubation period of COVID-19 infection. EClinicalMedicine 2020, 21, 100331. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Tu, L.; Zhu, P.; Mu, M.; Wang, R.; Yang, P.; Wang, X.; Hu, C.; Ping, R.; Hu, P.; et al. Clinical Features of 85 Fatal Cases of COVID-19 from Wuhan. A Retrospective Observational Study. Am. J. Respir. Crit. Care Med. 2020, 201, 1372–1379. [Google Scholar] [CrossRef] [Green Version]
- Long, B.; Brady, W.J.; Bridwell, R.E.; Ramzy, M.; Montrief, T.; Singh, M.; Gottlieb, M. Electrocardiographic manifestations of COVID-19. Am. J. Emerg. Med. 2021, 41, 96–103. [Google Scholar] [CrossRef]
- Atri, D.; Siddiqi, H.K.; Lang, J.P.; Nauffal, V.; Morrow, D.A.; Bohula, E.A. COVID-19 for the Cardiologist: Basic Virology, Epidemiology, Cardiac Manifestations, and Potential Therapeutic Strategies. JACC Basic Transl. Sci. 2020, 5, 518–536. [Google Scholar] [CrossRef]
- Chen, X.; Li, R.; Pan, Z.; Qian, C.; Yang, Y.; You, R.; Zhao, J.; Liu, P.; Gao, L.; Li, Z.; et al. Human monoclonal antibodies block the binding of SARS-CoV-2 spike protein to angiotensin converting enzyme 2 receptor. Cell. Mol. Immunol. 2020, 17, 647–649. [Google Scholar] [CrossRef]
- D’Aloia, A.; Faggiano, P.; Brentana, L.; Boldini, A.; Curnis, A.; Bontempi, L.; Dei Cas, L. Recurrent ventricular fibrillation during a febrile illness and hyperthermia in a patient with dilated cardiomyopathy and automatic implantable cardioverter defibrillator. An example of reversible electrical storm. Int. J. Cardiol. 2005, 103, 207–208. [Google Scholar] [CrossRef]
- Dinckal, M. Incessant monomorphic ventricular tachycardia during febrile illness in a patient with Brugada syndrome: Fatal electrical storm. Europace 2003, 5, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Vonderlin, N.; Siebermair, J.; Kaya, E.; Köhler, M.; Rassaf, T.; Wakili, R. Critical inflammatory mechanisms underlying arrhythmias. Herz 2019, 44, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Schett, G.; Sticherling, M.; Neurath, M.F. COVID-19: Risk for cytokine targeting in chronic inflammatory diseases? Nat. Rev. Immunol. 2020, 20, 271–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19). JAMA 2020, 324, 782. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Miyoshi, S.; Fukuda, K.; Nishiyama, N.; Ikegami, Y.; Tanimoto, K.; Murata, M.; Takahashi, E.; Shimoda, K.; Hirano, T.; et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J. Mol. Cell. Cardiol. 2007, 43, 710–716. [Google Scholar] [CrossRef]
- Alí, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, Inflammation, and Arrhythmias: Role for Interleukin-6 Molecular Mechanisms. Front. Physiol. 2019, 9, 1866. [Google Scholar] [CrossRef]
- Kao, Y.H.; Chen, Y.C.; Cheng, C.C.; Lee, T.I.; Chen, Y.J.; Chen, S.A. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit. Care Med. 2010, 38, 217–222. [Google Scholar] [CrossRef]
- Monnerat, G.; Alarcón, M.L.; Vasconcellos, L.R.; Hochman-Mendez, C.; Brasil, G.; Bassani, R.A.; Casis, O.; Malan, D.; Travassos, L.H.; Sepúlveda, M.; et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016, 7, 13344. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.M.; Jones, T.H. Testosterone: A vascular hormone in health and disease. J. Endocrinol. 2013, 217, R47–R71. [Google Scholar] [CrossRef] [Green Version]
- Pugh, P. Acute haemodynamic effects of testosterone in men with chronic heart failure. Eur. Heart J. 2003, 24, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Malkin, C.J.; Morris, P.D.; Pugh, P.J.; English, K.M.; Channer, K.S. Effect of testosterone therapy on QT dispersion in men with heart failure. Am. J. Cardiol. 2003, 92, 1241–1243. [Google Scholar] [CrossRef]
