
The Role of Sphingolipid Signaling in Oxidative Lung Injury and Pathogenesis of Bronchopulmonary Dysplasia

 and
and
Abstract
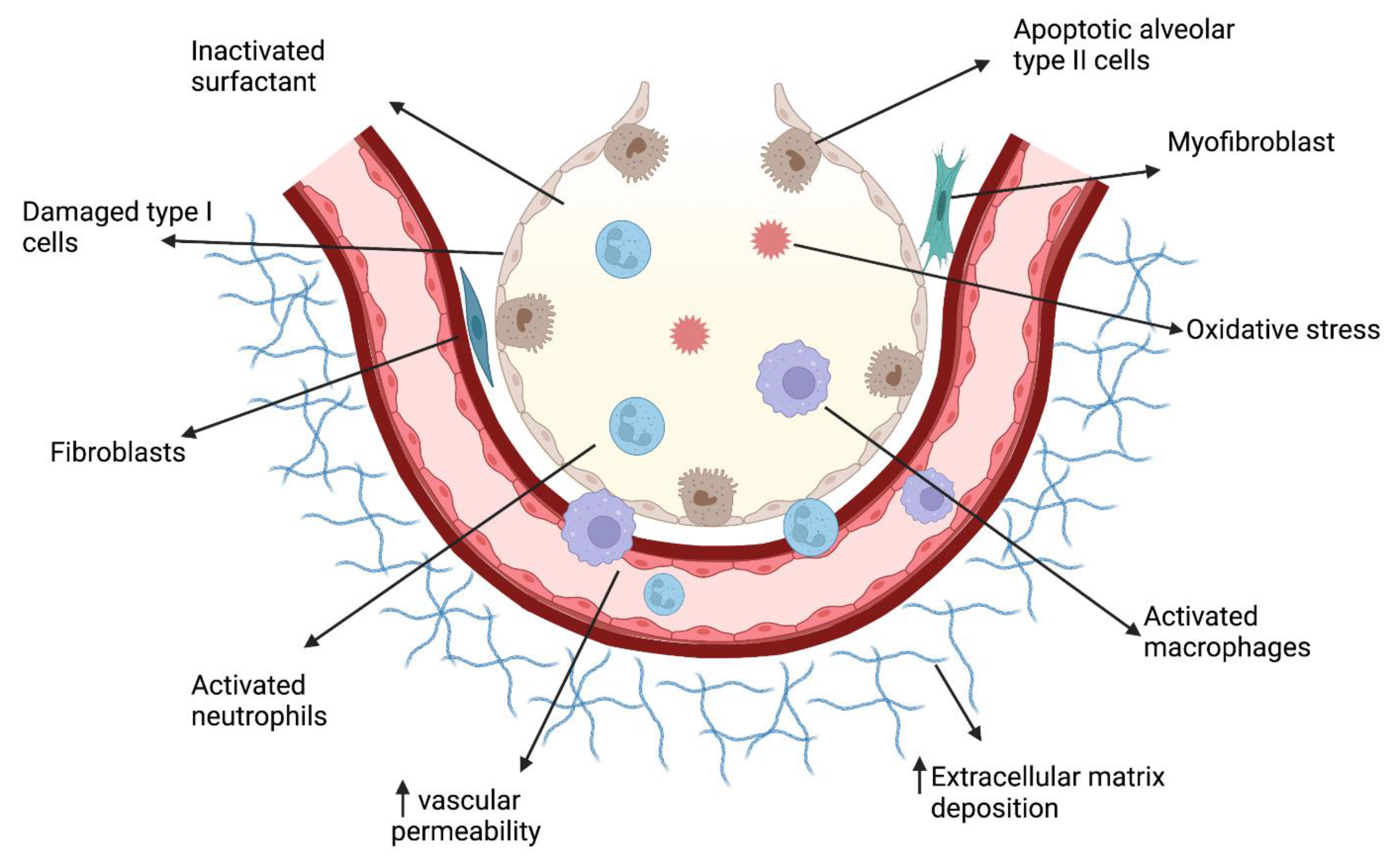
:1. Introduction
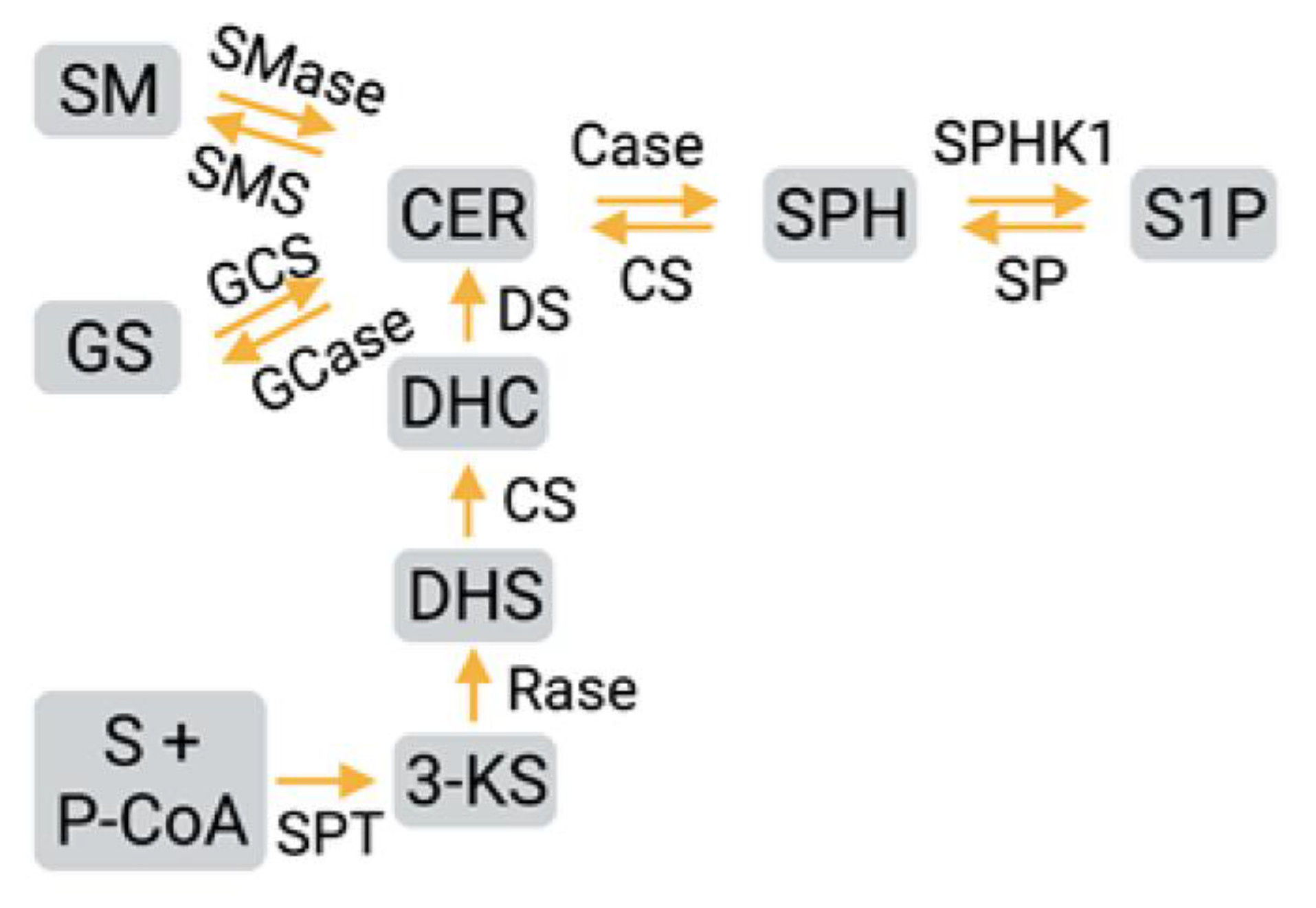
2. Sphingosine-1-phosphate Synthesis, Metabolism, and Signaling
3. Sphingolipids in Diseases
4. Ceramide, SphK1/S1P/S1PR1 Signaling Axis in BPD Pathogenesis
5. Reactive Oxygen Species and Antioxidant Defense Mechanisms
6. Oxidative Stress and Mitochondrial Injury
7. Mitochondrial Oxidative Stress in BPD
8. Mitochondrial Organelle Dynamics in BPD
9. Mitochondrial Biogenesis in BPD
10. Cellular Respiration in BPD
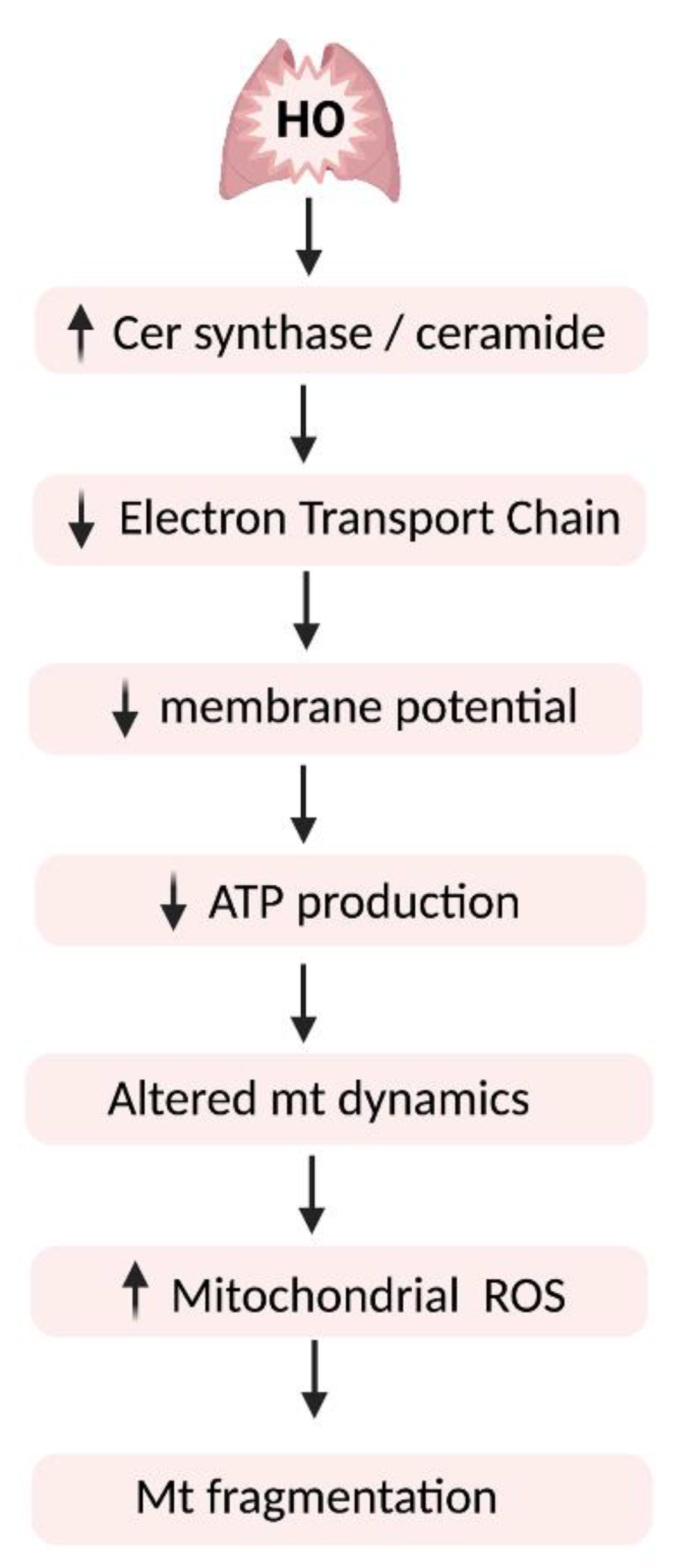
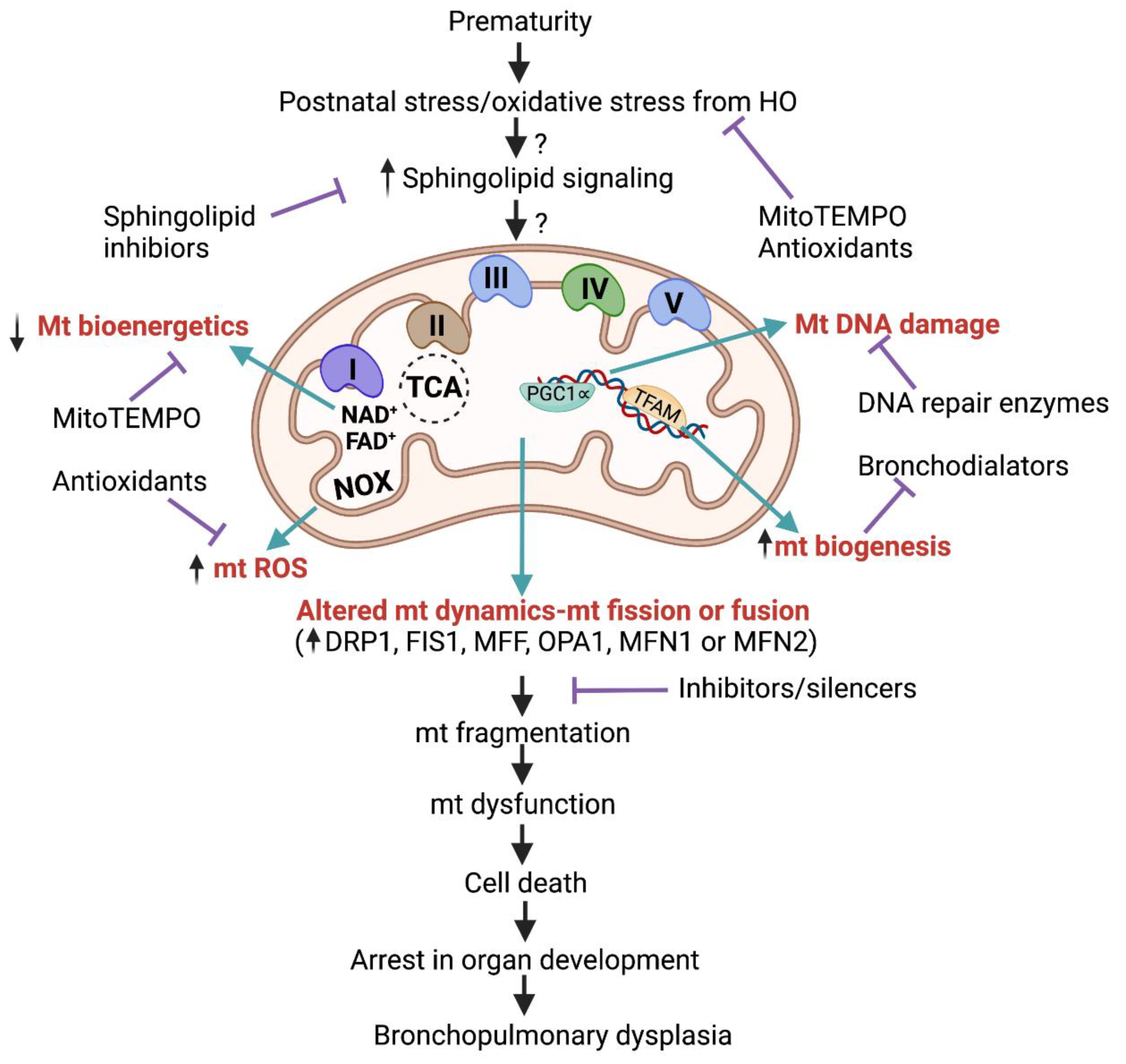
11. Sphingolipid Signaling in Hyperoxic Mitochondrial Injury
12. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Northway, W.H.; Rosan, R.C.; Porter, D.Y. Pulmonary Disease Following Respirator Therapy of Hyaline-Membrane Disease: Bronchopulmonary Dysplasia. N. Engl. J. Med. 1967, 276, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Cuevas Guaman, M.; Dahm, P.H.; Welty, S.E. The Challenge of Accurately Describing the Epidemiology of Bronchopulmonary Dysplasia (BPD) Based on the Various Current Definitions of BPD. Pediatr. Pulmonol. 2021, 56, 3527–3532. [Google Scholar] [CrossRef] [PubMed]
- Shennan, A.T.; Dunn, M.S.; Ohlsson, A.; Lennox, K.; Hoskins, E.M. Abnormal Pulmonary Outcomes in Premature Infants: Prediction from Oxygen Requirement in the Neonatal Period. Pediatrics 1988, 82, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.J. The New BPD: An Arrest of Lung Development. Pediatr. Res. 1999, 46, 641–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, M.; Turgut, M.; Taskin, E.; Cekmen, M.; Aygun, A.D. Nitric Oxide Levels and Antioxidant Enzyme Activities in Jaundices of Premature Infants. Cell Biochem. Funct. 2004, 22, 339–342. [Google Scholar] [CrossRef]
- Björklund, L.J.; Ingimarsson, J.; Curstedt, T.; John, J.; Robertson, B.; Werner, O.; Vilstrup, C.T. Manual Ventilation with a Few Large Breaths at Birth Compromises the Therapeutic Effect of Subsequent Surfactant Replacement in Immature Lambs. Pediatr. Res. 1997, 42, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Bancalari, E. Bronchopulmonary Dysplasia: Clinical Perspective. Birth Defects Res. A Clin. Mol. Teratol. 2014, 100, 134–144. [Google Scholar] [CrossRef]
- Bamat, N.A.; Zhang, H.; McKenna, K.J.; Morris, H.; Stoller, J.Z.; Gibbs, K. The Clinical Evaluation of Severe Bronchopulmonary Dysplasia. Neoreviews 2020, 21, e442–e453. [Google Scholar] [CrossRef]
- McEvoy, C.T.; Jain, L.; Schmidt, B.; Abman, S.; Bancalari, E.; Aschner, J.L. Bronchopulmonary Dysplasia: NHLBI Workshop on the Primary Prevention of Chronic Lung Diseases. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 3), S146–S153. [Google Scholar] [CrossRef] [Green Version]
- Ambalavanan, N.; Cotten, C.M.; Page, G.P.; Carlo, W.A.; Murray, J.C.; Bhattacharya, S.; Mariani, T.J.; Cuna, A.C.; Faye-Petersen, O.M.; Kelly, D.; et al. Integrated Genomic Analyses in Bronchopulmonary Dysplasia. J. Pediatr. 2015, 166, 531–537. [Google Scholar] [CrossRef] [Green Version]
- De Paepe, M.E.; Greco, D.; Mao, Q. Angiogenesis-Related Gene Expression Profiling in Ventilated Preterm Human Lungs. Exp. Lung Res. 2010, 36, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, K.; Shibata, A.; Yokota, T.; Koda, T.; Nagasaka, M.; Yagi, M.; Takeshima, Y.; Yamada, H.; Iijima, K.; Morioka, I. Association of a Vascular Endothelial Growth Factor Polymorphism with the Development of Bronchopulmonary Dysplasia in Japanese Premature Newborns. Sci. Rep. 2014, 4, 4459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, P.M.; Pham, C.; Jang, K.L. Heritability of Bronchopulmonary Dysplasia, Defined According to the Consensus Statement of the National Institutes of Health. Pediatrics 2008, 122, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughon, M.M.; Langer, J.C.; Bose, C.L.; Smith, P.B.; Ambalavanan, N.; Kennedy, K.A.; Stoll, B.J.; Buchter, S.; Laptook, A.R.; Ehrenkranz, R.A.; et al. Prediction of Bronchopulmonary Dysplasia by Postnatal Age in Extremely Premature Infants. Am. J. Respir. Crit. Care Med. 2011, 183, 1715–1722. [Google Scholar] [CrossRef]
- Ito, M.; Tamura, M.; Namba, F. Neonatal Research Network of Japan Role of Sex in Morbidity and Mortality of Very Premature Neonates. Pediatr. Int. 2017, 59, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, K.K.; Dagle, J.M.; Kelsey, K.; Momany, A.M.; Murray, J.C. Genetic Associations of Surfactant Protein D and Angiotensin-Converting Enzyme with Lung Disease in Preterm Neonates. J. Perinatol. 2012, 32, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Hadchouel, A.; Durrmeyer, X.; Bouzigon, E.; Incitti, R.; Huusko, J.; Jarreau, P.-H.; Lenclen, R.; Demenais, F.; Franco-Montoya, M.-L.; Layouni, I.; et al. Identification of SPOCK2 as a Susceptibility Gene for Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2011, 184, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Watterberg, K.L.; Demers, L.M.; Scott, S.M.; Murphy, S. Chorioamnionitis and Early Lung Inflammation in Infants in Whom Bronchopulmonary Dysplasia Develops. Pediatrics 1996, 97, 210–215. [Google Scholar] [CrossRef]
- Kramer, B.W. Antenatal Inflammation and Lung Injury: Prenatal Origin of Neonatal Disease. J. Perinatol. 2008, 28 (Suppl. 1), S21–S27. [Google Scholar] [CrossRef]
- Ballard, A.R.; Mallett, L.H.; Pruszynski, J.E.; Cantey, J.B. Chorioamnionitis and Subsequent Bronchopulmonary Dysplasia in Very-Low-Birth Weight Infants: A 25-Year Cohort. J. Perinatol. 2016, 36, 1045–1048. [Google Scholar] [CrossRef]
