Do Androgens Modulate the Pathophysiological Pathways of Inflammation? Appraising the Contemporary Evidence


Abstract
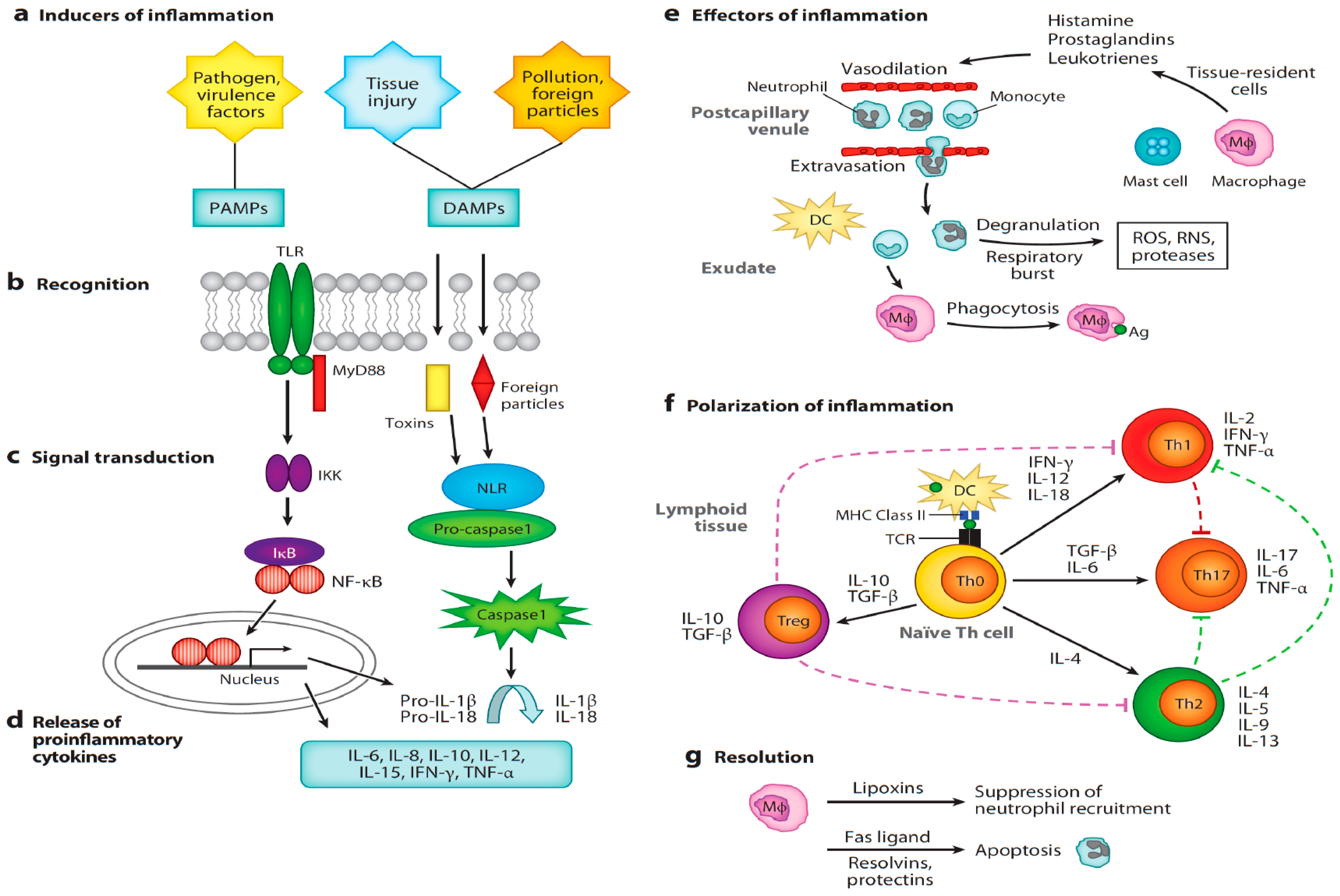
:1. Introduction
2. Inflammatory Biomarkers Are Elevated in Men with Testosterone Deficiency and Reduced with Testosterone Therapy
3. Role of Androgens in Chronic Inflammatory Diseases
3.1. Diabetes
3.2. Coronary Artery Disease
3.3. Psoriasis
3.4. Rheumatoid Arthritis
3.5. Crohn’s Disease
3.6. Airway Diseases
3.7. Multiple Sclerosis
3.8. Systemic Lupus Erythematosus (SLE)
4. Sex Steroid Hormones Modulate the Pathophysiology of Inflammation
4.1. General Overview of Androgen-Mediated Suppression of Inflammation
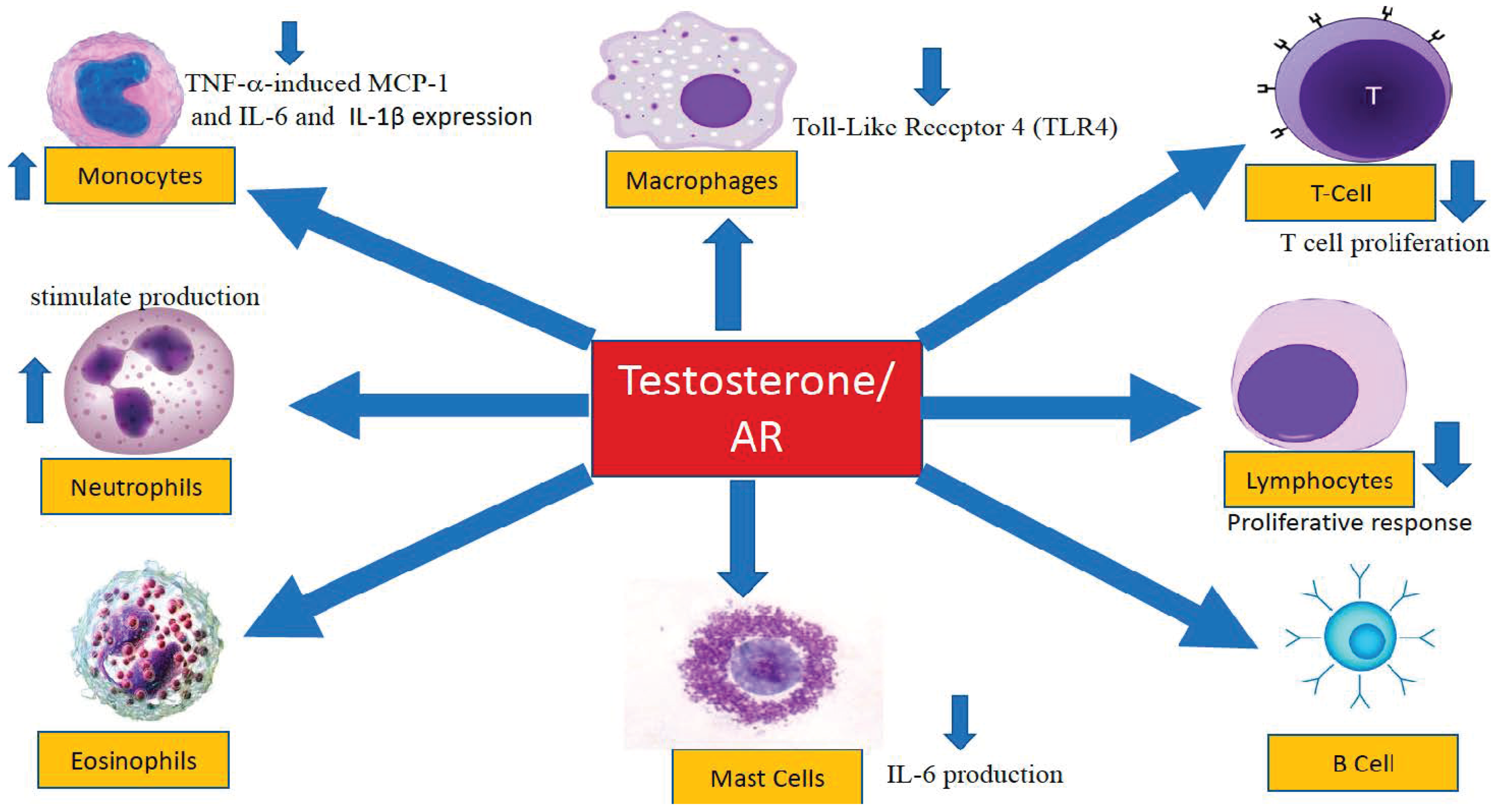
4.2. The Influence of Androgens on Specific Components of the Immune System
4.2.1. Antigen Recognition
4.2.2. Neutrophils
4.2.3. Monocytes and Adipocyte Chemoattraction
4.2.4. Macrophages
4.2.5. Mast Cells
4.2.6. Humoral Immunity
5. Discussion
Author Contributions
Conflicts of Interest
Abbreviations
| C-reactive protein | CRP |
| Tumor necrosis factor-α | TNF-α |
| Interleukin-1β | IL-1β |
| Interleukin-6 | IL-6 |
| Interleukin-10 | IL-10 |
| Non-small cell lung cancer | NSCLC |
| High-density lipoprotein | HDL |
| Coronary artery disease | CAD |
| Psoriasis area and reduced severity index | PASI |
| Physician global assessment for psoriasis | PGA |
| Androgen receptor | AR |
| 5α-dihydrotestosterone | 5α-DHT |
| Group 2 innate lymphoid cells | ILC2 |
| 5-lipoxygenase | 5-LO |
| Type 2 extracellular receptor kinase | ERK2 |
| Chronic obstructive pulmonary disease | COPD |
| Experimental autoimmune encephalomyelitis | EAE |
| Interferon-γ | IFN-γ |
| Multiple sclerosis | MS |
| Blood peripheral mononuclear cells | PBMCs |
| Tumor growth factor β1 | TGFβ1 |
| Natural killer cells | NK |
| Brain-derived neurotrophic factor | BDNF |
| Platelet-derived growth factor | PDGF-BB |
| Systemic lupus erythematosus | SLE |
| Pathogen-associated molecular patterns | PAMPs |
| Damage-associated molecular patterns | DAMPs |
| Toll-like receptors | TLRs |
| Nuclear factor kappa-light-chain-enhancer | |
| of activated B cells | NF-κB |
| Lipopolysaccharides | LPS |
| Inducible nitric oxide synthase | iNOS |
| Inducible nitric oxide | iNO |
| Experimental autoimmune orchitis | EAO |
| Androgen receptor knockout mouse model | ARKO |
| Monocyte chemoattractant protein-1 | MCP-1 |
| Receptor for activated C kinase 1 | RACK1 |
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Ashley, N.T.; Weil, Z.M.; Nelson, R.J. Inflammation: Mechanisms, Costs, and Natural Variation. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 385–406. [Google Scholar] [CrossRef]
- Miligkos, M.; Papamichael, K.; Vande Casteele, N.; Mantzaris, G.J.; Giles, A.; Levesque, B.G.; Zintzaras, E. Efficacy and safety profile of anti-tumor necrosis factor-α versus anti-integrin agents for the treatment of Crohn’s disease: A network meta-analysis of indirect comparisons. Clin. Ther. 2016, 38, 1342–1358. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Bernasconi, C.; Aassi, M.; Molina, J.F.; Epis, O.M. Treatment of Rheumatoid Arthritis With Anti–Tumor Necrosis Factor or Tocilizumab Therapy as First Biologic Agent in a Global Comparative Observational Study. Arthritis Care Res. 2017, 69, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Krisam, J.; Stremmel, W.; Gauss, A. Real-World Outcomes of Vedolizumab Therapy in Ulcerative Colitis and Crohn’s Disease at a Tertiary Referral Center. Dig. Dis. 2018, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Hernandez, J.G.; Rebollo, N.; Munoz, F.; Martin-Suarez, A.; Calvo, M.V. Therapeutic drug monitoring of tumour necrosis factor inhibitors in the management of chronic inflammatory diseases. Ann. Clin. Biochem. 2018. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Anti-inflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; CANTOS Trial Group. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H.O. Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med. 1990, 323, 1228–1233. [Google Scholar] [CrossRef]
- O’Callaghan, D.S.; O’Donnell, D.; O’Connell, F.; O’Byrne, K.J. The Role of Inflammation in the Pathogenesis of Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 2024–2036. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Basaria, S.; Ble, A.; Lauretani, F.; Bandinelli, S.; Ceda, G.P.; Valenti, G.; Ling, S.M.; Ferrucci, L. Correlation between testosterone and the inflammatory marker soluble interleukin-6 receptor in older men. J. Clin. Endocrinol. Metab. 2006, 91, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Kupelian, V.; Chiu, G.R.; Araujo, A.B.; Williams, R.E.; Clark, R.V.; McKinlay, J.B. Association of sex hormones and C-reactive protein levels in men. Clin. Endocrinol. 2010, 72, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Bobjer, J.; Naumovska, M.; Giwercman, Y.L.; Giwercman, A. High prevalence of androgen deficiency and abnormal lipid profile in infertile men with non-obstructive azoospermia. Int. J. Androl. 2012, 35, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Tsilidis, K.K.; Rohrmann, S.; McGlynn, K.A.; Nyante, S.J.; Lopez, D.S.; Bradwin, G.; Feinleib, M.; Joshu, C.E.; Kanarek, N.; Nelson, W.G.; et al. Association between endogenous sex steroid hormones and inflammatory biomarkers in US men. Andrology 2013, 1, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burney, B.O.; Hayes, T.G.; Smiechowska, J.; Cardwell, G.; Papusha, V.; Bhargava, P.; Konda, B.; Auchus, R.J.; Garcia, J.M. Low testosterone levels and increased inflammatory markers in patients with cancer and relationship with cachexia. J. Clin. Endocrinol. Metab. 2012, 97, E700–E709. [Google Scholar] [CrossRef] [PubMed]
- Tremellen, K.; McPhee, N.; Pearce, K. Metabolic endotoxaemia related inflammation is associated with hypogonadism in overweight men. Basic Clin. Androl. 2017, 27, 5. [Google Scholar] [CrossRef] [PubMed]
- Tremellen, K.; McPhee, N.; Pearce, K.; Benson, S.; Schedlowski, M.; Engler, H. Endotoxin-initiated inflammation reduces testosterone production in men of reproductive age. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E206–E213. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.K.; Liu, P.Y.; Williams, A.J.; Nakhla, S.; Ly, L.P.; Handelsman, D.J.; Celermajer, D.S. Prospective study of effect of androgens on serum inflammatory markers in men. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1136–1141. [Google Scholar] [CrossRef]
- Malkin, C.J.; Pugh, P.J.; Jones, R.D.; Kapoor, D.; Channer, K.S.; Jones, T.H. The effect of testosterone replacement on endogenous inflammatory cytokinesandlipid profiles in hypogonadalmen. J. Clin. Endocrinol. Metab. 2004, 89, 3313–3318. [Google Scholar] [CrossRef]
- Corrales, J.J.; Almeida, M.; Burgo, R.; Mories, M.T.; Miralles, J.M.; Orfao, A. Androgen-replacement therapy depresses the ex vivo production of inflammatory cytokines by circulating antigen-presenting cells in aging type-2 diabetic men with partial androgen deficiency. J. Endocrinol. 2006, 189, 595–604. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, D.; Clarke, S.; Stanworth, R.; Channer, K.S.; Jones, T.H. The effect of testosterone replacement therapy on adipocytokines and C-reactive protein in hypogonadal men with type 2 diabetes. Eur. J. Endocrinol. 2007, 156, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakhai-Pour, H.R.; Grobbee, D.E.; Emmelot-Vonk, M.H.; Bots, M.L.; Verhaar, H.J.; van der Schouw, Y.T. Oral testosterone supplementation and chronic low-grade inflammation in elderly men: A 26-week randomized, placebo-controlled trial. Am. Heart J. 2007, 154, 1228. [Google Scholar] [CrossRef] [PubMed]
- Kalinchenko, S.Y.; Tishova, Y.A.; Mskhalaya, G.J.; Gooren, L.J.; Giltay, E.J.; Saad, F. Effects of testosterone supplementation on markers of the metabolic syndrome and inflammation in hypogonadal men with the metabolic syndrome: The double-blinded placebo-controlled Moscow study. Clin. Endocrinol. 2010, 73, 602–612. [Google Scholar] [CrossRef]