- Schwartz, J.B.; Volterrani, M.; Caminiti, G.; Marazzi, G.; Fini, M.; Rosano, G.M.C.; Iellamo, F. Effects of testosterone on the Q-T Interval in older men and older women with chronic heart failure. Int. J. Androl. 2011, 34, e415–e421. [Google Scholar] [CrossRef]
- Townsend, E.A.; Miller, V.M.; Prakash, Y.S. Sex Differences and Sex Steroids in Lung Health and Disease. Endocr. Rev. 2012, 33, 1–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarotsky, V.; Huang, M.-Y.; Carman, W.; Morgentaler, A.; Singhal, P.K.; Coffin, D.; Jones, T.H. Systematic literature review of the risk factors, comorbidities, and consequences of hypogonadism in men. Andrology 2014, 2, 819–834. [Google Scholar] [CrossRef]
- Jones, T.H.; Kelly, D.M. Randomized controlled trials—Mechanistic studies of testosterone and the cardiovascular system. Asian J. Androl. 2018, 20, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Chatterjee, K.; Beale, C.; Poole-Wilson, P.A.; Collins, P. Testosterone relaxes rabbit coronary arteries and aorta. Circulation 1995, 91, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Tep-Areenan, P.; Kendall, D.A.; Randall, M.D. Testosterone-induced vasorelaxation in the rat mesenteric arterial bed is mediated predominantly via potassium channels. Br. J. Pharmacol. 2002, 135, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.M.; Sudhir, K.; Hutchison, S.J.; Ko, E.; Amidon, T.M.; Collins, P.; Chatterjee, K. Testosterone induces dilation of canine coronary conductance and resistance arteries in vivo. Circulation 1996, 94, 2614–2619. [Google Scholar] [CrossRef]
- Yildiz, O.; Seyrek, M.; Gul, H.; Un, I.; Yildirim, V.; Ozal, E.; Uzun, M.; Bolu, E. Testosterone relaxes human internal mammary artery in vitro. J. Cardiovasc. Pharmacol. 2005, 45, 580–585. [Google Scholar] [CrossRef]
- Murphy, J.G.; Khalil, R.A. Decreased [Ca2+]i during inhibition of coronary smooth muscle contraction by 17β-estradiol, progesterone, and testosterone. J. Pharmacol. Exp. Ther. 1999, 291, 44–52. [Google Scholar]
- English, K.M.; Jones, R.D.; Jones, T.H.; Morice, A.H.; Channer, K.S. Aging reduces the responsiveness of coronary arteries from male Wistar rats to the vasodilatory action of testosterone. Clin. Sci. 2000, 99, 77–82. [Google Scholar] [CrossRef]
- English, K.M.; Jones, R.D.; Jones, T.H.; Morice, A.H.; Channer, K.S. Testosterone acts as a coronary vasodilator by a calcium antagonistic action. J. Endocrinol. Investig. 2002, 25, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Perusquía, M.; Hernández, R.; Morales, M.A.; Campos, M.G.; Villalón, C.M. Role of endothelium in the vasodilating effect of progestins and androgens on the rat thoracic aorta. Gen. Pharmacol. 1996, 27, 181–185. [Google Scholar] [CrossRef]
- Honda, H.; Unemoto, T.; Kogo, H. Different Mechanisms for Testosterone-Induced Relaxation of Aorta Between Normotensive and Spontaneously Hypertensive Rats. Hypertension 1999, 34, 1232–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, A.Q.; Stallone, J.N. Testosterone-induced relaxation of rat aorta is androgen structure specific and involves K+ channel activation. J. Appl. Physiol. 2001, 91, 2742–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.D.; English, K.M.; Pugh, P.J.; Morice, A.H.; Jones, T.H.; Channer, K.S. Pulmonary vasodilatory action of testosterone: Evidence of a calcium antagonistic action. J. Cardiovasc. Pharmacol. 2002, 39, 814–823. [Google Scholar] [CrossRef]