- Lahra, M.M.; Beeby, P.J.; Jeffery, H.E. Intrauterine Inflammation, Neonatal Sepsis, and Chronic Lung Disease: A 13-Year Hospital Cohort Study. Pediatrics 2009, 123, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Watterberg, K.L.; Gerdes, J.S.; Cole, C.H.; Aucott, S.W.; Thilo, E.H.; Mammel, M.C.; Couser, R.J.; Garland, J.S.; Rozycki, H.J.; Leach, C.L.; et al. Prophylaxis of Early Adrenal Insufficiency to Prevent Bronchopulmonary Dysplasia: A Multicenter Trial. Pediatrics 2004, 114, 1649–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darlow, B.A.; Graham, P.J.; Rojas-Reyes, M.X. Vitamin A Supplementation to Prevent Mortality and Short- and Long-Term Morbidity in Very Low Birth Weight Infants. Cochrane Database Syst. Rev. 2016, CD000501. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C.; Chua, R.; Tepper, R. Effect of Dexamethasone on Pulmonary Inflammation and Pulmonary Function of Ventilator-Dependent Infants with Bronchopulmonary Dysplasia. Am. Rev. Respir. Dis. 1991, 143, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Halliday, H.L. Clinical Trials of Postnatal Corticosteroids: Inhaled and Systemic. Biol. Neonate 1999, 76 (Suppl. 1), 29–40. [Google Scholar] [CrossRef]
- Slaughter, J.L.; Stenger, M.R.; Reagan, P.B. Variation in the Use of Diuretic Therapy for Infants with Bronchopulmonary Dysplasia. Pediatrics 2013, 131, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Harijith, A.; Pendyala, S.; Reddy, N.M.; Bai, T.; Usatyuk, P.V.; Berdyshev, E.; Gorshkova, I.; Huang, L.S.; Mohan, V.; Garzon, S.; et al. Sphingosine Kinase 1 Deficiency Confers Protection against Hyperoxia-Induced Bronchopulmonary Dysplasia in a Murine Model: Role of S1P Signaling and Nox Proteins. Am. J. Pathol. 2013, 183, 1169–1182. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, V.; Ha, A.W.; Dong, Y.; Reddy, N.M.; Ebenezer, D.L.; Kanteti, P.; Reddy, S.P.; Usha Raj, J.; Lei, Z.; Maienschein-Cline, M.; et al. Expression Profiling of Genes Regulated by Sphingosine Kinase1 Signaling in a Murine Model of Hyperoxia Induced Neonatal Bronchopulmonary Dysplasia. BMC Genom. 2017, 18, 664. [Google Scholar] [CrossRef] [Green Version]
- Ha, A.W.; Sudhadevi, T.; Ebenezer, D.L.; Fu, P.; Berdyshev, E.V.; Ackerman, S.J.; Natarajan, V.; Harijith, A. Neonatal Therapy with PF543, a Sphingosine Kinase 1 Inhibitor, Ameliorates Hyperoxia-Induced Airway Remodeling in a Murine Model of Bronchopulmonary Dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L497–L512. [Google Scholar] [CrossRef]
- Ha, A.W.; Bai, T.; Ebenezer, D.L.; Sethi, T.; Sudhadevi, T.; Mangio, L.A.; Garzon, S.; Pryhuber, G.S.; Natarajan, V.; Harijith, A. Sphingosine Kinase 1 Regulates Lysyl Oxidase through STAT3 in Hyperoxia-Mediated Neonatal Lung Injury. Thorax 2021, 77, 47–57. [Google Scholar] [CrossRef]
- Sudhadevi, T.; Jafri, A.; Ha, A.W.; Basa, P.; Thomas, J.M.; Fu, P.; Wary, K.; Mehta, D.; Natarajan, V.; Harijith, A. Hyperoxia-Induced S1P1 Signaling Reduced Angiogenesis by Suppression of TIE-2 Leading to Experimental Bronchopulmonary Dysplasia. Cell Biochem. Biophys. 2021, 79, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaho, V.A.; Hla, T. Regulation of Mammalian Physiology, Development, and Disease by the Sphingosine 1-Phosphate and Lysophosphatidic Acid Receptors. Chem. Rev. 2011, 111, 6299–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haughey, N.J.; Bandaru, V.V.R.; Bae, M.; Mattson, M.P. Roles for Dysfunctional Sphingolipid Metabolism in Alzheimer’s Disease Neuropathogenesis. Biochim. Biophys. Acta 2010, 1801, 878–886. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.A.; Knotts, T.A.; Wang, L.-P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A Role for Ceramide, but Not Diacylglycerol, in the Antagonism of Insulin Signal Transduction by Saturated Fatty Acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular Ceramide Synthesis and Protein Kinase Czeta Activation Play an Essential Role in Palmitate-Induced Insulin Resistance in Rat L6 Skeletal Muscle Cells. Biochem. J. 2004, 382, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 6492. [Google Scholar] [CrossRef]
- Yang, Y.; Uhlig, S. The Role of Sphingolipids in Respiratory Disease. Ther. Adv. Respir. Dis. 2011, 5, 325–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibboel, J.; Joza, S.; Reiss, I.; de Jongste, J.C.; Post, M. Amelioration of Hyperoxia-Induced Lung Injury Using a Sphingolipid-Based Intervention. Eur. Respir. J. 2013, 42, 776–784. [Google Scholar] [CrossRef]
- van Mastrigt, E.; Zweekhorst, S.; Bol, B.; Tibboel, J.; van Rosmalen, J.; Samsom, J.N.; Kroon, A.A.; de Jongste, J.C.; Reiss, I.K.M.; Post, M.; et al. Ceramides in Tracheal Aspirates of Preterm Infants: Marker for Bronchopulmonary Dysplasia. PLoS ONE 2018, 13, e0185969. [Google Scholar] [CrossRef] [PubMed]
- Muehlbacher, T.; Bassler, D.; Bryant, M.B. Evidence for the Management of Bronchopulmonary Dysplasia in Very Preterm Infants. Children 2021, 8, 298. [Google Scholar] [CrossRef]
- Wang, J.; Dong, W. Oxidative Stress and Bronchopulmonary Dysplasia. Gene 2018, 678, 177–183. [Google Scholar] [CrossRef] [PubMed]
- de Faria Poloni, J.; Chapola, H.; Feltes, B.C.; Bonatto, D. The Importance of Sphingolipids and Reactive Oxygen Species in Cardiovascular Development. Biol. Cell 2014, 106, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Salton, F.; Braga, L.; Wade, B.; Confalonieri, P.; Volpe, M.C.; Baratella, E.; Maiocchi, S.; Confalonieri, M. The History and Mystery of Alveolar Epithelial Type II Cells: Focus on Their Physiologic and Pathologic Role in Lung. Int. J. Mol. Sci. 2021, 22, 2566. [Google Scholar] [CrossRef] [PubMed]
- Sarode, P.; Mansouri, S.; Karger, A.; Schaefer, M.B.; Grimminger, F.; Seeger, W.; Savai, R. Epithelial Cell Plasticity Defines Heterogeneity in Lung Cancer. Cell Signal. 2020, 65, 109463. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Sullards, M.C.; Merrill, A.H. An Introduction to Sphingolipid Metabolism and Analysis by New Technologies. Neuromolecular Med. 2010, 12, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Merrill, A.H.; Wang, M.D.; Park, M.; Sullards, M.C. (Glyco)Sphingolipidology: An Amazing Challenge and Opportunity for Systems Biology. Trends Biochem. Sci. 2007, 32, 457–468. [Google Scholar] [CrossRef]
- Goetzl, E.J.; An, S. Diversity of Cellular Receptors and Functions for the Lysophospholipid Growth Factors Lysophosphatidic Acid and Sphingosine 1-Phosphate. FASEB J. 1998, 12, 1589–1598. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Functions of the Multifaceted Family of Sphingosine Kinases and Some Close Relatives. J. Biol. Chem. 2007, 282, 2125–2129. [Google Scholar] [CrossRef] [Green Version]