- Traish, A.M.; Haider, A.; Doros, G.; Saad, F. Long-term testosterone therapy in hypogonadal men ameliorates elements of the metabolic syndrome: An observational, long-term registry study. Int. J. Clin. Pract. 2014, 68, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Haider, A.; Gooren, L. Hypogonadal men with psoriasis benefit from long-term testosterone replacement therapy—A series of 15 case reports. Andrologia 2016, 48, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.; Haider, A.; Saad, F.; Kurtz, W.; Doros, G.; Fijak, M.; Vignozzi, L.; Gooren, L. Testosterone therapy in men with Crohn’s disease improves the clinical course of the disease: Data from long-term observational registry study. Horm. Mol. Biol. Clin. Investig. 2015, 22, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Balleari, E.; Giusti, M.; Monachesi, M.; Accardo, S. Sex hormone status of male patients with rheumatoid arthritis: Evidence of low serum concentrations of testosterone at baseline and after human chorionic gonadotropin stimulation. Arthritis Rheumatol. 1988, 31, 1314–1317. [Google Scholar] [CrossRef]
- Tengstrand, B.; Carlström, K.; Hafström, I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J. Rheumatol. 2009, 36, 887–892. [Google Scholar] [CrossRef]
- Tengstrand, B.; Carlstrom, K.; Hafstrom, I. Bioavailable testosterone in men with rheumatoid arthritis- high frequency of hypogonadism. Rheumatology 2002, 41, 285–289. [Google Scholar] [CrossRef]
- Spector, T.D.; Ollier, W.; Perry, L.A.; Ailman, A.J.; Thompson, P.W.; Edwards, A. Free and serum testosterone levels in 276 males: A comparative study of rheumatoid arthritis, ankylosing spondylitis, and healthy controls. Clin. Rheumatol. 1989, 8, 37–41. [Google Scholar] [CrossRef]
- Jimenez-Balderas, F.J.; Tapia-Serrano, R.; Fonseca, M.E.; Arellano, J.; Beltran, A.; Yanez, P.; Camargo-Coronel, A.; Fraga, A. High frequency of association of rheumatic/autoimmune diseases and untreated male hypogonadism with severe testicular dysfunction. Arthritis Res. 2001, 3, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, Y.; Tan, A.; Yang, X.; Zhang, H.; Zhang, S.; Wu, C.; Lu, Z.; Wang, M.; Liao, M.; et al. Endogenous sex hormones and C-reactive protein in healthy Chinese men. Clin. Endocrinol. 2013, 78, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, S.A.; Johnson-Levonas, A.O.; Lin, J.; Shah, A.K.; Meehan, A.G. Elevated high sensitivity C-reactive protein levels in aging men with low testosterone. Aging Male 2010, 13, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, F.; Lepault, F.; Homo-Delarche, F.; Bach, J.F.; Dardenne, M. Influence of castration, alone or combined with thymectomy, on the development of diabetes in the nonobese diabetic mouse. Endocrinology 1991, 129, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Dalal, M.; Kim, S.; Voskuhl, R.R. Testosterone therapy ameliorates experimental autoimmune encephalomyelitis and induces a T helper 2 bias in the autantigen-specific T lymphocyte response. J. Immunol. 1997, 159, 3–6. [Google Scholar] [PubMed]
- Bebo, B.F., Jr.; Schuster, J.C.; Vandenbark, A.A.; Offner, H. Androgens alter the cytokine profile and reduce encephalitogenicity of myelin-reactive T cells. J. Immunol. 1999, 162, 35–40. [Google Scholar] [PubMed]
- Duan, R.S.; Link, H.; Xiao, B.G. Dehydroepiandrosterone therapy ameliorates experimental autoimmune myasthenia gravis in Lewis rats. J. Clin. Immunol. 2003, 23, 100–106. [Google Scholar] [CrossRef]
- Gold, S.M.; Voskuhl, R.R. Estrogen and testosterone therapies in multiple sclerosis. Prog. Brain Res. 2009, 175, 239–251. [Google Scholar] [Green Version]
- Dhindsa, S.; Ghanim, H.; Batra, M.; Kuhadiya, N.D.; Abuaysheh, S.; Sandhu, S.; Green, K.; Makdissi, A.; Hejna, J.; Chaudhuri, A.; et al. Insulin Resistance and Inflammation in Hypogonadotropic Hypogonadism and Their Reduction After Testosterone Replacement in Men with Type 2 Diabetes. Diabetes Care 2016, 39, 82–91. [Google Scholar] [CrossRef]
- Hackett, G. Type 2 Diabetes and Testosterone Therapy. World J. Men’s Health 2018, 26. in press. [Google Scholar] [CrossRef]
- Hackett, G.; Cole, N.; Mulay, A.; Strange, R.C.; Ramachandran, S. Long term testosterone therapy in type 2 diabetes is associated with reduced mortality without improvement in conventional cardiovascular risk factors. BJU Int. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Shigehara, K.; Konaka, H.; Kato, Y.; Iijima, M.; Nakashima, K.; Kawaguchi, S.; Nohara, T.; Izumi, K.; Namiki, M.; Mizokami, A. Effect of testosterone replacement therapy on sexual function and glycemic control among hypogonadal men with type 2 diabetes mellitus. Int. J. Impot. Res. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.; Matsumoto, M.; Tamura, Y. Potential application of testosterone replacement therapy as treatment for obesity and type 2 diabetes in men. Steroids 2018, 138, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Groti, K.; Žuran, I.; Antonič, B.; Foršnarič, L.; Pfeifer, M. The impact of testosterone replacement therapy on glycemic control, vascular function, and components of the metabolic syndrome in obese hypogonadal men with type 2 diabetes. Aging Male 2018, 21, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Saad, F. Testosterone Therapy and Glucose Homeostasis in Men with Testosterone Deficiency (Hypogonadism). Adv. Exp. Med. Biol. 2017, 1043, 527–558. [Google Scholar] [PubMed]
- Haider, A.; Haider, K.S.; Saad, F. Remission of type 2 diabetes in a hypogonadal man under long-term testosterone therapy. Endocrinol. Diabetes Metab. Case Rep. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Wickramatilake, C.M.; Mohideen, M.R.; Withanawasam, B.P.; Pathirana, C. Testosterone and high-sensitive C-reactive protein in coronary artery disease patients awaiting coronary artery bypass graft. Andrologia 2015, 47, 493–498. [Google Scholar] [CrossRef]
- Kloner, R.A.; Carson, C., 3rd; Dobs, A.; Kopecky, S.; Mohler, E.R., 3rd. Testosterone and Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 67, 545–557. [Google Scholar] [CrossRef]
- Pongkan, W.; Chattipakorn, S.C.; Chattipakorn, N. Chronic testosterone replacement exerts cardioprotection against cardiac ischemia-reperfusion injury by attenuating mitochondrial dysfunction in testosterone-deprived rats. PLoS ONE 2015, 10, e0122503. [Google Scholar] [CrossRef]
- Etminan, M.; Skeldon, S.C.; Goldenberg, S.L.; Carleton, B.; Brophy, J.M. Testosterone therapy and risk of myocardial infarction: A pharmacoepidemiologic study. Pharmacotherapy 2015, 35, 72–78. [Google Scholar] [CrossRef]
- Nettleship, J.E.; Jones, R.D.; Channer, K.S.; Jones, T.H. Testosterone and coronary artery disease. Front. Horm. Res. 2009, 37, 91–107. [Google Scholar] [PubMed]
- Traish, A.M.; Haider, A.; Haider, K.S.; Doros, G.; Saad, F. Long-Term Testosterone Therapy Improves Cardiometabolic Function and Reduces Risk of Cardiovascular Disease in Men with Hypogonadism: A Real-Life Observational Registry Study Setting Comparing Treated and Untreated (Control) Groups. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, T.C.; An, J.; Jacobsen, S.J.; Niu, F.; Sidney, S.; Quesenberry, C.P.; VanDenEeden, S.K. Association of Testosterone Replacement With Cardiovascular Outcomes Among Men With Androgen Deficiency. JAMA Intern. Med. 2017, 177, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; May, H.T.; Lappé, D.L.; Bair, T.; Le, V.; Carlquist, J.F.; Muhlestein, J.B. Impact of Testosterone Replacement Therapy on Myocardial Infarction, Stroke, and Death in Men with Low Testosterone Concentrations in an Integrated Health Care System. Am. J. Cardiol. 2016, 117, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Oni, O.A.; Gupta, K.; Chen, G.; Sharma, M.; Dawn, B.; Sharma, R.; Parashara, D.; Savin, V.J.; Ambrose, J.A.; et al. Normalization of testosterone level is associated with reduced incidence of myocardial infarction and mortality in men. Eur. Heart J. 2015, 36, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baillargeon, J.; Al Snih, S.; Raji, M.A.; Urban, R.J.; Sharma, G.; Sheffield-Moore, M.; Lopez, D.S.; Baillargeon, G.; Kuo, Y.F. Hypogonadism and the risk of rheumatic autoimmune disease. Clin. Rheumatol. 2016, 35, 2983–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisk, G.H.; Howard, R.P.; Fay, K. Rheumatoid arthritis. I. Clinical effects of testosterone and pregnenolone therapy. Can. Med. Assoc. J. 1950, 63, 342–344. [Google Scholar] [PubMed]
- Ganesan, K.; Balachandran, C.; Manohar, B.M.; Puvanakrishnan, R. Effects of testosterone, estrogen and progesterone on TNF-α mediated cellular damage in rat arthritic synovial fibroblasts. Rheumatol. Int. 2012, 32, 3181–3188. [Google Scholar] [CrossRef]