- Rowell, K.O.; Hall, J.; Pugh, P.J.; Jones, T.H.; Channer, K.S.; Jones, R.D. Testosterone acts as an efficacious vasodilator in isolated human pulmonary arteries and veins: Evidence for a biphasic effect at physiological and supra-physiological concentrations. J. Endocrinol. Investig. 2009, 32, 718–723. [Google Scholar] [CrossRef]
- Seyrek, M.; Yildiz, O.; Ulusoy, H.B.; Yildirim, V. Testosterone Relaxes Isolated Human Radial Artery by Potassium Channel Opening Action. J. Pharmacol. Sci. 2007, 103, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairrão, E.; Álvarez, E.; Santos-Silva, A.J.; Verde, I. Potassium channels are involved in testosterone-induced vasorelaxation of human umbilical artery. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008, 376, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Cairrão, E.; Santos-Silva, A.J.; Verde, I. PKG is involved in testosterone-induced vasorelaxation of human umbilical artery. Eur. J. Pharmacol. 2010, 640, 94–101. [Google Scholar] [CrossRef]
- Han, D.H.; Chae, M.R.; Jung, J.H.; So, I.; Park, J.K.; Lee, S.W. Effect of testosterone on potassium channel opening in human corporal smooth muscle cells. J. Sex. Med. 2008, 5, 822–832. [Google Scholar] [CrossRef]
- Crews, J.K.; Khalil, R.A. Gender-specific inhibition of Ca2+ entry mechanisms of arterial vasoconstriction by sex hormones. Clin. Exp. Pharmacol. Physiol. 1999, 26, 707–715. [Google Scholar] [CrossRef]
- Hu, Z.; Ma, R.; Gong, J. Investigation of testosterone-mediated non-transcriptional inhibition of Ca2+ in vascular smooth muscle cells. Biomed. Rep. 2016, 4, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Jones, R.D.; Jones, T.H.; Channer, K.S.; Peers, C. Selective Inhibition of L-Type Ca2+ Channels in A7r5 Cells by Physiological Levels of Testosterone. Endocrinology 2006, 147, 2675–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perusquía, M.; Herrera, N.; Ferrer, M.; Stallone, J.N. Antihypertensive effects of androgens in conscious, spontaneously hypertensive rats. J. Steroid Biochem. Mol. Biol. 2017, 167, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Crews, J.K.; Khalil, R.A. Antagonistic Effects of 17β-Estradiol, Progesterone, and Testosterone on Ca2+ Entry Mechanisms of Coronary Vasoconstriction. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1034–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.D.; English, K.M.; Jones, T.H.; Channer, K.S. Testosterone-induced coronary vasodilatation occurs via a non-genomic mechanism: Evidence of a direct calcium antagonism action. Clin. Sci. 2004, 107, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Rosas, M.B.; Cobos-Puc, L.E.; Muñoz-Islas, E.; González-Hernández, A.; Sánchez-López, A.; Villalón, C.M.; Maassenvandenbrink, A.; Centurión, D. Pharmacological evidence that Ca2+ channels and, to a lesser extent, K+ channels mediate the relaxation of testosterone in the canine basilar artery. Steroids 2011, 76, 409–415. [Google Scholar] [CrossRef]
- Perusquía, M.; Navarrete, E.; González, L.; Villalón, C.M. The modulatory role of androgens and progestins in the induction of vasorelaxation in human umbilical artery. Life Sci. 2007, 81, 993–1002. [Google Scholar] [CrossRef]