- Pyne, S.; Pyne, N.J. Sphingosine 1-Phosphate Signalling in Mammalian Cells. Biochem. J. 2000, 349, 385–402. [Google Scholar] [CrossRef]
- Matsushita, K.; Morrell, C.N.; Lowenstein, C.J. Sphingosine 1-Phosphate Activates Weibel-Palade Body Exocytosis. Proc. Natl. Acad. Sci. USA 2004, 101, 11483–11487. [Google Scholar] [CrossRef] [Green Version]
- Sudhadevi, T.; Ha, A.W.; Ebenezer, D.L.; Fu, P.; Putherickal, V.; Natarajan, V.; Harijith, A. Advancements in Understanding the Role of Lysophospholipids and Their Receptors in Lung Disorders Including Bronchopulmonary Dysplasia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158685. [Google Scholar] [CrossRef] [PubMed]
- Lone, M.A.; Hülsmeier, A.J.; Saied, E.M.; Karsai, G.; Arenz, C.; von Eckardstein, A.; Hornemann, T. Subunit Composition of the Mammalian Serine-Palmitoyltransferase Defines the Spectrum of Straight and Methyl-Branched Long-Chain Bases. Proc. Natl. Acad. Sci. USA 2020, 117, 15591–15598. [Google Scholar] [CrossRef] [PubMed]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive Sphingolipids: Metabolism and Function. J. Lipid Res. 2009, 50 (Supplement), S91–S96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and Their Metabolism in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Ohanian, J.; Ohanian, V. Sphingolipids in Mammalian Cell Signalling. Cell Mol. Life Sci. 2001, 58, 2053–2068. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-Phosphate Signaling and Its Role in Disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Pitson, S.M.; Moretti, P.A.B.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of Sphingosine Kinase 1 by ERK1/2-Mediated Phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of Histone Acetylation in the Nucleus by Sphingosine-1-Phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-Phosphate Produced by Sphingosine Kinase 2 in Mitochondria Interacts with Prohibitin 2 to Regulate Complex IV Assembly and Respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, A.; Nishi, T.; Hisano, Y.; Fukui, H.; Yamaguchi, A.; Mochizuki, N. The Sphingolipid Transporter Spns2 Functions in Migration of Zebrafish Myocardial Precursors. Science 2009, 323, 524–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, P.A.; Dudek, S.M.; Ma, S.-F.; Garcia, J.G.N. Transactivation of Sphingosine 1-Phosphate Receptors Is Essential for Vascular Barrier Regulation. Novel Role for Hyaluronan and CD44 Receptor Family. J. Biol. Chem. 2006, 281, 34381–34393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S.S.; Mahadevan, K.; Bandi, V.; Zheng, Z.; Smith, C.W.; Rumbaut, R.E. Neutrophils, Nitric Oxide, and Microvascular Permeability in Severe Sepsis. Chest 2005, 128, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Claus, R.A.; Bunck, A.C.; Bockmeyer, C.L.; Brunkhorst, F.M.; Lösche, W.; Kinscherf, R.; Deigner, H.-P. Role of Increased Sphingomyelinase Activity in Apoptosis and Organ Failure of Patients with Severe Sepsis. FASEB J. 2005, 19, 1719–1721. [Google Scholar] [CrossRef] [Green Version]
- Göggel, R.; Winoto-Morbach, S.; Vielhaber, G.; Imai, Y.; Lindner, K.; Brade, L.; Brade, H.; Ehlers, S.; Slutsky, A.S.; Schütze, S.; et al. PAF-Mediated Pulmonary Edema: A New Role for Acid Sphingomyelinase and Ceramide. Nat. Med. 2004, 10, 155–160. [Google Scholar] [CrossRef]
- Sanchez, T.; Skoura, A.; Wu, M.T.; Casserly, B.; Harrington, E.O.; Hla, T. Induction of Vascular Permeability by the Sphingosine-1-Phosphate Receptor-2 (S1P2R) and Its Downstream Effectors ROCK and PTEN. Arter. Thromb. Vasc. Biol. 2007, 27, 1312–1318. [Google Scholar] [CrossRef] [Green Version]
- Roviezzo, F.; D’Agostino, B.; Brancaleone, V.; De Gruttola, L.; Bucci, M.; De Dominicis, G.; Orlotti, D.; D’Aiuto, E.; De Palma, R.; Rossi, F.; et al. Systemic Administration of Sphingosine-1-Phosphate Increases Bronchial Hyperresponsiveness in the Mouse. Am. J. Respir. Cell Mol. Biol. 2010, 42, 572–577. [Google Scholar] [CrossRef]
- Magnussen, H. Inhalation Therapy for Bronchial Asthma: Strategies and Targets. Curr. Opin. Pulm. Med. 2003, 9 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef]
- Park, S.-J.; Im, D.-S. Blockage of Sphingosine-1-Phosphate Receptor 2 Attenuates Allergic Asthma in Mice. Br. J. Pharmacol. 2019, 176, 938–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, P.S.; Bektas, M.; Olivera, A.; Gonzalez-Espinosa, C.; Proia, R.L.; Rivera, J.; Milstien, S.; Spiegel, S. Transactivation of Sphingosine-1-Phosphate Receptors by FcepsilonRI Triggering Is Required for Normal Mast Cell Degranulation and Chemotaxis. J. Exp. Med. 2004, 199, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmé, H.; Jendrossek, V.; Riehle, A.; von Kürthy, G.; Berger, J.; Schwarz, H.; Weller, M.; Kolesnick, R.; Gulbins, E. Host Defense against Pseudomonas Aeruginosa Requires Ceramide-Rich Membrane Rafts. Nat. Med. 2003, 9, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.P.; Pier, G.B. Localization of Cystic Fibrosis Transmembrane Conductance Regulator to Lipid Rafts of Epithelial Cells Is Required for Pseudomonas Aeruginosa-Induced Cellular Activation. J. Immunol. 2004, 172, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodlie, M.; McKean, M.C.; Johnson, G.E.; Gray, J.; Fisher, A.J.; Corris, P.A.; Lordan, J.L.; Ward, C. Ceramide Is Increased in the Lower Airway Epithelium of People with Advanced Cystic Fibrosis Lung Disease. Am. J. Respir. Crit. Care Med. 2010, 182, 369–375. [Google Scholar] [CrossRef]
- Teichgräber, V.; Ulrich, M.; Endlich, N.; Riethmüller, J.; Wilker, B.; De Oliveira-Munding, C.C.; van Heeckeren, A.M.; Barr, M.L.; von Kürthy, G.; Schmid, K.W.; et al. Ceramide Accumulation Mediates Inflammation, Cell Death and Infection Susceptibility in Cystic Fibrosis. Nat. Med. 2008, 14, 382–391. [Google Scholar] [CrossRef]