- Cutolo, M.; Montagna, P.; Brizzolara, R.; Sulli, A.; Seriolo, B.; Villaggio, B.; Triolo, P.; Clerico, P.; Soldano, S. Sex hormones modulate the effects of Leflunomide on cytokine production by cultures of differentiated monocyte/macrophages and synovial macrophages from rheumatoid arthritis patients. J. Autoimmun. 2009, 32, 254–260. [Google Scholar] [CrossRef]
- Cutolo, M.; Sulli, A.; Capellino, S.; Villaggio, B.; Montagna, P.; Pizzorni, C.; Paolino, S.; Seriolo, B.; Felli, L.; Straub, R.H. Anti-TNF and sex hormones. Ann. N. Y. Acad. Sci. 2006, 1069, 391–400. [Google Scholar] [CrossRef]
- Pope, J.E.; Joneja, M.; Hong, P. Anti-androgen treatment of prostatic carcinoma may be a risk factor for development of rheumatoid arthritis. J. Rheumatol. 2002, 29, 2459–2462. [Google Scholar] [PubMed]
- Cutolo, M.; Seriolo, B.; Villaggio, B.; Pizzorni, C.; Craviotto, C.; Sulli, A. Androgens and estrogens modulate the immune and inflammatory responses in rheumatoid arthritis. Ann. N. Y. Acad. Sci. 2002, 966, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M. Sex hormone adjuvant therapy in rheumatoid arthritis. Rheum. Dis. Clin. North Am. 2000, 26, 881–895. [Google Scholar] [CrossRef]
- Hall, G.M.; Larbre, J.P.; Spector, T.D.; Perry, L.A.; Da Silva, J.A. A randomized trial of testosterone therapy in males with rheumatoid arthritis. Br. J. Rheumatol. 1996, 35, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Wimer, B.M.; Sloan, M.M. Remission of Felty’s syndrome with long-term testosterone therapy. JAMA 1973, 223, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Laffont, S.; Blanquart, E.; Savignac, M.; Cenac, C.; Laverny, G.; Metzger, D.; Girard, J.P.; Belz, G.T.; Pelletier, L.; Seillet, C.; Guery, J.C. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Cephus, J.Y.; Stier, M.T.; Fuseini, H.; Yung, J.A.; Toki, S.; Bloodworth, M.H.; Zhou, W.; Goleniewska, K.; Zhang, J.; Garon, S.L.; et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep. 2017, 21, 2487–2499. [Google Scholar] [CrossRef]
- Montaño, L.M.; Espinoza, J.; Flores-Soto, E.; Chávez, J.; Perusquía, M. Androgens are bronchoactive drugs that act by relaxing airway smooth muscle and preventing bronchospasm. J. Endocrinol. 2014, 222, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Canguven, O.; Albayrak, S. Do low testosterone levels contribute to the pathogenesis of asthma? Med. Hypotheses 2011, 76, 585–588. [Google Scholar] [CrossRef]
- Kamischke, A.; Kemper, D.E.; Castel, M.A.; Lüthke, M.; Rolf, C.; Behre, H.M.; Magnussen, H.; Nieschlag, E. Testosterone levels in men with chronic obstructive pulmonary disease with or without glucocorticoid therapy. Eur. Respir. J. 1998, 11, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Baillargeon, J.; Urban, R.J.; Zhang, W.; Zaiden, M.F.; Javed, Z.; Sheffield-Moore, M.; Kuo, Y.F.; Sharma, G. Testosterone replacement therapy and hospitalization rates in men with COPD. Chron. Respir. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Atlantis, E.; Fahey, P.; Cochrane, B.; Wittert, G.; Smith, S. Endogenous testosterone level and testosterone supplementation therapy in chronic obstructive pulmonary disease (COPD): A systematic review and meta-analysis. BMJ Open 2013, 3, e003127. [Google Scholar] [CrossRef] [PubMed]
- Samaras, N.; Samaras, D.; Lang, P.O.; Bridevaux, P.O.; Gex, G.; Janssens, J.P.; Pichard, C. Chronic obstructive pulmonary disease: Risk and benefit of testosterone therapy. Rev. Med. Suisse 2012, 8, 2224–2227. [Google Scholar] [PubMed]
- Velema, M.S.; Kwa, B.H.; de Ronde, W. Should androgenic anabolic steroids be considered in the treatment regime of selected chronic obstructive pulmonary disease patients? Curr. Opin. Pulm. Med. 2012, 18, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Svartberg, J. Androgens and chronic obstructive pulmonary disease. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Puhan, M.A.; Schünemann, H.J. Testosterone Supplementation during Respiratory Rehabilitation. Am. J. Respir. Crit. Care Med. 2005, 172, 399. [Google Scholar] [CrossRef] [PubMed]
- Casaburi, R.; Bhasin, S.; Cosentino, L.; Porszasz, J.; Somfay, A.; Lewis, M.I.; Fournier, M.; Storer, T.W. Effects of testosterone and resistance training in men with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2004, 170, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Creutzberg, E.C.; Casaburi, R. Endocrinological disturbances in chronic obstructive pulmonary disease. Eur. Respir. J. Suppl. 2003, 46, 76s–80s. [Google Scholar] [CrossRef]
- Gold, S.M.; Chalifoux, S.; Giesser, B.S.; Voskuhl, R.R. Immune modulation and increased neurotrophic factor production in multiple sclerosis patients treated with testosterone. J. Neuroinflamm. 2008, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Gold, S.M.; Voskuhl, R.R. Testosterone replacement therapy for the treatment of neurological and neuropsychiatric disorders. Curr. Opin. Investig. Drugs 2006, 7, 625–630. [Google Scholar]
- Collongues, N.; Patte-Mensah, C.; De Seze, J.; Mensah-Nyagan, A.G.; Derfuss, T. Testosterone and estrogen in multiple sclerosis: From pathophysiology to therapeutics. Expert Rev. Neurother. 2018, 18, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Ziehn, M.O.; Avedisian, A.A.; Dervin, S.M.; Umeda, E.A.; O’Dell, T.J.; Voskuhl, R.R. Therapeutic testosterone administration preserves excitatory synaptic transmission in the hippocampus during autoimmune demyelinating disease. J. Neurosci. 2012, 32, 12312–12324. [Google Scholar] [CrossRef] [PubMed]
- Sicotte, N.L.; Giesser, B.S.; Tandon, V.; Klutch, R.; Steiner, B.; Drain, A.E.; Shattuck, D.W.; Hull, L.; Wang, H.J.; Elashoff, R.M.; et al. Testosterone treatment in multiple sclerosis: A pilot study. Arch. Neurol. 2007, 64, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Pakpoor, J.; Goldacre, R.; Goldacre, M.J. Associations between clinically diagnosed testicular hypofunction and systemic lupus erythematosus: A record linkage study. Clin. Rheumatol. 2018, 37, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.; Wallace, D.J.; Shinada, S.; Kalunian, K.C.; Forbess, L.; Braunstein, G.D.; Weisman, M.H. Testosterone patches in the management of patients with mild/moderate systemic lupus erythematosus. Rheumatology 2008, 47, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Yamauchi, K.; Sato, R.; Masuda, T.; Sawai, T.; Inoue, H. Klinefelter’s syndrome associated with systemic lupus erythematosus and autoimmune hepatitis. Mod. Rheumatol. 2006, 16, 305–308. [Google Scholar] [CrossRef]
- Olsen, N.J.; Kovacs, W.J. Case report: Testosterone treatment of systemic lupus erythematosus in a patient with Klinefelter’s syndrome. Am. J. Med. Sci. 1995, 310, 158–160. [Google Scholar] [CrossRef]
- Bizzarro, A.; Valentini, G.; Di Martino, G.; DaPonte, A.; De Bellis, A.; Iacono, G. Influence of testosterone therapy on clinical and immunological features of autoimmune diseases associated with Klinefelter’s syndrome. J. Clin. Endocrinol. Metab. 1987, 64, 32–36. [Google Scholar] [CrossRef]
- Amor, B.; Dougados, M.; Benhamou, L.; Kuhn, J.M.; Laudat, M.H. Failure of androgen therapy in a flare-up of acute disseminated lupus erythematosus. Presse Med. 1983, 12, 1726. [Google Scholar]
- Costello, M.J.; Singer, J.I. Lupus erythematosus, chronic disseminated type, showing a dramatic response to testosteronetherapy. AMA Arch. Derm. Syphilol. 1952, 65, 631–632. [Google Scholar]
- Yang, Y.M.; Lv, X.Y.; Huang, W.D.; Xu, Z.R.; Wu, L.J. Study of androgen and atherosclerosis in old-age male. J. Zhejiang Univ. Sci. B 2005, 6, 931–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoodi, G.; Amirezadegan, A.; Borumand, M.A.; Dehkori, M.R.; Kazemisaeid, A.; Yaminisharif, A. The relationship between level of androgenic hormones and coronary artery disease in men. Cardiovasc. J. Afr. 2007, 18, 362–366. [Google Scholar] [PubMed]