- Navarro-Dorado, J.; Orensanz, L.M.; Recio, P.; Bustamante, S.; Benedito, S.; Martínez, A.C.; García-Sacristán, A.; Prieto, D.; Hernández, M. Mechanisms involved in testosterone-induced vasodilatation in pig prostatic small arteries. Life Sci. 2008, 83, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, P.A.; Cairrão, E.; Maia, C.J.; Verde, I. Long- and short-term effects of androgens in human umbilical artery smooth muscle. Clin. Exp. Pharmacol. Physiol. 2013, 40, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Stanetić, K.; Stanetić, B.; Petrović, V.; Marković, B.; Kević, V.; Todorović, N.; Stanetić, M. The Influence of Different Risk Factors on COVID-19 Outcomes in Adult Patients—An Observational-Descriptive Study. Acta Med. Acad. 2021, 50, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary Angiotensin-Converting Enzyme 2 in Hypertensive Patients May Be Increased by Olmesartan, an Angiotensin II Receptor Blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, L.M.M.; Flores-Soto, E. COVID-19 y su asociación con los inhibidores de la enzima convertidora de angiotensina y los antagonistas de los receptores para angiotensina II. Rev. Fac. Med. UNAM 2020, 63, 30–34. [Google Scholar]
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of “inflame-aging”. Inflamm. Res. 2020, 69, 825–839. [Google Scholar] [CrossRef]
- Eshak, N.; Abdelnabi, M.; Beltagy, A. Inflamm-aging: The missing link to COVID-19 age-related mortality? Southwest Respir. Crit. Care Chron. 2020, 8, 66–67. [Google Scholar] [CrossRef]
- Stout, M.B.; Justice, J.N.; Nicklas, B.J.; Kirkland, J.L. Physiological Aging: Links Among Adipose Tissue Dysfunction, Diabetes, and Frailty. Physiology 2017, 32, 9–19. [Google Scholar] [CrossRef]
- Golan, R.; Scovell, J.M.; Ramasamy, R. Age-related testosterone decline is due to waning of both testicular and hypothalamic-pituitary function. Aging Male 2015, 18, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Dabaja, A.A.; Bryson, C.F.; Schlegel, P.N.; Paduch, D.A. The effect of hypogonadism and testosterone-enhancing therapy on alkaline phosphatase and bone mineral density. BJU Int. 2015, 115, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Darbandi, M.; Darbandi, S.; Agarwal, A.; Sengupta, P.; Durairajanayagam, D.; Henkel, R.; Sadeghi, M.R. Reactive oxygen species and male reproductive hormones. Reprod. Biol. Endocrinol. 2018, 16, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiers, J.G.; Chen, H.J.; Sernia, C.; Lavidis, N.A. Activation of the hypothalamic-pituitary-adrenal stress axis induces cellular oxidative stress. Front. Neurosci. 2015, 8, 456. [Google Scholar] [CrossRef] [Green Version]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Hardy, M.P.; Gao, H.-B.; Dong, Q.; Ge, R.; Wang, Q.; Chai, W.R.; Feng, X.; Sottas, C. Stress hormone and male reproductive function. Cell Tissue Res. 2005, 322, 147–153. [Google Scholar] [CrossRef]
- Hwang, T.I.; Liao, T.-L.; Lin, J.-F.; Lin, Y.-C.; Lee, S.-Y.; Lai, Y.-C.; Kao, S.-H. Low-dose testosterone treatment decreases oxidative damage in TM3 Leydig cells. Asian J. Androl. 2011, 13, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintana, H.; Pongkan, W.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Testosterone replacement attenuates cognitive decline in testosterone-deprived lean rats, but not in obese rats, by mitigating brain oxidative stress. Age 2015, 37, 84. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Li, G.; Liu, C.; Gao, H.; Wang, H.; Liu, W.; Chen, M.; Shang, Y.; Wang, L.; Shi, J.; et al. Autophagy regulates testosterone synthesis by facilitating cholesterol uptake in Leydig cells. J. Cell Biol. 2018, 217, 2103–2119. [Google Scholar] [CrossRef] [Green Version]