- Bodas, M.; Min, T.; Mazur, S.; Vij, N. Critical Modifier Role of Membrane-Cystic Fibrosis Transmembrane Conductance Regulator-Dependent Ceramide Signaling in Lung Injury and Emphysema. J. Immunol. 2011, 186, 602–613. [Google Scholar] [CrossRef]
- Becker, K.A.; Riethmüller, J.; Lüth, A.; Döring, G.; Kleuser, B.; Gulbins, E. Acid Sphingomyelinase Inhibitors Normalize Pulmonary Ceramide and Inflammation in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 716–724. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Huang, L.S.; Watanabe, S.; Joshi, N.; Williams, K.J.N.; Chi, M.; Lu, Z.; Harijith, A.; Yeldandi, A.; et al. The Sphingosine Kinase 1 Inhibitor, PF543, Mitigates Pulmonary Fibrosis by Reducing Lung Epithelial Cell MtDNA Damage and Recruitment of Fibrogenic Monocytes. Int. J. Mol. Sci. 2020, 21, 5595. [Google Scholar] [CrossRef]
- Huang, L.S.; Berdyshev, E.; Mathew, B.; Fu, P.; Gorshkova, I.A.; He, D.; Ma, W.; Noth, I.; Ma, S.-F.; Pendyala, S.; et al. Targeting Sphingosine Kinase 1 Attenuates Bleomycin-Induced Pulmonary Fibrosis. FASEB J. 2013, 27, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- MacRitchie, N.; Volpert, G.; Al Washih, M.; Watson, D.G.; Futerman, A.H.; Kennedy, S.; Pyne, S.; Pyne, N.J. Effect of the Sphingosine Kinase 1 Selective Inhibitor, PF-543 on Arterial and Cardiac Remodelling in a Hypoxic Model of Pulmonary Arterial Hypertension. Cell Signal. 2016, 28, 946–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Berka, V.; Song, A.; Sun, K.; Wang, W.; Zhang, W.; Ning, C.; Li, C.; Zhang, Q.; Bogdanov, M.; et al. Elevated Sphingosine-1-Phosphate Promotes Sickling and Sickle Cell Disease Progression. J. Clin. Investig. 2014, 124, 2750–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xia, Y.; Yan, W.; Zhang, H.; Zhou, F.; Zhao, S.; Wang, W.; Zhu, D.; Xin, C.; Lee, Y.; et al. Sphingosine 1-Phosphate Signaling Contributes to Cardiac Inflammation, Dysfunction, and Remodeling Following Myocardial Infarction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H250–H261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for Sphingosine Kinase 1 in Colon Carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Okajima, F. Role of Sphingosine 1-Phosphate in Anti-Atherogenic Actions of High-Density Lipoprotein. World J. Biol. Chem. 2010, 1, 327–337. [Google Scholar] [CrossRef]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating Sphingolipid Biomarkers in Models of Type 1 Diabetes. J. Lipid Res. 2011, 52, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and Development of an Oral Drug to Treat Multiple Sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Filipenko, I.; Schwalm, S.; Reali, L.; Pfeilschifter, J.; Fabbro, D.; Huwiler, A.; Zangemeister-Wittke, U. Upregulation of the S1P3 Receptor in Metastatic Breast Cancer Cells Increases Migration and Invasion by Induction of PGE2 and EP2/EP4 Activation. Biochim. Biophys. Acta 2016, 1861, 1840–1851. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B. Sphingolipid Metabolism in Cancer Signalling and Therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Guan, H.; Song, L.; Cai, J.; Huang, Y.; Wu, J.; Yuan, J.; Li, J.; Li, M. Sphingosine Kinase 1 Regulates the Akt/FOXO3a/Bim Pathway and Contributes to Apoptosis Resistance in Glioma Cells. PLoS ONE 2011, 6, e19946. [Google Scholar] [CrossRef]
- Penaranda, C.; Tang, Q.; Ruddle, N.H.; Bluestone, J.A. Prevention of Diabetes by FTY720-Mediated Stabilization of Peri-Islet Tertiary Lymphoid Organs. Diabetes 2010, 59, 1461–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imasawa, T.; Koike, K.; Ishii, I.; Chun, J.; Yatomi, Y. Blockade of Sphingosine 1-Phosphate Receptor 2 Signaling Attenuates Streptozotocin-Induced Apoptosis of Pancreatic Beta-Cells. Biochem. Biophys. Res. Commun. 2010, 392, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idzko, M.; Hammad, H.; van Nimwegen, M.; Kool, M.; Müller, T.; Soullié, T.; Willart, M.A.M.; Hijdra, D.; Hoogsteden, H.C.; Lambrecht, B.N. Local Application of FTY720 to the Lung Abrogates Experimental Asthma by Altering Dendritic Cell Function. J. Clin. Investig. 2006, 116, 2935–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinas, N.R.; Lopes, C.T.; Palma, P.V.; Oshima, C.T.; Bueno, V. Lung Tumor Development in the Presence of Sphingosine 1-Phosphate Agonist FTY720. Pathol. Oncol. Res. 2009, 15, 549–554. [Google Scholar] [CrossRef]
- Comi, G.; Hartung, H.-P.; Bakshi, R.; Williams, I.M.; Wiendl, H. Benefit-Risk Profile of Sphingosine-1-Phosphate Receptor Modulators in Relapsing and Secondary Progressive Multiple Sclerosis. Drugs 2017, 77, 1755–1768. [Google Scholar] [CrossRef]
- Sammani, S.; Moreno-Vinasco, L.; Mirzapoiazova, T.; Singleton, P.A.; Chiang, E.T.; Evenoski, C.L.; Wang, T.; Mathew, B.; Husain, A.; Moitra, J.; et al. Differential Effects of Sphingosine 1-Phosphate Receptors on Airway and Vascular Barrier Function in the Murine Lung. Am. J. Respir. Cell Mol. Biol. 2010, 43, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Suzuki, K.; Uechi, M.; Kurihara, E.; Goto, K.; Sakai, H.; Misawa, M. Downregulation of Sphingosine-1-Phosphate Receptors in Bronchial Smooth Muscle of Mouse Experimental Asthma. Pharmacol. Res. 2010, 62, 357–363. [Google Scholar] [CrossRef]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The Sphingosine Kinase 1/Sphingosine-1-Phosphate Pathway in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef]
- Xu, Y.; Krause, A.; Limberis, M.; Worgall, T.S.; Worgall, S. Low Sphingosine-1-Phosphate Impairs Lung Dendritic Cells in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 48, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.-K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, E2064. [Google Scholar] [CrossRef] [Green Version]