- Coumbe, A.G.; Pritzker, M.R.; Duprez, D.A. Cardiovascular risk and psoriasis: Beyond the traditional risk factors. Am. J. Med. 2014, 127, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Horreau, C.; Pouplard, C.; Brenaut, E.; Barnetche, T.; Misery, L.; Cribier, B.; Jullien, D.; Aractingi, S.; Aubin, F.; Joly, P.; et al. Cardiovascular morbidity and mortality in psoriasis and psoriatic arthritis: A systematic literature review. J. Eur. Acad. Dermatol. Venereol. 2013, 27 (Suppl. 3), 12–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Krueger, J.G. The immunopathogenesis of psoriasis. Dermatol. Clin. 2015, 33, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J. Inhibition of interleukin-17, interleukin-23 and the TH17 cell pathway in the treatment of psoriatic arthritis and psoriasis. Curr. Opin. Rheumatol. 2015, 27, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Balleari, E.; Giusti, M.; Intra, E.; Accardo, S. Androgen replacement therapy in male patients with rheumatoid arthritis. Arthritis Rheum. 1991, 34, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booji, A.; Biewenga-Booji, C.M.; Huber-Bruning, O.; Cornelis, C.; Jacobs, J.W.; Bijlsma, J.W. Androgens as adjuvant treatment in postmenopausal female patients with rheumatoid arthritis. Ann. Rheum. Dis. 1996, 55, 811–815. [Google Scholar] [CrossRef]
- Melgert, B.N.; Ray, A.R.; Hylkema, M.N.; Timens, W.; Postma, D.S. Are There Reasons Why Adult Asthma is More Common in Females? Curr. Allergy Asthma Rep. 2007, 7, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Salam, M.T.; Wenten, M.; Gilliland, F.D. Endogenous and exogenous sex steroid hormones and asthma and wheeze in young women. J. Allergy Clin. Immunol. 2006, 117, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Postma, D.S.; Kerstjens, H.A. Perimenstrual asthma: A syndrome without known cause or cure. J. Allergy Clin. Immunol. 2003, 112, 271–282. [Google Scholar] [CrossRef]
- Mileva, Z.; Maleeva, A. The serum testosterone level of patients with bronchial asthma treated with corticosteroids and untreated. Vutr. Boles. 1988, 27, 29–32. [Google Scholar] [PubMed]
- Kwon, H.L.; Belanger, K.; Holford, T.R.; Bracken, M.B. Effect of fetal sex on airway lability in pregnant women with asthma. Am. J. Epidemiol. 2006, 163, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Adachi, Y.; Hasegawa, K.; Morimoto, M. Less sensitivity for late airway inflammation in males than females in BALB/c mice. Scand. J. Immunol. 2003, 57, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Th2 cytokines and asthma: An introduction. Respir. Res. 2001, 2, 64–65. [Google Scholar] [CrossRef]
- Huber, S.A.; Pfaeffle, B. Differential Th1 and Th2 cell responses in male and female BALB/c mice infected with coxsackievirus group B type 3. J. Virol. 1994, 68, 5126–5132. [Google Scholar]
- Lund, S.J.; Portillo, A.; Cavagnero, K.; Baum, R.E.; Naji, L.H.; Badrani, J.H.; Mehta, A.; Croft, M.; Broide, D.H.; Doherty, T.A. Leukotriene C4 potentiates IL-33-Induced Group 2 Innate Lymphoid Cell Activation and Lung Inflammation. J. Immunol. 2017, 199, 1096–1104. [Google Scholar] [CrossRef]
- Pergola, C.; Dodt, G.; Rossi, A.; Neunhoeffer, E.; Lawrenz, B.; Northoff, H.; Samuelsson, B.; Radmark, O.; Sautebin, L.; Werz, O. ERK-mediated regulation of leukotriene biosynthesis by androgens: A molecular basis for gender differences in inflammation and asthma. Proc. Natl. Acad. Sci. USA 2008, 105, 19881–19886. [Google Scholar] [CrossRef] [Green Version]
- Johnston, N.W.; Mandhane, P.J.; Dai, J.; Duncan, J.M.; Green, J.M.; Lambert, K.; Sears, M.R. Attenuation of the September epidemic of asthma exacerbations in children: A randomized, controlled trial of montelukast added to usual therapy. Pediatrics 2007, 120, e702–e712. [Google Scholar] [CrossRef] [PubMed]
- Semple, P.D.; Beastall, G.H.; Watson, W.S.; Hume, R. Serum testosterone depression associated with hypoxia in respiratory failure. Clin. Sci. 1980, 58, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Palaszynski, K.M.; Loo, K.K.; Ashouri, J.F.; Liu, H.B.; Voskuhl, R.R. Androgens are protective in experimental autoimmune encephalomyelitis: Implications for multiple sclerosis. J. Neuroimmunol. 2004, 146, 144–152. [Google Scholar] [CrossRef]
- Liva, S.M.; Voskuhl, R.R. Testosterone acts directly on CD4+ T lymphocytes to increase IL-10 production. J. Immunol. 2001, 167, 2060–2067. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Mok, C.C. Development of systemic lupus erythematosus in a male-to-female transsexual: The role of sex hormones revisited. Lupus 2013, 22, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Gubbels Bupp, M.R.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Hemmrich, G.; Klostermeier, U.C.; L´ opez-Quintero, J.A.; Miller, D.J.; Rahn, T.; Weiss, Y.; Bosch, T.C.; Rosenstiel, P. Defining the origins of the NOD-like receptor system at the base of animal evolution. Mol. Biol. Evol. 2001, 28, 1687–1702. [Google Scholar] [CrossRef] [PubMed]
- Proell, M.; Riedel, S.J.; Fritz, J.H.; Rojas, A.M.; Schwarzenbacher, R. The Nod-like receptor (NLR) family: A tale of similarities and differences. PLoS ONE 2008, 3, e2199. [Google Scholar] [CrossRef]
- Roach, J.C.; Glusman, G.; Rowen, L.; Kaur, A.; Purcell, M.K.; Smith, K.D.; Hood, L.E.; Aderem, A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 9577–9582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitzmann, M.; Erren, M.; Kamischke, A.; Simoni, M.; Nieschlag, E. Endogenous progesterone and the exogenous progestin norethisterone enanthate are associated with a proinflammatory profile in healthy men. J. Clin. Endocrinol. Metab. 2005, 90, 6603–6608. [Google Scholar] [CrossRef]
- Guler, N.; Batyraliev, T.; Dulger, H.; Ozkara, C.; Tuncer, M.; Aslan, S.; Okut, H.; Agirbasli, M. The effects of short term (3 weeks) testosterone treatment on serum inflammatory markers in men undergoing coronary artery stenting. Int. J. Cardiol. 2006, 109, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Nettleship, J.E.; Pugh, P.J.; Channer, K.S.; Jones, T.; Jones, R.D. Inverse relationship between serum levels of interleukin-1beta and testosterone in men with stable coronary artery disease. Horm. Metab. Res. 2007, 39, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Corrales, J.J.; Almeida, M.; Miralles, J.M.; Orfao, A. Persistence of androgenic effects on the production of proinflammatory cytokines by circulating antigen-presenting cells after withdrawal of testosterone treatment in aging type 2 diabetic men with partial androgen deficiency. Fertil. Steril. 2009, 92, 311–319. [Google Scholar] [CrossRef]
- Gilliver, S.C. Sex steroids as inflammatory regulators. J. Steroid Biochem. Mol. Biol. 2010, 120, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Viselli, S.M.; Reese, K.R.; Fan, J.; Kovacs, W.J.; Olsen, N.J. Androgens alter B cell development in normal male mice. Cell. Immunol. 1997, 182, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Mantalaris, A.; Panoskaltsis, N.; Sakai, Y.; Bourne, P.; Chang, C.; Messing, E.M.; Wu, J.H. Localization of androgen receptor expression in human bone marrow. J. Pathol. 2001, 193, 361–366. [Google Scholar] [CrossRef]
- Chen, W.; Beck, I.; Schober, W.; Brockow, K.; Effner, R.; Buters, J.T.; Behrendt, H.; Ring, J. Human mast cells express androgen receptors but treatment with testosterone exerts no influence on IgE-independent mast cell degranulation elicited by neuromuscular blocking agents. Exp. Dermatol. 2010, 19, 302–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.J.; Lai, K.P.; Zeng, W.; Chuang, K.H.; Altuwaijri, S.; Chang, C. Androgen receptor influences on body defense system via modulation of innate and adaptiveimmune systems: Lessons from conditional AR knockout mice. Am. J. Pathol. 2012, 181, 1504–1512. [Google Scholar] [CrossRef]
- Freeman, B.M.; Mountain, D.J.; Brock, T.C.; Chapman, J.R.; Kirkpatrick, S.S.; Freeman, M.B.; Klein, F.A.; Grandas, O.H. Low testosterone elevates interleukin family cytokines in a rodent model: A possible mechanism for the potentiation of vascular disease in androgen-deficient males. J. Surg. Res. 2014, 190, 319–327. [Google Scholar] [CrossRef]
- Chen, C.W.; Jian, C.Y.; Lin, P.H.; Chen, C.C.; Lieu, F.K.; Soong, C.; Hsieh, C.C.; Wan, C.Y.; Idova, G.; Hu, S.; et al. Role of testosterone in regulating induction of TNF-α in rat spleen via ERK signaling pathway. Steroids 2016, 111, 148–154. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, M.Y.; Guo, D.J.; Wan, C.W.; Lau, C.C.; Chan, C.O.; Mok, D.K.W.; Chan, S.W. Gui-ling-gao (turtle jelly), a traditional Chinese functional food, exerts anti-inflammatory effects by inhibiting iNOS and pro-inflammatory cytokine expressions in splenocytes isolated from BALB/c mice. J. Funct. Foods 2013, 5, 625–632. [Google Scholar] [CrossRef]