- Kahn, B.E.; Brannigan, R.E. Obesity and male infertility. Curr. Opin. Urol. 2017, 27, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ding, Z. Obesity, a serious etiologic factor for male subfertility in modern society. Reproduction 2017, 154, R123–R131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Pugh, T.D.; Stebler, B.; Ershler, W.B.; Keller, E.T. Orchiectomy Increases Bone Marrow Interleukin-6 Levels in Mice. Calcif. Tissue Int. 1998, 62, 219–226. [Google Scholar] [CrossRef]
- Freeman, B.M.; Mountain, D.J.; Brock, T.C.; Chapman, J.R.; Kirkpatrick, S.S.; Freeman, M.B.; Klein, F.A.; Grandas, O.H. Low testosterone elevates interleukin family cytokines in a rodent model: A possible mechanism for the potentiation of vascular disease in androgen-deficient males. J. Surg. Res. 2014, 190, 319–327. [Google Scholar] [CrossRef]
- Malkin, C.J.; Pugh, P.J.; Jones, R.D.; Kapoor, D.; Channer, K.S.; Jones, T.H. The Effect of Testosterone Replacement on Endogenous Inflammatory Cytokines and Lipid Profiles in Hypogonadal Men. J. Clin. Endocrinol. Metab. 2004, 89, 3313–3318. [Google Scholar] [CrossRef] [Green Version]
- Fijak, M.; Schneider, E.; Klug, J.; Bhushan, S.; Hackstein, H.; Schuler, G.; Wygrecka, M.; Gromoll, J.; Meinhardt, A. Testosterone Replacement Effectively Inhibits the Development of Experimental Autoimmune Orchitis in Rats: Evidence for a Direct Role of Testosterone on Regulatory T Cell Expansion. J. Immunol. 2011, 186, 5162–5172. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; Von Schonfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.-C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Raisky, J.; Ratliff, S.M.; Liu, J.; Kardia, S.L.R.; Turner, S.T.; Mosley, T.H.; Zhao, W. Intrinsic and extrinsic epigenetic age acceleration are associated with hypertensive target organ damage in older African Americans. BMC Med. Genom. 2019, 12, 141. [Google Scholar] [CrossRef]
- Joyce, B.T.; Gao, T.; Zheng, Y.; Ma, J.; Hwang, S.-J.; Liu, L.; Nannini, D.; Horvath, S.; Lu, A.T.; Bai Allen, N.; et al. Epigenetic Age Acceleration Reflects Long-Term Cardiovascular Health. Circ. Res. 2021, 129, 770–781. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Tignanelli, C.; Kuchel, G.A.; Melzer, D.; Beckman, K.B.; Levine, M.E. Biological Aging Predicts Vulnerability to COVID-19 Severity in UK Biobank Participants. J. Gerontol. Ser. A 2021, 76, e133–e141. [Google Scholar] [CrossRef]
- Berezina, T.N.; Rybtsov, S. Acceleration of Biological Aging and Underestimation of Subjective Age Are Risk Factors for Severe COVID-19. Biomedicines 2021, 9, 913. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montaño, L.M.; Sommer, B.; Solís-Chagoyán, H.; Romero-Martínez, B.S.; Aquino-Gálvez, A.; Gomez-Verjan, J.C.; Calixto, E.; González-Avila, G.; Flores-Soto, E. Could Lower Testosterone in Older Men Explain Higher COVID-19 Morbidity and Mortalities? Int. J. Mol. Sci. 2022, 23, 935. https://doi.org/10.3390/ijms23020935
Montaño LM, Sommer B, Solís-Chagoyán H, Romero-Martínez BS, Aquino-Gálvez A, Gomez-Verjan JC, Calixto E, González-Avila G, Flores-Soto E. Could Lower Testosterone in Older Men Explain Higher COVID-19 Morbidity and Mortalities? International Journal of Molecular Sciences. 2022; 23(2):935. https://doi.org/10.3390/ijms23020935
Chicago/Turabian StyleMontaño, Luis M., Bettina Sommer, Héctor Solís-Chagoyán, Bianca S. Romero-Martínez, Arnoldo Aquino-Gálvez, Juan C. Gomez-Verjan, Eduardo Calixto, Georgina González-Avila, and Edgar Flores-Soto. 2022. "Could Lower Testosterone in Older Men Explain Higher COVID-19 Morbidity and Mortalities?" International Journal of Molecular Sciences 23, no. 2: 935. https://doi.org/10.3390/ijms23020935