- Yura, Y.; Masui, A.; Hamada, M. Inhibitors of Ceramide- and Sphingosine-Metabolizing Enzymes as Sensitizers in Radiotherapy and Chemotherapy for Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 2062. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, S.; Hartmann, D.; Fuchs, S.; Birod, K.; Ferreiròs, N.; Schreiber, Y.; Zivkovic, A.; Geisslinger, G.; Grösch, S.; Stark, H. Inhibitors of Specific Ceramide Synthases. Biochimie 2012, 94, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Klumpp, M.; Habig, M.; Rovina, P.; Billich, A.; Baumruker, T.; Oberhauser, B.; Bornancin, F. Targeting Ceramide Metabolism with a Potent and Specific Ceramide Kinase Inhibitor. Mol. Pharmacol. 2008, 74, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Lone, M.A.; Santos, T.; Alecu, I.; Silva, L.C.; Hornemann, T. 1-Deoxysphingolipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Saied, E.M.; Arenz, C. Stereoselective Synthesis of Novel Sphingoid Bases Utilized for Exploring the Secrets of Sphinx. Int. J. Mol. Sci. 2021, 22, 8171. [Google Scholar] [CrossRef]
- Garofalo, K.; Penno, A.; Schmidt, B.P.; Lee, H.-J.; Frosch, M.P.; von Eckardstein, A.; Brown, R.H.; Hornemann, T.; Eichler, F.S. Oral L-Serine Supplementation Reduces Production of Neurotoxic Deoxysphingolipids in Mice and Humans with Hereditary Sensory Autonomic Neuropathy Type 1. J. Clin. Investig. 2011, 121, 4735–4745. [Google Scholar] [CrossRef] [Green Version]
- Steiner, R.; Saied, E.M.; Othman, A.; Arenz, C.; Maccarone, A.T.; Poad, B.L.J.; Blanksby, S.J.; von Eckardstein, A.; Hornemann, T. Elucidating the Chemical Structure of Native 1-Deoxysphingosine. J. Lipid Res. 2016, 57, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Sakagami, H.; Kishino, K.; Amano, O.; Kanda, Y.; Kunii, S.; Yokote, Y.; Oizumi, H.; Oizumi, T. Cell Death Induced by Nutritional Starvation in Mouse Macrophage-like RAW264.7 Cells. Anticancer Res. 2009, 29, 343–347. [Google Scholar]
- Zuellig, R.A.; Hornemann, T.; Othman, A.; Hehl, A.B.; Bode, H.; Güntert, T.; Ogunshola, O.O.; Saponara, E.; Grabliauskaite, K.; Jang, J.-H.; et al. Deoxysphingolipids, Novel Biomarkers for Type 2 Diabetes, Are Cytotoxic for Insulin-Producing Cells. Diabetes 2014, 63, 1326–1339. [Google Scholar] [CrossRef] [Green Version]
- Othman, A.; Bianchi, R.; Alecu, I.; Wei, Y.; Porretta-Serapiglia, C.; Lombardi, R.; Chiorazzi, A.; Meregalli, C.; Oggioni, N.; Cavaletti, G.; et al. Lowering Plasma 1-Deoxysphingolipids Improves Neuropathy in Diabetic Rats. Diabetes 2015, 64, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.; Lee, P.-T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z.; Lin, W.-W.; Wang, L.; Bellen, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to α-Synuclein Gain. Cell Metab. 2018, 28, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alecu, I.; Othman, A.; Penno, A.; Saied, E.M.; Arenz, C.; von Eckardstein, A.; Hornemann, T. Cytotoxic 1-Deoxysphingolipids Are Metabolized by a Cytochrome P450-Dependent Pathway. J. Lipid Res. 2017, 58, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowler, R.P.; Jacobson, S.; Cruickshank, C.; Hughes, G.J.; Siska, C.; Ory, D.S.; Petrache, I.; Schaffer, J.E.; Reisdorph, N.; Kechris, K. Plasma Sphingolipids Associated with Chronic Obstructive Pulmonary Disease Phenotypes. Am. J. Respir. Crit. Care Med. 2015, 191, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, S.; Merrill, A.H. Sphingolipid Metabolism and Cell Growth Regulation. FASEB J. 1996, 10, 1388–1397. [Google Scholar] [CrossRef]
- Agrons, G.A.; Courtney, S.E.; Stocker, J.T.; Markowitz, R.I. From the Archives of the AFIP: Lung Disease in Premature Neonates: Radiologic-Pathologic Correlation. Radiographics 2005, 25, 1047–1073. [Google Scholar] [CrossRef]
- Brody, J.S.; Kagan, H.; Manalo, A. Lung Lysyl Oxidase Activity: Relation to Lung Growth. Am. Rev. Respir. Dis. 1979, 120, 1289–1295. [Google Scholar] [CrossRef]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted Pulmonary Vasculature and Decreased Vascular Endothelial Growth Factor, Flt-1, and TIE-2 in Human Infants Dying with Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Laviña, B.; Siemsen, B.L.; Muhl, L.; Hofmann, J.J.; Ebarasi, L.; Nyström, S.; Rymo, S.; et al. The Sphingosine-1-Phosphate Receptor S1PR1 Restricts Sprouting Angiogenesis by Regulating the Interplay between VE-Cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Choi, A.M.K. Regulation of Autophagy in Oxygen-Dependent Cellular Stress. Curr. Pharm. Des. 2013, 19, 2747–2756. [Google Scholar] [CrossRef]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and Physiological Role of Reactive Oxygen Species—The Good, the Bad and the Ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, C.; Virion, A.; Ohayon, R.; Kaniewski, J.; Dème, D.; Pommier, J. Mechanism of Hydrogen Peroxide Formation Catalyzed by NADPH Oxidase in Thyroid Plasma Membrane. J. Biol. Chem. 1991, 266, 3739–3743. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; ALSalamat, H.A.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [Green Version]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.J.; Lin, S.W.; Pacifici, R.E. Protein Damage and Degradation by Oxygen Radicals. IV. Degradation of Denatured Protein. J. Biol. Chem. 1987, 262, 9914–9920. [Google Scholar] [CrossRef]
- Dean, R.T.; Roberts, C.R.; Jessup, W. Fragmentation of Extracellular and Intracellular Polypeptides by Free Radicals. Prog. Clin. Biol. Res. 1985, 180, 341–350. [Google Scholar]
- Uchida, K.; Shiraishi, M.; Naito, Y.; Torii, Y.; Nakamura, Y.; Osawa, T. Activation of Stress Signaling Pathways by the End Product of Lipid Peroxidation. 4-Hydroxy-2-Nonenal Is a Potential Inducer of Intracellular Peroxide Production. J. Biol. Chem. 1999, 274, 2234–2242. [Google Scholar] [CrossRef] [Green Version]
- Boveris, A.; Chance, B. The Mitochondrial Generation of Hydrogen Peroxide. General Properties and Effect of Hyperbaric Oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Schindler, L.; Smyth, L.C.D.; Bernhagen, J.; Hampton, M.B.; Dickerhof, N. Macrophage Migration Inhibitory Factor (MIF) Enhances Hypochlorous Acid Production in Phagocytic Neutrophils. Redox Biol. 2021, 41, 101946. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.X.L.; Gozal, D. Reactive Oxygen Species and the Brain in Sleep Apnea. Respir. Physiol. Neurobiol. 2010, 174, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox Family NADPH Oxidases: Molecular Mechanisms of Activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Ufer, C.; Wang, C.C.; Borchert, A.; Heydeck, D.; Kuhn, H. Redox Control in Mammalian Embryo Development. Antioxid. Redox Signal. 2010, 13, 833–875. [Google Scholar] [CrossRef]