- Ali, T.; Kaitha, S.; Mahmood, S.; Ftesi, A.; Stone, J.; Bronze, M.S. Clinical use of anti-TNF therapy and increased risk of infections. Drug Healthc. Patient Saf. 2013, 5, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Li, D.W.; Xiang, L.; Chang, J.J.; Xia, Z.L.; Han, E.J. Neuroprotective effects of an aqueous extract of Futokadsura stem in an Aβ-induced Alzheimer’s disease-like rat model. Chin. J. Physiol. 2015, 58, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pugh, T.D.; Stebler, B.; Ershler, W.B.; Keller, E.T. Orchiectomy Increases Bone Marrow Interleukin-6 Levels in Mice. Calcif. Tissue Int. 1998, 62, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Steffens, J.P.; Herrera, B.S.; Coimbra, L.S.; Stephens, D.N.; Rossa, C., Jr.; Spolidorio, L.C.; Kantarci, A.; Van Dyke, T.E. Testosterone regulates bone response to inflammation. Horm. Metab. Res. 2014, 46, 193–200. [Google Scholar] [CrossRef]
- Lammers, A.J.; de Porto, A.P.; de Boer, O.J.; Florquin, S.; van der Poll, T. The role of TLR2 in the host response to pneumococcal pneumonia in absence of the spleen. BMC Infect. Dis. 2012, 12, 139. [Google Scholar] [CrossRef]
- Le, M.Q.; Kim, M.S.; Song, Y.S.; Ryu, H.W.; Oh, S.R.; Yoon, D.Y. 6-O-Veratroyl catalpol suppresses pro-inflammatory cytokines via regulation of extracellular signal-regulated kinase and nuclear factor-κB in human monocytic cells. Biochimie 2015, 119, 52–59. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, J.; Xin, W.; Li, Y.; Ni, L.; Ma, X.; Zhang, D.; Zhang, D.; Zhang, T.; Du, G. Anti-inflammation effect of methyl salicylate 2-O-β-D-lactoside on adjuvant induced-arthritis rats and lipopolysaccharide (LPS)-treated murine macrophages RAW264.7 cells. Int. Immunopharmacol. 2015, 25, 88–95. [Google Scholar] [CrossRef]
- Gong, Z.; Zhou, J.; Li, H.; Gao, Y.; Xu, C.; Zhao, S.; Chen, Y.; Cai, W.; Wu, J. Curcumin suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Mol. Nutr. Food Res. 2015, 59, 2132–2142. [Google Scholar] [CrossRef]
- Fijak, M.; Schneider, E.; Klug, J.; Bhushan, S.; Hackstein, H.; Schuler, G.; Wygrecka, M.; Gromoll, J.; Meinhardt, A. Testosterone replacement effectively inhibits the development of experimental autoimmune orchitis in rats: Evidence for a direct role of testosterone on regulatory T cell expansion. J. Immunol. 2011, 186, 5162–5172. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Lin, J.; Fu, L.; Mei, Y.F.; Peng, G.; Tan, X.; Wang, D.M.; Wang, W.; Li, Y.G. Physiological testosterone stimulates tissue plasminogen activator and tissue factor pathway inhibitor and inhibits plasminogen activator inhibitor type 1 release in endothelial cells. Biochem. Cell Biol. 2007, 85, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Qiu, W.B.; Mei, Y.F.; Wang, D.M.; Li, Y.G.; Tan, X.R. Testosterone alleviates tumor necrosis factor-alpha-mediated tissue factor pathway inhibitor downregulation via suppression of nuclear factor-kappa B in endothelial cells. Asian J. Androl. 2009, 11, 266–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norata, G.D.; Cattaneo, P.; Poletti, A.; Catapano, A.L. The androgen derivative 5alpha-androstane-3beta, 17beta-diol inhibits tumor necrosis factor alpha and lipopolysaccharide induced inflammatory response in human endothelial cells and in mice aorta. Atherosclerosis 2010, 212, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Tibolla, G.; Seccomandi, P.M.; Poletti, A.; Catapano, A.L. Dihydrotestosterone decreases tumor necrosis factor-alpha and lipopolysaccharide-induced inflammatory response in human endothelial cells. J. Clin. Endocrinol. Metab. 2006, 91, 546–554. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.; Milano, S.; Barbera, C.; Di Bella, G.; La Rosa, M.; Ferlazzo, V.; Farruggio, R.; Miceli, D.M.; Miele, M.; Castagnetta, L.; et al. Sex hormones modulate inflammatory mediators produced by macrophages. Ann. N. Y. Acad. Sci. 1999, 876, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Liang, J.; Han, X.; Hou, Y. In vivo modulation of the circulating lymphocyte subsets and monocytes by androgen. Int. Immunopharmacol. 2003, 3, 1853–1860. [Google Scholar] [CrossRef]
- Quintar, A.A.; Roth, F.D.; De Paul, A.L.; Aoki, A.; Maldonado, C.A. Toll-like receptor 4 in rat prostate: Modulation by testosterone and acute bacterial infection in epithelial and stromal cells. Biol. Reprod. 2006, 75, 664–672. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Chuang, K.H.; Altuwaijri, S.; Li, G.; Lai, J.J.; Chu, C.Y.; Lai, K.P.; Lin, H.Y.; Hsu, J.W.; Keng, P.; Wu, M.C.; et al. Neutropenia with impaired host defense against microbial infection in mice lacking androgen receptor. J. Exp. Med. 2009, 206, 1181–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morooka, N.; Ueguri, K.; Yee, K.K.L.; Yanase, T.; Sato, T. Androgen-receptor system improves chronic inflammatory conditions by suppressing monocyte chemoattractant protein-1 gene expression in adipocytes via transcriptional regulation. Biochem. Biophys. Res. Commun. 2016, 477, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Surette, M.E.; Nadeau, M.; Borgeat, P.; Gosselin, J. Priming of human peripheral blood mononuclear cells with lipopolysaccharides for enhanced arachidonic acid release and leukotriene synthesis. J. Leukoc. Biol. 1996, 59, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Pergola, C.; Rogge, A.; Dodt, G.; Northoff, H.; Weinigel, C.; Barz, D.; Rådmark, O.; Sautebin, L.; Werz, O. Testosterone suppresses phospholipase D, causing sex differences in leukotriene biosynthesis in human monocytes. FASEB J. 2011, 25, 3377–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, J.; Borgeat, P. Epstein-Barr virus modulates 5-lipoxygenase product synthesis in human peripheral blood mononuclear cells. Blood 1997, 89, 2122–2130. [Google Scholar] [PubMed]
- Rai, U. Sex steroid hormones modulate the activation of murine peritoneal macrophages: Receptor mediated modulation. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1998, 119, 199–204. [Google Scholar]
- Kanda, N.; Tsuchida, T.; Tamaki, K. Testosterone inhibits immunoglobulin production by human peripheral blood mononuclear cells. Clin. Exp. Immunol. 1996, 106, 410–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, N.; Tsuchida, T.; Tamaki, K. Testosterone suppresses anti-DNA antibody production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1997, 40, 1703–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagi, M.; Morishita, M.; Iwamoto, Y. Effects of sex hormones on production of prostaglandin E2 by human peripheral monocytes. J. Periodontol. 1993, 64, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Capellino, S.; Montagna, P.; Ghiorzo, P.; Sulli, A.; Villaggio, B. Sex hormone modulation of cell growth and apoptosis of the human monocytic/macrophage cell line. Arthritis Res. Ther. 2005, 7, R1124–R1132. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Galbiati, V.; Papale, A.; Kummer, E.; Pinto, A.; Serafini, M.M.; Guaita, A.; Spezzano, R.; Caruso, D.; Marinovich, M.; et al. Role of androgens in dhea-induced rack1 expression and cytokine modulation in monocytes. Immun. Ageing 2016, 29, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Corrales, J.J.; Almeida, M.; Martín-Martín, L.; Miralles, J.M.; Orfao, A. Testosterone replacement therapy in hypogonadal men is associated with increased expression of LAMP-2 (CD107b) by circulating monocytes and dendritic cells. Clin. Endocrinol. 2014, 80, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, M.P.; Meydani, M.; Lichtenstein, A.H.; Schaefer, E.J.; Dillard, A.; Lamon-Fava, S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J. Endocrinol. 2010, 206, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, T.C.; Van Alten, P.J.; Greager, J.A.; Walter, R.J. Steroid sex hormones regulate the release of tumor necrosis factor by macrophages. Cell. Immunol. 1995, 160, 43–49. [Google Scholar] [CrossRef]
- Friedl, R.; Brunner, M.; Moeslinger, T.; Spieckermann, P.G. Testosterone inhibits expression of inducible nitric oxide synthase in murine macrophages. Life Sci. 2000, 68, 417–429. [Google Scholar] [CrossRef]
- Da Silva, E.Z.; Jamur, M.C.; Oliver, C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014, 62, 698–738. [Google Scholar] [CrossRef]