- Saugstad, O.D. Oxygen and Oxidative Stress in Bronchopulmonary Dysplasia. J. Perinat. Med. 2010, 38, 571–577. [Google Scholar] [CrossRef]
- Huizing, M.J.; Cavallaro, G.; Moonen, R.M.; González-Luis, G.E.; Mosca, F.; Vento, M.; Villamor, E. Is the C242T Polymorphism of the CYBA Gene Linked with Oxidative Stress-Associated Complications of Prematurity? Antioxid. Redox Signal. 2017, 27, 1432–1438. [Google Scholar] [CrossRef]
- San José, G.; Fortuño, A.; Beloqui, O.; Díez, J.; Zalba, G. NADPH Oxidase CYBA Polymorphisms, Oxidative Stress and Cardiovascular Diseases. Clin. Sci. 2008, 114, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Pendyala, S.; Gorshkova, I.A.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of Nox4 and Nox2 in Hyperoxia-Induced Reactive Oxygen Species Generation and Migration of Human Lung Endothelial Cells. Antioxid. Redox Signal. 2009, 11, 747–764. [Google Scholar] [CrossRef] [Green Version]
- Auten, R.L.; Mason, S.N.; Auten, K.M.; Brahmajothi, M. Hyperoxia Impairs Postnatal Alveolar Epithelial Development via NADPH Oxidase in Newborn Mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L134–L142. [Google Scholar] [CrossRef] [Green Version]
- Pendyala, S.; Moitra, J.; Kalari, S.; Kleeberger, S.R.; Zhao, Y.; Reddy, S.P.; Garcia, J.G.N.; Natarajan, V. Nrf2 Regulates Hyperoxia-Induced Nox4 Expression in Human Lung Endothelium: Identification of Functional Antioxidant Response Elements on the Nox4 Promoter. Free Radic. Biol. Med. 2011, 50, 1749–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.-Y.; Kleeberger, S.R. Mitochondrial Biology in Airway Pathogenesis and the Role of NRF2. Arch. Pharm. Res. 2020, 43, 297–320. [Google Scholar] [CrossRef] [PubMed]
- Xuefei, Y.; Xinyi, Z.; Qing, C.; Dan, Z.; Ziyun, L.; Hejuan, Z.; Xindong, X.; Jianhua, F. Effects of Hyperoxia on Mitochondrial Homeostasis: Are Mitochondria the Hub for Bronchopulmonary Dysplasia? Front. Cell Dev. Biol. 2021, 9, 642717. [Google Scholar] [CrossRef] [PubMed]
- Dani, C.; Cecchi, A.; Bertini, G. Role of Oxidative Stress as Physiopathologic Factor in the Preterm Infant. Minerva Pediatr. 2004, 56, 381–394. [Google Scholar] [PubMed]
- Kaarteenaho-Wiik, R.; Kinnula, V.L. Distribution of Antioxidant Enzymes in Developing Human Lung, Respiratory Distress Syndrome, and Bronchopulmonary Dysplasia. J. Histochem. Cytochem. 2004, 52, 1231–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xue, T.; Ye, H.; Sang, C.; Wu, S.; Li, S. Study of the Common Activating Mechanism of Apoptosis and Epithelial-to-Mesenchymal Transition in Alveolar Type II Epithelial Cells. Respir. Physiol. Neurobiol. 2021, 284, 103584. [Google Scholar] [CrossRef]
- Freeman, B.A.; Panus, P.C.; Matalon, S.; Buckley, B.J.; Baker, R.R. Oxidant Injury to the Alveolar Epithelium: Biochemical and Pharmacologic Studies. Res. Rep. Health Eff. Inst. 1993, 1–30; discussion 31–39. [Google Scholar]
- Frank, L.; Groseclose, E.E. Preparation for Birth into an O2-Rich Environment: The Antioxidant Enzymes in the Developing Rabbit Lung. Pediatr. Res. 1984, 18, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbach, H. Alveolar Epithelial Type II Cell: Defender of the Alveolus Revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef]
- Li, Z.; Fang, F.; Xu, F. Effects of Different States of Oxidative Stress on Fetal Rat Alveolar Type II Epithelial Cells in Vitro and ROS-induced Changes in Wnt Signaling Pathway Expression. Mol. Med. Rep. 2013, 7, 1528–1532. [Google Scholar] [CrossRef]
- Caldeira, D.D.A.F.; Weiss, D.J.; Rocco, P.R.M.; Silva, P.L.; Cruz, F.F. Mitochondria in Focus: From Function to Therapeutic Strategies in Chronic Lung Diseases. Front. Immunol. 2021, 12, 782074. [Google Scholar] [CrossRef] [PubMed]
- Moos, W.H.; Faller, D.V.; Glavas, I.P.; Harpp, D.N.; Kamperi, N.; Kanara, I.; Kodukula, K.; Mavrakis, A.N.; Pernokas, J.; Pernokas, M.; et al. Pathogenic Mitochondrial Dysfunction and Metabolic Abnormalities. Biochem. Pharmacol. 2021, 193, 114809. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.J.P.; Tibboel, D.; de Kleer, I.M.; Reiss, I.K.M.; Rottier, R.J. The Future of Bronchopulmonary Dysplasia: Emerging Pathophysiological Concepts and Potential New Avenues of Treatment. Front. Med. 2017, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.M.; Abrantes, M.A.; Hasan, J.; Aranda, J.V.; Beharry, K.D. Reactive Oxygen Species, Biomarkers of Microvascular Maturation and Alveolarization, and Antioxidants in Oxidative Lung Injury. React. Oxyg. Species 2018, 6, 373–388. [Google Scholar] [CrossRef]
- Ratner, V.; Starkov, A.; Matsiukevich, D.; Polin, R.A.; Ten, V.S. Mitochondrial Dysfunction Contributes to Alveolar Developmental Arrest in Hyperoxia-Exposed Mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Berkelhamer, S.K.; Kim, G.A.; Radder, J.E.; Wedgwood, S.; Czech, L.; Steinhorn, R.H.; Schumacker, P.T. Developmental Differences in Hyperoxia-Induced Oxidative Stress and Cellular Responses in the Murine Lung. Free Radic. Biol. Med. 2013, 61, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Ten, V.S.; Ratner, V. Mitochondrial Bioenergetics and Pulmonary Dysfunction: Current Progress and Future Directions. Paediatr. Respir. Rev. 2020, 34, 37–45. [Google Scholar] [CrossRef]
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.K.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in Lung Biology and Pathology: More than Just a Powerhouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L962–L974. [Google Scholar] [CrossRef] [Green Version]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-Targeted Antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Kim, G.A.; Taylor, J.M.; Gugino, S.F.; Farrow, K.N.; Schumacker, P.T.; Berkelhamer, S.K. Mouse Lung Development and NOX1 Induction during Hyperoxia Are Developmentally Regulated and Mitochondrial ROS Dependent. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L369–L377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roper, J.M.; Mazzatti, D.J.; Watkins, R.H.; Maniscalco, W.M.; Keng, P.C.; O’Reilly, M.A. In Vivo Exposure to Hyperoxia Induces DNA Damage in a Population of Alveolar Type II Epithelial Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L1045–L1054. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, J.; Olave, N.; Ballinger, S.W.; Ambalavanan, N. Vascular Endothelial Mitochondrial Function Predicts Death or Pulmonary Outcomes in Preterm Infants. Am. J. Respir. Crit. Care Med. 2017, 196, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Ruchko, M.; Gorodnya, O.; LeDoux, S.P.; Alexeyev, M.F.; Al-Mehdi, A.-B.; Gillespie, M.N. Mitochondrial DNA Damage Triggers Mitochondrial Dysfunction and Apoptosis in Oxidant-Challenged Lung Endothelial Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L530–L535. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pan, X.; Wang, L.; Yu, G. Alveolar Cells under Mechanical Stressed Niche: Critical Contributors to Pulmonary Fibrosis. Mol. Med. 2020, 26, 95. [Google Scholar] [CrossRef]
- Gebb, S.A.; Decoux, A.; Waggoner, A.; Wilson, G.L.; Gillespie, M.N. Mitochondrial DNA Damage Mediates Hyperoxic Dysmorphogenesis in Rat Fetal Lung Explants. Neonatology 2013, 103, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Cheresh, P.; Williams, D.; Cheng, Y.; Ridge, K.; Schumacker, P.T.; Weitzman, S.; Bohr, V.A.; Kamp, D.W. Mitochondria-Targeted Ogg1 and Aconitase-2 Prevent Oxidant-Induced Mitochondrial DNA Damage in Alveolar Epithelial Cells. J. Biol. Chem. 2014, 289, 6165–6176. [Google Scholar] [CrossRef] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial Oxidative Stress Causes Mitochondrial Fragmentation via Differential Modulation of Mitochondrial Fission-Fusion Proteins. FEBS J. 2011, 278, 941–954. [Google Scholar] [CrossRef]
- Dai, Y.; Yu, B.; Ai, D.; Yuan, L.; Wang, X.; Huo, R.; Fu, X.; Chen, S.; Chen, C. Mitochondrial Fission-Mediated Lung Development in Newborn Rats with Hyperoxia-Induced Bronchopulmonary Dysplasia With Pulmonary Hypertension. Front. Pediatr. 2020, 8, 619853. [Google Scholar] [CrossRef]
- Ma, C.; Beyer, A.M.; Durand, M.; Clough, A.V.; Zhu, D.; Norwood Toro, L.; Terashvili, M.; Ebben, J.D.; Hill, R.B.; Audi, S.H.; et al. Hyperoxia Causes Mitochondrial Fragmentation in Pulmonary Endothelial Cells by Increasing Expression of Pro-Fission Proteins. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 622–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, R.-J.; Jing, X.; Michalkiewicz, T.; Afolayan, A.J.; Wu, T.-J.; Konduri, G.G. Attenuation of Endoplasmic Reticulum Stress by Caffeine Ameliorates Hyperoxia-Induced Lung Injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L586–L598. [Google Scholar] [CrossRef] [PubMed]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 to Regulate Mitochondrial Dynamics during Stress. Mol. Cell Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Sun, Y.; Cai, Q.; Zhao, X.; Liu, Z.; Xue, X.; Fu, J. Hyperoxia Exposure Arrests Alveolarization in Neonatal Rats via PTEN-induced Putative Kinase 1-Parkin and Nip3-like Protein X-mediated Mitophagy Disorders. Int. J. Mol. Med. 2020, 46, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Carraway, M.S.; Suliman, H.B.; Kliment, C.; Welty-Wolf, K.E.; Oury, T.D.; Piantadosi, C.A. Mitochondrial Biogenesis in the Pulmonary Vasculature during Inhalational Lung Injury and Fibrosis. Antioxid. Redox Signal. 2008, 10, 269–275. [Google Scholar] [CrossRef]
- Wei, G.; Sun, R.; Xu, T.; Kong, S.; Zhang, S. Aminophylline Promotes Mitochondrial Biogenesis in Human Pulmonary Bronchial Epithelial Cells. Biochem. Biophys. Res. Commun. 2019, 515, 31–36. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, Y.; Liu, Y.; Shi, J.; Cheng, Z. Montelukast Promotes Mitochondrial Biogenesis via CREB/PGC-1α in Human Bronchial Epithelial Cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 4234–4239. [Google Scholar] [CrossRef]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and Mechanism of Mitochondrial Electron Transport Chain. Biomed. J. 2018, 41, 9–20. [Google Scholar] [CrossRef]
- Rosenbaum, R.M.; Wittner, M.; Lenger, M. Mitochondrial and Other Ultrastructural Changes in Great Alveolar Cells of Oxygen-Adapted and Poisoned Rats. Lab. Investig. 1969, 20, 516–528. [Google Scholar]
- Hendricks-Muñoz, K.D.; Xu, J.; Voynow, J.A. Tracheal Aspirate VEGF and Sphingolipid Metabolites in the Preterm Infant with Later Development of Bronchopulmonary Dysplasia. Pediatr. Pulmonol. 2018, 53, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Das, K.C. Hyperoxia Decreases Glycolytic Capacity, Glycolytic Reserve and Oxidative Phosphorylation in MLE-12 Cells and Inhibits Complex I and II Function, but Not Complex IV in Isolated Mouse Lung Mitochondria. PLoS ONE 2013, 8, e73358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; White, C.W.; Chang, L.Y.; Schneider, B.K.; Allen, C.B. Glutamine Protects Mitochondrial Structure and Function in Oxygen Toxicity. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L779–L791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resseguie, E.A.; Staversky, R.J.; Brookes, P.S.; O’Reilly, M.A. Hyperoxia Activates ATM Independent from Mitochondrial ROS and Dysfunction. Redox Biol. 2015, 5, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Gardner, P.R.; Nguyen, D.D.; White, C.W. Aconitase Is a Sensitive and Critical Target of Oxygen Poisoning in Cultured Mammalian Cells and in Rat Lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252. [Google Scholar] [CrossRef] [Green Version]
- Otsubo, C.; Bharathi, S.; Uppala, R.; Ilkayeva, O.R.; Wang, D.; McHugh, K.; Zou, Y.; Wang, J.; Alcorn, J.F.; Zuo, Y.Y.; et al. Long-Chain Acylcarnitines Reduce Lung Function by Inhibiting Pulmonary Surfactant. J. Biol. Chem. 2015, 290, 23897–23904. [Google Scholar] [CrossRef] [Green Version]
- Dennery, P.A.; Carr, J.; Peterson, A.; Yao, H. The role of mitochondrial fatty acid use in neonatal lung injury and repair. Trans. Am. Clin. Climatol. Assoc. 2018, 129, 195–201. [Google Scholar]