- Gonçalves, B.F.; Campos, S.G.; Costa, C.F.; Scarano, W.R.; Góes, R.M.; Taboga, S.R. Key participants of the tumor microenvironment of the prostate: An approach of the structural dynamic of cellular elements and extracellular matrix components during epithelial-stromal transition. Acta Histochem. 2015, 117, 4–13. [Google Scholar] [CrossRef]
- Guhl, S.; Artuc, M.; Zuberbier, T.; Babina, M. Testosterone exerts selective anti-inflammatory effects on human skin mast cells in a cell subset dependent manner. Exp. Dermatol. 2012, 21, 878–880. [Google Scholar] [CrossRef] [Green Version]
- Coletta, R.D.; Reynolds, M.A.; Martelli-Junior, H.; Graner, E.; Almeida, O.P.; Sauk, J.J. Testosterone stimulates proliferation and inhibits interleukin 6 production of noral and hereditary gingival fibromatosis fibroblasts. Oral Microbiol. Immunol. 2002, 17, 186–192. [Google Scholar] [CrossRef]
- Keller, E.T.; Chang, C.; Ershler, W.B. Inhibition of NFkappaB activity through maintenance of IkappaBalpha levels contributes to dihydrotestosterone-mediated repression of the interleukin-6 promoter. J. Biol. Chem. 1996, 271, 26267–26275. [Google Scholar] [CrossRef] [PubMed]
- Altuwaijri, S.; Chuang, K.H.; Lai, K.P.; Lai, J.J.; Lin, H.Y.; Young, F.M.; Bottaro, A.; Tsai, M.Y.; Zeng, W.P.; Chang, H.C.; et al. Susceptibility to autoimmunity and B cell resistance to apoptosis in mice lacking androgen receptor in B cells. Mol. Endocrinol. 2009, 23, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Kupperman, J.; Newell, M.K. Hormonal regulation of CD4+ T-cell responses in coxsackievirus B3-induced myocarditis in mice. J. Virol. 1999, 73, 4689–4695. [Google Scholar]
- Huber, S.A.; Kupperman, J.; Newell, M.K. Estradiol prevents and testosterone promotes Fas-dependent apoptosis in CD4+ Th2 cells by altering Bcl 2 expression. Lupus 1999, 8, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Kissick, H.T.; Sanda, M.G.; Dunn, L.K.; Pellegrini, K.L.; On, S.T.; Noel, J.K.; Arredouani, M.S. Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 9887–9892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignozzi, L.; Morelli, A.; Sarchielli, E.; Comeglio, P.; Filippi, S.; Cellai, I.; Maneschi, E.; Semi, S.; Gacci, M.; Carini, M.; et al. Testosterone protects from metabolic syndrome-associated prostate inflammation: An experimental study in rabbit. J. Endocrinol. 2012, 212, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Vignozzi, L.; Gacci, M.; Cellai, I.; Santi, R.; Corona, G.; Morelli, A.; Rastrelli, G.; Comeglio, P.; Sebastanelli, A.; Maneschi, E.; et al. Fat boosts, while androgen receptor activation counteracts, BPH associated prostate inflammation. Prostate 2013, 73, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Vignozzi, L.; Cellai, I.; Santi, R.; Lombardelli, L.; Morelli, A.; Comeglio, P.; Filippi, S.; Logiodice, F.; Carini, M.; Nesi, G.; et al. Anti-inflammatory effect of androgen receptor activation in human benign prostatic hyperplasia cells. J. Endocrinol. 2012, 211, 31–43. [Google Scholar]
- Haider, A.; Gooren, L.J.; Padungtod, P.; Saad, F. Improvement of the metabolic syndrome and of non-alcoholic liver steatosis upon treatment of hypogonadal elderly men with parenteral testosterone undecanoate. Exp. Clin. Endocrinol. Diabetes 2010, 118, 167–1671. [Google Scholar] [CrossRef]
- Carneiro, F.S.; Webb, R.C.; Tostes, R.C. Emerging role for TNF-α in erectile dysfunction. J. Sex Med. 2010, 7, 3823–3834. [Google Scholar] [CrossRef]
- Anderson, H.D.; Rahmutula, D. Gardner DG Tumor necrosis factor-alpha inhibits endothelial nitric-oxide synthase gene promoter activity in bovine aortic endothelial cells. J. Biol. Chem. 2004, 279, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Perrella, M.A.; Burnett, J.C., Jr.; Lee, M.E. Tumor necrosis factor down-regulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res. 1993, 73, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Filippi, S.; Vignozzi, L.; Morelli, A.; Chavalmane, A.K.; Sarchielli, E.; Fibbi, B.; Saad, F.; Sandner, P.; Ruggiano, P.; Vannelli, G.B.; et al. Testosterone partially ameliorates metabolic profile and erectile responsiveness to PDE5 inhibitors in an animal model of male metabolic syndrome. J. Sex Med. 2009, 6, 3274–3288. [Google Scholar] [CrossRef] [PubMed]
- Vignozzi, L.; Morelli, A.; Filippi, S.; Comeglio, P.; Chavalmane, A.K.; Marchetta, M.; Toce, M.; Yehiely-Cohen, R.; Vannelli, G.B.; Adorini, L.; et al. Farnesoid X receptor activation improves erectile function in animal models of metabolic syndrome and diabetes. J. Sex Med. 2011, 8, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Maneschi, E.; Morelli, A.; Filippi, S.; Cellai, I.; Comeglio, P.; Mazzanti, B.; Mello, T.; Calcagno, A.; Sarchielli, E.; Vignozzi, L.; et al. Testosterone treatment improves metabolic syndrome-induced adipose tissue derangements. J. Endocrinol. 2012, 215, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maneschi, E.; Vignozzi, L.; Morelli, A.; Mello, T.; Filippi, S.; Cellai, I.; Comeglio, P.; Sarchielli, E.; Calcagno, A.; Mazzaniti, B.; et al. FXR activation normalizes insulin sensitivity in visceral preadipocytes of a rabbit model of Met S. J. Endocrinol. 2013, 218, 215–231. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Study Design | Major Findings | Authors Comments |
|---|---|---|---|
| Maggio et al., 2006 [11] | This study included 497 male participants, aged 65 years and older, excluded 22 men taking glucocorticoids, exogenous androgens, or antibiotic treatment and eight who had been recently hospitalized. The final analysis included 467 men (mean age, 74.8 ± 7.0 years; range, 65–97 years). | Older men tended to have higher levels of IL-6, TNF-α, IL-1β, and C-reactive protein (CRP), but the levels of soluble IL-6 receptor (sIL-6r) were not different. Levels of sIL-6r were inversely related to total testosterone and bioavailable testosterone. IL-6 was not associated with total testosterone or bioavailable testosterone. After adjusting for age and multiple confounders, the negative correlation between sIL-6r and testosterone and bioavailable testosterone was maintained. No independent association of testosterone or bio-availability with TNF-α or IL-1β CRP was found. | Testosterone was significantly, inversely and independently associated with sIL-6r, but not with IL-6, IL-1β, TNF-α, or CRP. Study findings suggest that testosterone may decrease inflammation by reducing the production of sIL-6r. |
| Kupelian et al., 2010 [12] | Cross-sectional observational survey of a random sample of 2301 racially and ethnically diverse men aged 30–79 years. Analyses were conducted on 1559 men, with complete data on CRP and sex hormone levels. | About 40% of men were overweight with body mass index (BMI) of 25.0–29.9 and one-third were obese (BMI ≥ 30). Over half of the analysis samples reported the use of anti-inflammatories or other medications that could affect CRP levels. One-third of men were at moderate cardiovascular risk (CRP levels 9.5–28.5 nmol/L) while almost 20% were at high risk (CRP >28.5 nmol/L). Strong correlation between testosterone/SHBG (sex hormone binding globulin) and CRP. | A robust inverse correlation of testosterone and sex hormone-binding globulin with CRP levels. These findings provide evidence supporting the hypothesis that modulation of inflammatory processes is a potential pathway by which androgens could affect cardiometabolic risk and associated conditions such as metabolic syndrome, diabetes, and cardiovascular disease. |
| Bobjer et al., 2012 [13] | Sub-fertile hypogonadal men (n = 20) and eugonadal men (n = 20) (mean age 37 years, standard deviation (SD) = 4.3 and age-matched controls (n = 20) were assessed for the levels of inflammatory markers. | Macrophage inflammatory proteins a and 1b (MIP1a & MIP1b and TNF-α all showed negative associations with testosterone levels, while MIP1a and TNF-α also showed negative association with calculated free testosterone (cFT) levels. Compared with men exhibiting normal testosterone and cFT levels, TNF-α levels were higher in men with subnormal levels of testosterone and cFT. Also, MIP1 levels were higher in men with subnormal levels of testosterone. | Higher levels of proinflammatory biomarkers were noted in young men with reduced testosterone, even in the absence of concurrent metabolic disease. These findings suggest that testosterone levels are directly associated with low grade systemic inflammation, which may contribute to the development of several adverse health effects previously associated with androgen deficiency. It appears that TNF-α and MIP1 are strongly associated with low testosterone in men not suffering from serious systemic disease. |