- Chiantia, S.; London, E. Sphingolipids and Membrane Domains: Recent Advances. Handb. Exp. Pharmacol. 2013, 215, 33–55. [Google Scholar] [CrossRef]
- Smith, W.L.; Merrill, A.H. Sphingolipid Metabolism and Signaling Minireview Series. J. Biol. Chem. 2002, 277, 25841–25842. [Google Scholar] [CrossRef] [Green Version]
- Young, M.M.; Kester, M.; Wang, H.-G. Sphingolipids: Regulators of Crosstalk between Apoptosis and Autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.X.; Zou, A.-P.; Li, P.-L. Ceramide-Induced Activation of NADPH Oxidase and Endothelial Dysfunction in Small Coronary Arteries. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H605–H612. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct Effect of Ceramide on the Mitochondrial Electron Transport Chain Leads to Generation of Reactive Oxygen Species. Role of Mitochondrial Glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldkorn, T.; Chung, S.; Filosto, S. Lung Cancer and Lung Injury: The Dual Role of Ceramide. Handb. Exp. Pharmacol. 2013, 216, 93–113. [Google Scholar] [CrossRef] [Green Version]
- Morad, S.A.F.; Cabot, M.C. Ceramide-Orchestrated Signalling in Cancer Cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Husari, A.W.; Dbaibo, G.S.; Bitar, H.; Khayat, A.; Panjarian, S.; Nasser, M.; Bitar, F.F.; El-Sabban, M.; Zaatari, G.; Mroueh, S.M. Apoptosis and the Activity of Ceramide, Bax and Bcl-2 in the Lungs of Neonatal Rats Exposed to Limited and Prolonged Hyperoxia. Respir. Res. 2006, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Roszczyc-Owsiejczuk, K.; Zabielski, P. Sphingolipids as a Culprit of Mitochondrial Dysfunction in Insulin Resistance and Type 2 Diabetes. Front. Endocrinol. 2021, 12, 635175. [Google Scholar] [CrossRef]
- Knupp, J.; Martinez-Montañés, F.; Van Den Bergh, F.; Cottier, S.; Schneiter, R.; Beard, D.; Chang, A. Sphingolipid Accumulation Causes Mitochondrial Dysregulation and Cell Death. Cell Death Differ. 2017, 24, 2044–2053. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, M.; Ogretmen, B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers 2021, 13, 2475. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Saada, A. Ceramide and the Mitochondrial Respiratory Chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Zhang, J. Autophagy and Mitophagy in Cellular Damage Control. Redox Biol. 2013, 1, 19–23. [Google Scholar] [CrossRef] [Green Version]
- El Bawab, S.; Roddy, P.; Qian, T.; Bielawska, A.; Lemasters, J.J.; Hannun, Y.A. Molecular Cloning and Characterization of a Human Mitochondrial Ceramidase. J. Biol. Chem. 2000, 275, 21508–21513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular Compartmentalization of Ceramide Metabolism: MAM (Mitochondria-Associated Membrane) and/or Mitochondria? Biochem. J. 2004, 382, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabu, T.; Shimuzu, A.; Yamashita, M. A Novel Mitochondrial Sphingomyelinase in Zebrafish Cells. J. Biol. Chem. 2009, 284, 20349–20363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, V.; Eisner, V.; Chiong, M.; Criollo, A.; Moraga, F.; Garcia, A.; Härtel, S.; Jaimovich, E.; Zorzano, A.; Hidalgo, C.; et al. Changes in Mitochondrial Dynamics during Ceramide-Induced Cardiomyocyte Early Apoptosis. Cardiovasc. Res. 2008, 77, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarlo, L.; Manganelli, V.; Garofalo, T.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Sorice, M. Association of Fission Proteins with Mitochondrial Raft-like Domains. Cell Death Differ. 2010, 17, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.E.; Tippetts, T.S.; Brassfield, E.S.; Tucker, B.J.; Ockey, A.; Swensen, A.C.; Anthonymuthu, T.S.; Washburn, T.D.; Kane, D.A.; Prince, J.T.; et al. Mitochondrial Fission Mediates Ceramide-Induced Metabolic Disruption in Skeletal Muscle. Biochem. J. 2013, 456, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Gong, J.; Peterson, A.L.; Lu, X.; Zhang, P.; Dennery, P.A. Fatty Acid Oxidation Protects against Hyperoxia-Induced Endothelial Cell Apoptosis and Lung Injury in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2019, 60, 667–677. [Google Scholar] [CrossRef]
- Vyas-Read, S.; Wang, W.; Kato, S.; Colvocoresses-Dodds, J.; Fifadara, N.H.; Gauthier, T.W.; Helms, M.N.; Carlton, D.P.; Brown, L.A.S. Hyperoxia Induces Alveolar Epithelial-to-Mesenchymal Cell Transition. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L326–L340. [Google Scholar] [CrossRef] [Green Version]



 indicate inhibition.
indicate inhibition.
indicate inhibition.
indicate inhibition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specificity | Drugs | Disease Models |
|---|---|---|
| S1P1 modulators | Fingolimod (FTY720) | Asthma [93] Lung cancer [94] |
| Siponimod | Multiple sclerosis [95] | |
| NIBR0213 | BPD [31] | |
| SB-649146 | ALI [96] | |
| S1P2 modulators | JTE-013 | Asthma [97] Pulmonary hypertension [98] Cystic fibrosis [99] |
| S1P3 modulators | BML-241 | Asthma [97] |
| SK1 modulators | PF543 | BPD [29,30] Pulmonaryfibrosis [79,100] Cancer [101] |
| SK2 modulators | ABC294640 (Opaganib) | Cancer [102] |
| CERS inhibitors | CERS 2, 4, and 6 | Cancer [103] |
| Cerk inhibitors | NVP-231 | Cancer [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, J.M.; Sudhadevi, T.; Basa, P.; Ha, A.W.; Natarajan, V.; Harijith, A. The Role of Sphingolipid Signaling in Oxidative Lung Injury and Pathogenesis of Bronchopulmonary Dysplasia. Int. J. Mol. Sci. 2022, 23, 1254. https://doi.org/10.3390/ijms23031254
Thomas JM, Sudhadevi T, Basa P, Ha AW, Natarajan V, Harijith A. The Role of Sphingolipid Signaling in Oxidative Lung Injury and Pathogenesis of Bronchopulmonary Dysplasia. International Journal of Molecular Sciences. 2022; 23(3):1254. https://doi.org/10.3390/ijms23031254
Chicago/Turabian StyleThomas, Jaya M., Tara Sudhadevi, Prathima Basa, Alison W. Ha, Viswanathan Natarajan, and Anantha Harijith. 2022. "The Role of Sphingolipid Signaling in Oxidative Lung Injury and Pathogenesis of Bronchopulmonary Dysplasia" International Journal of Molecular Sciences 23, no. 3: 1254. https://doi.org/10.3390/ijms23031254