| Tsilidis et al., 2013 [14] | Data from 809 adult men in the National Health and Nutrition Examination Survey 1999–2004 were analyzed by geometric means and 95% confidence intervals for CRP and white blood cell (WBC) concentrations by sex steroid hormones and sex hormone binding globulin (SHBG), using weighted linear regression models. | Total and calculated free estradiol (E2) were positively associated with both CRP and WBC concentrations. SHBG concentrations were inversely associated with WBC count, but not with CRP. These cross-sectional findings are consistent with the hypothesis that higher androgen and lower estrogen concentrations may have an anti-inflammatory effect in men. | Testosterone and cFT are modestly inversely associated with CRP concentrations, and that total and calculated free estradiol are modestly positively associated with CRP and WBC. |
| Burney et al., 2012 [15] | Patients with cancer cachexia (n = 45) and cancer without cachexia (n = 50), as well as non-cancer controls (n = 45). Total testosterone, bioavailable testosterone, CRP, and IL-6 were measured in plasma. Functional performance was assessed by the Eastern Cooperative Oncology Group and Karnofsky Performance Scales, and sexual function was assessed by the International Index of Erectile Function (IIEF). | Low testosterone-levels were noted in more than 70% of cancer cachexia cases. Total testosterone was lower in cancer cachexia compared to cancer without cachexia. Cancer cachexia patients had lower bioavailable testosterone (BAT) and grip strength, IIEF scores, appendicular lean body mass, and fat mass, and higher IL-6 and CRP compared to controls. | Patients with cancer cachexia have lower grip strength, testosterone, fat mass, and a lean body mass with higher bone resorption, high sensitivity-CRP (hs-CRP), and IL-6, and poor functional status and erectile function, suggesting that cancer cachexia patients exhibited higher degrees of inflammation. |
| Tremellen et al., 2017 [16,17] | Men (n = 50) aged between 21 and 50 years (mean 35.1 ± 6.8 years) recruited from a private fertility clinic. BMI, waist circumference and % body fat, inflammatory status (serum CRP, IL-1β, IL-6, TNF-α, and lipopolysaccharide-binding protein (LBP)) and testicular endocrine function (serum testosterone, estradiol, anti-Mullerian hormone, luteinizing hormone (LH) and follicle stimulating hormone (FSH) were measured. | The mean (± SD) age, BMI, percentage body fat, and waist circumference of participants was 35.1 ± 6.8 years, 26.96 ± 3.5 kg/m2, 23.6 ± 6%, and 93.2 ± 9.5 cm, respectively. Inflammatory status and all three measures of adiposity, were positively correlated with serum IL-6 and CRP, but not with IL-1β or TNF-α. CRP was an overall marker of inflammation, and a positive correlation was observed between both CRP and the cytokines IL1β and IL-6, but not with TNF-α, CRP was negatively correlated with total testosterone (r = −0.471, p = 0.001,) but not calculated free testosterone (r = −0.238, p = 0.11). A significant negative relationship between serum IL-6 and testosterone (r = −0.516, p < 0.001), was observed. | Male adiposity was associated with both metabolic endotoxemia and an increase in serum IL-6, with this heightened inflammatory response being associated with a decline in both Leydig (testosterone) and Sertoli cell (anti-Mullerian hormone) function. |
| Study | Type of Study | Main Findings | Comments |
|---|---|---|---|
| Ng et al., 2002 [18] | Three-month, randomized, double-blind, placebo-controlled clinical trials. In one group, 18 men were assigned to 5α-DHT, and 19 men were assigned to a placebo. In a second group, 20 men were assigned to recombinant human chorionic gonadotropin (rhCG), and 20 men were assigned to a placebo. | Median CRP levels were 1.65 and 1.50 mg/L, mean sICAM-1 levels were 228 and 224 ng/mL, and mean soluble vascular adhesion molecule-1 (sVCAM-1) levels were 861 and 895 ng/mL in placebo- and 5α-DHT-assigned treatment groups, respectively. rhCG had no significant effect on either CRP, sVCAM-1, or sICAM-1 compared with the placebo. | 5 α-DHT replacement in older men for three months had no significant effects on the serum levels of CRP, sICAM-1, or sVCAM-1. rhCG administration had no effect on serum inflammatory markers in older men, suggesting that the conversion of testosterone to estrogens may not have a significant effect on circulating ICAM-1, VCAM-1, or CRP in older men. |
| Malkin et al., 2004 [19] | A randomized, single-blind, placebo-controlled, crossover trial of testosterone therapy in hypogonadal men. | Testosterone therapy reduced serum cytokines TNF-α and IL-1β, and an increase in IL-10. The reduction in IL-1β only approached significance. The reduction of TNF-α and IL-1β was positively correlated. | This study suggests that testosterone therapy reduces inflammatory cytokine levels. |
| Corrales et al., 2006 [20] | Thirteen men >55 years with type-2 diabetes were enrolled in the study. Eight healthy men with neither diabetes mellitus, nor any of the components of the metabolic syndrome, of similar age, were studied in parallel as a control group. Analyses were performed at baseline and 1, 3, 6 and 12 months after treatment with 150 mg testosterone enanthate every two weeks in the 13 men with type-2 diabetes. | After testosterone therapy, the number of peripheral blood monocytes capable of spontaneously producing IL-1β, IL-6, and TNF-α became undetectable, both at the first follow-up time point and at the end of therapy. Also, the percentage of CD33hi myeloid dendritic cells (DCs), and that of plasmacytoid DCs capable of spontaneously producing IL-6 and TNF-α also became undetectable. Testosterone therapy was associated with a decreased number of TNF-α secreting plasmacytoid DCs at month 12. | It was suggested that testosterone therapy in men beneficially alters cytokine balance and may reduce inflammation. |
| Kapoor et al., 2007 [21] | Double-blind placebo-controlled crossover study of testosterone therapy in 20 hypogonadal men with type 2 diabetes and >30 years. | There was a significant inverse correlation between baseline IL-6, CRP, and total testosterone. | Baseline testosterone levels correlated inversely with IL-6 and CRP, suggesting that testosterone may regulate the expression of inflammatory cytokines. |
| Nakhai-Pour et al., 2007 [22] | A randomized, double-blind, placebo-controlled trial of 237 men with serum testosterone levels <13.7 nmol/L and aged 60 to 80 years were treated with testosterone undecanoate (TU) or placebo for 26 weeks. | Median hs-CRP was 2.20 vs. 2.00 mg/L in the testosterone and the placebo group, respectively. Neither baseline testosterone level, nor age, or baseline CRP level modified the effect of testosterone supplementation on CRP levels. | This study suggested that 26 weeks of T therapy had no effect on serum hs-CRP levels in elderly men. |
| Kalinchenko et al., 2010 [23] | This was a double-blinded, placebo-controlled phase III trial of 170 men aged 35–70. Testosterone therapy was followed up for 30 weeks (n = 113; or placebo, n = 71). One hundred- and five-men receiving testosterone and 65 receiving a placebo completed the trial. | Testosterone therapy reduced the levels of TNF-α and CRP significantly, but not those of IL-1β. Levels of IL-6 and IL-10 were not affected by testosterone. Changes in CRP were correlated with changes in total and free testosterone. | This study suggested that testosterone therapy reduced levels of markers of inflammation. |
| Traish et al., 2014 [24] | Long-term observational registry study, throughout a 5-year period. | Cumulative registry study of 255 men, aged between 33 and 69 years (mean 58.02 ± 6.30) with subnormal plasma total T levels (mean: 9.93 ± 1.38; range: 5.89–12.13 nmol/L), as well as at least mild symptoms of TD assessed by the Aging Males’ Symptoms Scale. Testosterone therapy was followed up to 60 months. | Long-term testosterone therapy markedly reduced the levels of CRP in hypogonadal men. |
| Saad et al., 2015 [25] | In a single-center, cumulative, prospective registry study, hypogonadal men with psoriasis were investigated. | Men (n = 15) with a previous diagnosis of psoriasis received testosterone therapy for up to 93 months. Scores on the Psoriasis Area and Severity Index and Physician Global Assessment for Psoriasis showed significant improvement for the first 24 months. These improvements were sustained with continued testosterone therapy. | Levels of CRP, a biochemical indicator of inflammation, declined significantly. |
| Nasser et al., 2015 [26] | This study was a cumulative, prospective, registry with an increasing number of men with Crohn’s disease receiving testosterone over time. In total, 92 men received parenteral testosterone undecanoate 1000 mg/12 weeks for up to 7 years. Fourteen men opted not to receive testosterone and served as a comparison group. | In men receiving testosterone, the Crohn’s Disease Activity Index declined from 239.36 ± 36.96 to 71.67 ± 3.26 at 84 months (p < 0.0001 vs. baseline). C-reactive protein levels decreased from 12.89 ± 8.64 to 1.78 ± 1.37 mg/L at 84 months (p < 0.0001 vs. baseline). Leukocyte count decreased from 11.93 ± 2.85 to 6.21 ± 1.01 × 109/L (p < 0.0001 at 84 months vs. baseline). | The authors suggested that normalizing serum testosterone in hypogonadal men with Crohn’s disease had a positive effect on the clinical course, also evidenced by biochemical parameters. |
| Chronic Inflammatory Disease | Reported Studies | General Comments from Various Studies |
|---|---|---|
| Type 2 Diabetes Mellitus | Dhindsa et al., 2016 [39] Hackett et al., 2018 [40] Hackett et al., 2018 [41] Shigehara et al., 2018 [42] Fink et al., 2018 [43] Groti et al., 2018 [44] Saad, 2017 [45] Haider et al., 2017 [46] | Testosterone treatment in hypogonadal diabetic men resulted in a significant fall in circulating concentrations of free fatty acids, C-reactive protein, interleukin-1β, tumor necrosis factor-α, and leptin (p < 0.05), concomitant with weight loss and an improvement in glycemic control, endothelial function, sexual function, and reduced mortality. |
| Coronary Artery and Vascular Diseases | Wickramatilake et al., 2014 [47] Kloner et al., 2016 [48] Pongkan et al., 2015 [49] Etminan et al., 2015 [50] Nettleship et al., 2009 [51] Traish et al., 2017 [52] Cheetham et al., 2017 [53] Anderson et al., 2016 [54] Sharma et al., 2015 [55] | An association between low testosterone levels and the presence of atherosclerosis, coronary artery disease, and coronary events was noted in a number of studies. Testosterone concentrations were lower, and highly-sensitive C-reactive protein levels were higher in coronary artery disease patients as compared with controls, and testosterone therapy may have a potential as a therapeutic agent in treating heart failure, angina, and myocardial ischemia, and testosterone therapy may exert cardioprotective effects. |
| Psoriasis | Saad et al., 2016 [25] | Testosterone therapy in hypogonadal men with psoriasis produced considerable improvements in scores of the Psoriasis Area and Severity Index and Physician Global Assessment for Psoriasis in the first 24 months, and these improvements were sustained thereafter. C-reactive protein levels declined significantly. |
| Rheumatoid Arthritis | Baillargeon et al., 2016 [56] Fisk et al., 1950 [57] Ganesan et al., 2012 [58] Cutolo et al., 2009 [59] Cutolo et al., 2006 [60] Pope et al., 2002 [61] Cutolo et al., 2002 [62] Cutolo et al., 2000 [63] Hall et al., 1996 [64] Wimer et al., 1973 [65] | Serum testosterone levels are inversely correlated with rheumatoid arthritis (RA) activity. Testosterone is significantly reduced in inflamed synovial tissue/fluids during active disease as a consequence of the inflammatory reaction, which supports a pro-inflammatory milieu in RA joints. Patients diagnosed with hypogonadism who were not treated with testosterone had an increased risk of developing any rheumatic autoimmune disease and RA, and lupus and testosterone therapy produced improvements in patients’ symptoms. |
| Crohn’s Disease | Nasser et al., 2015 [26] | Low circulating levels of testosterone are common in males with chronic obstructive pulmonary disease. It is possible that low anabolic hormones will reduce muscle mass and eventually result in a diminished muscle function. Normalizing serum testosterone in hypogonadal men with Crohn’s disease had a positive effect on the clinical course, also evidenced by biochemical parameters. |
| Airway Diseases | Laffont et al., 2017 [66] Cephus et al., 2017 [67] Montaño et al., 2014 [68] Canguven & Albayrak, 2011 [69] Kamischke et al., 1998 [70] Baillargeon et al., 2018 [71] Atlantis et al., 2013 [72] Samaras et al., 2012 [73] Velema et al., 2012 [74] Svartberg, 2010 [75] Puhan & Schünemann, 2005 [76] Casaburi et al., 2004 [77] Creutzberg & Casaburi, 2003 [78] | Testosterone therapy may slow disease progression in patients with chronic obstructive pulmonary disease (COPD). Androgens limits IL-33-driven lung inflammation through a cell-intrinsic inhibition of ILC2 expansion. In vivo, testosterone attenuated Alternaria-extract-induced IL-5+ and IL-13+ ILC2 numbers and lung eosinophils by intrinsically decreasing lung ILC2 numbers, as well as by decreasing expression of IL-33 and thymic stromal lymphopoietin (TSLP), ILC2-stimulating cytokines. Collectively, these findings provide a foundational understanding of sexual dimorphism in ILC2 function. |
| Multiple Sclerosis | Gold et al., 2008 [79] Gold & Voskuhl., 2009 [38] Gold & Voskuhl, 2006 [80] Collongues et al., 2018 [81] Ziehn et al., 2012 [82] Sicotte et al., 2007 [83] Dalal et al., 1997 [35] | Testosterone demonstrated an anti-inflammatory effect in multiple sclerosis (MS). Testosterone has an effect in protecting neurons in culture against glutamate-induced toxicity and oxidative stress, and it stimulates myelin formation and regeneration mediated through the neural androgen receptor (AR). Testosterone treatment has potential neuroprotective effects in men with relapsing-remitting MS. Testosterone induces anti-inflammatory as well as neuroprotective effects, and transdermal testosterone in male MS patients appears promising. |
| Systemic Lupus Erythematosus | Pakpoor et al., 2018 [84] Gordon et al., 2008 [85] Sasaki et al., 2006 [86] Olsen & Kovacs, 1995 [87] Bizzarro et al., 1987 [88] Amor et al., 1983 [89] Costello & Singer, 1952 [90] | Lupus erythematosus, chronic disseminated type, showing a dramatic response to testosterone therapy. Androgenic steroids can exert significant effects on immune parameters, and this suggests that the effects of androgens on the immune system may contribute to the sexual dimorphism of autoimmune diseases. Treatment with a high dose of methyltestosterone improved thrombocytopenia and symptoms, suggesting that methyltestosterone may have a clinical benefit in the treatment of patients with Klinefelter’s syndrome with a low level of testosterone accompanying immunological disorders. The positive association between testicular testosterone deficiency and systemic lupus erythematosus (SLE) supports the hypothesis that low testosterone levels may influence the development of male SLE. Of clinical importance, males with SLE are at increased risk of co-morbid testicular hypotestosteronemia, and this may warrant consideration in the management of patients with testosterone therapy. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traish, A.; Bolanos, J.; Nair, S.; Saad, F.; Morgentaler, A. Do Androgens Modulate the Pathophysiological Pathways of Inflammation? Appraising the Contemporary Evidence. J. Clin. Med. 2018, 7, 549. https://doi.org/10.3390/jcm7120549
Traish A, Bolanos J, Nair S, Saad F, Morgentaler A. Do Androgens Modulate the Pathophysiological Pathways of Inflammation? Appraising the Contemporary Evidence. Journal of Clinical Medicine. 2018; 7(12):549. https://doi.org/10.3390/jcm7120549
Chicago/Turabian StyleTraish, Abdulmaged, Jose Bolanos, Sunil Nair, Farid Saad, and Abraham Morgentaler. 2018. "Do Androgens Modulate the Pathophysiological Pathways of Inflammation? Appraising the Contemporary Evidence" Journal of Clinical Medicine 7, no. 12: 549. https://doi.org/10.3390/jcm7120549