KIFC1 Inhibitor CW069 Induces Apoptosis and Reverses Resistance to Docetaxel in Prostate Cancer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. DTX and CW069 Treatment
2.3. Western Blotting Analysis
2.4. qRT-PCR Analysis
2.5. RNA Interference
2.6. Cell Death ELISA
2.7. Statistical Analysis
3. Results
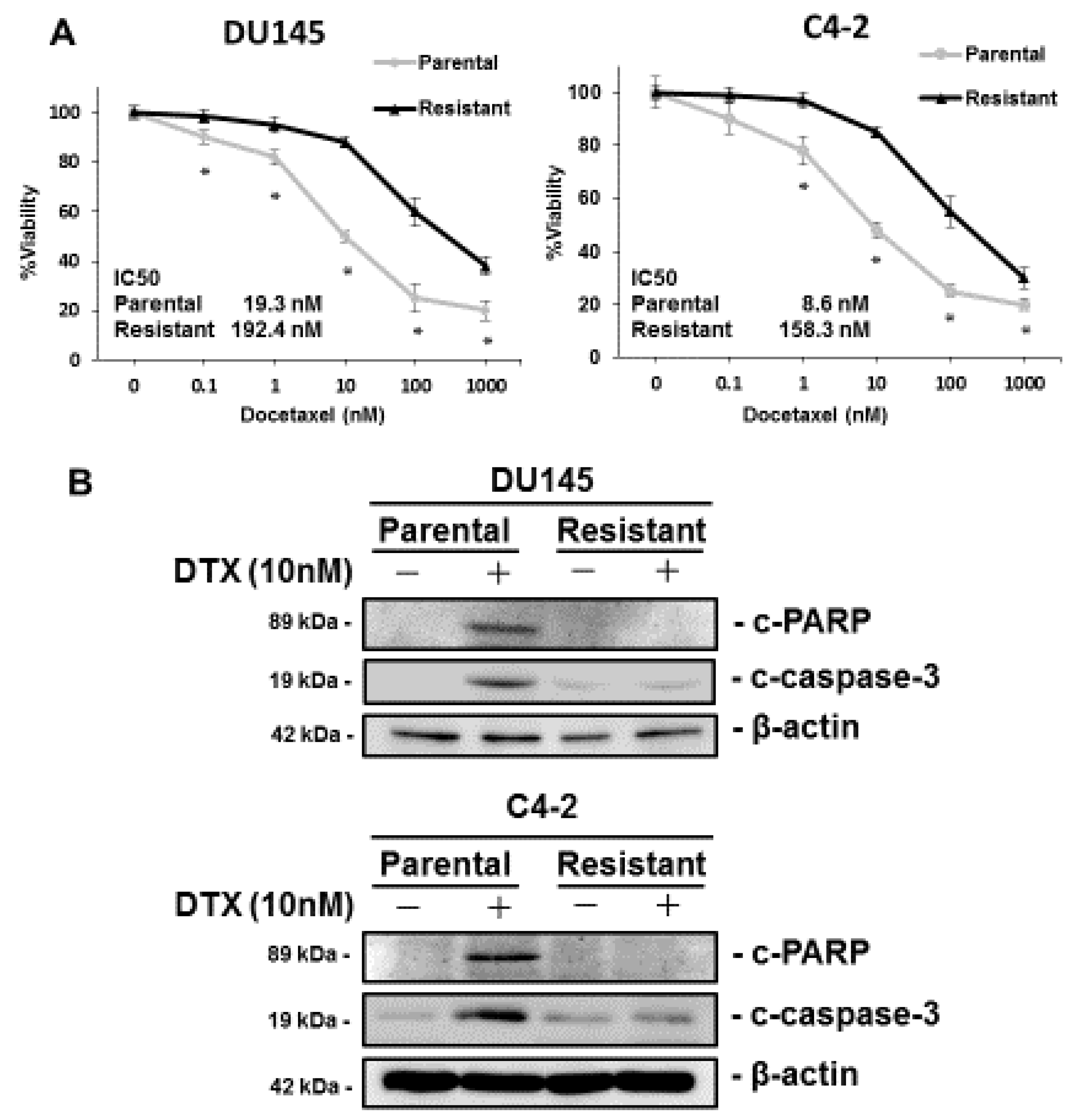
3.1. Characterization of DTX-Resistant PCa Cell Lines
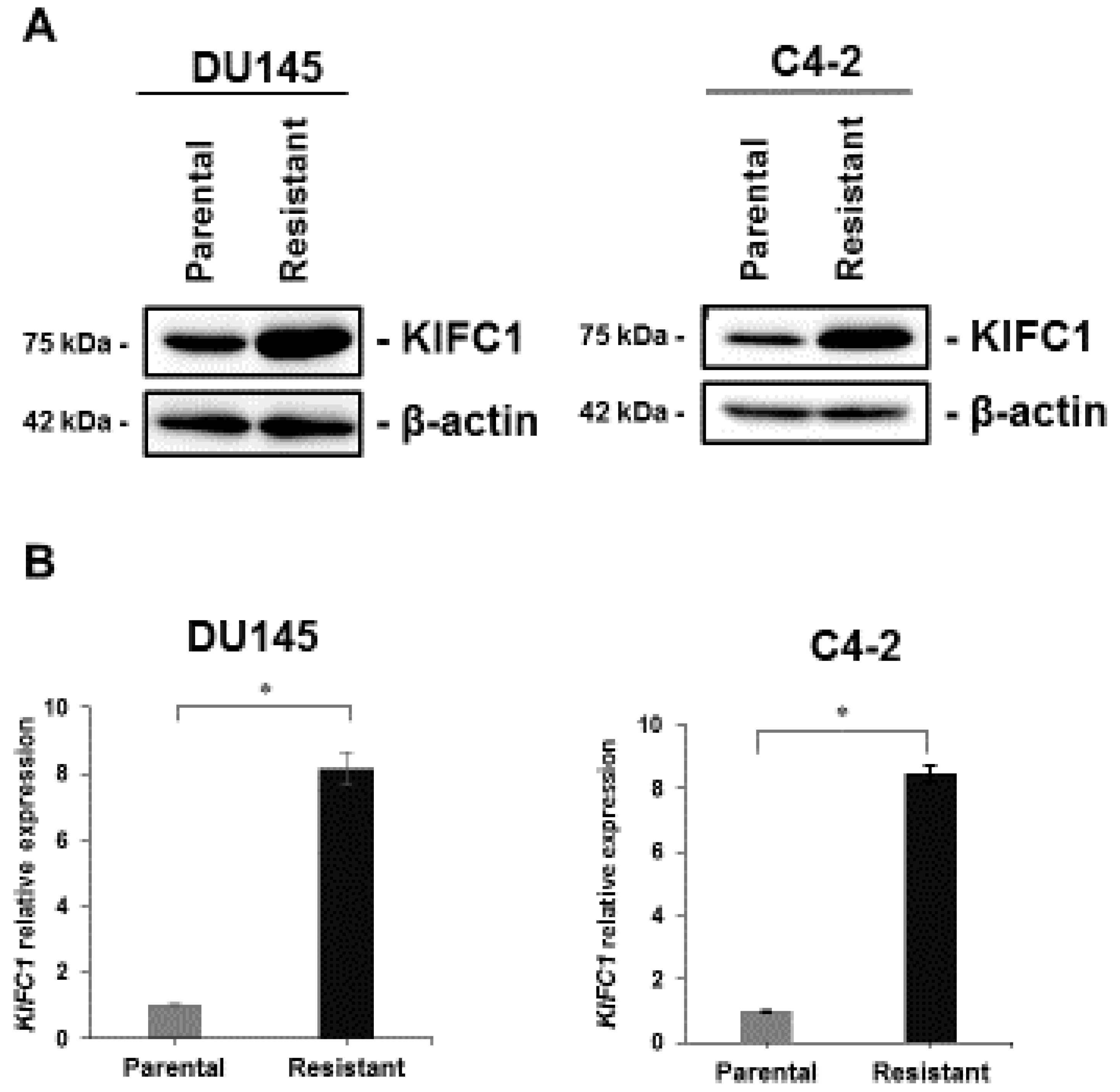
3.2. KIFC1 is Overexpressed in DTX-Resistant Cell Lines
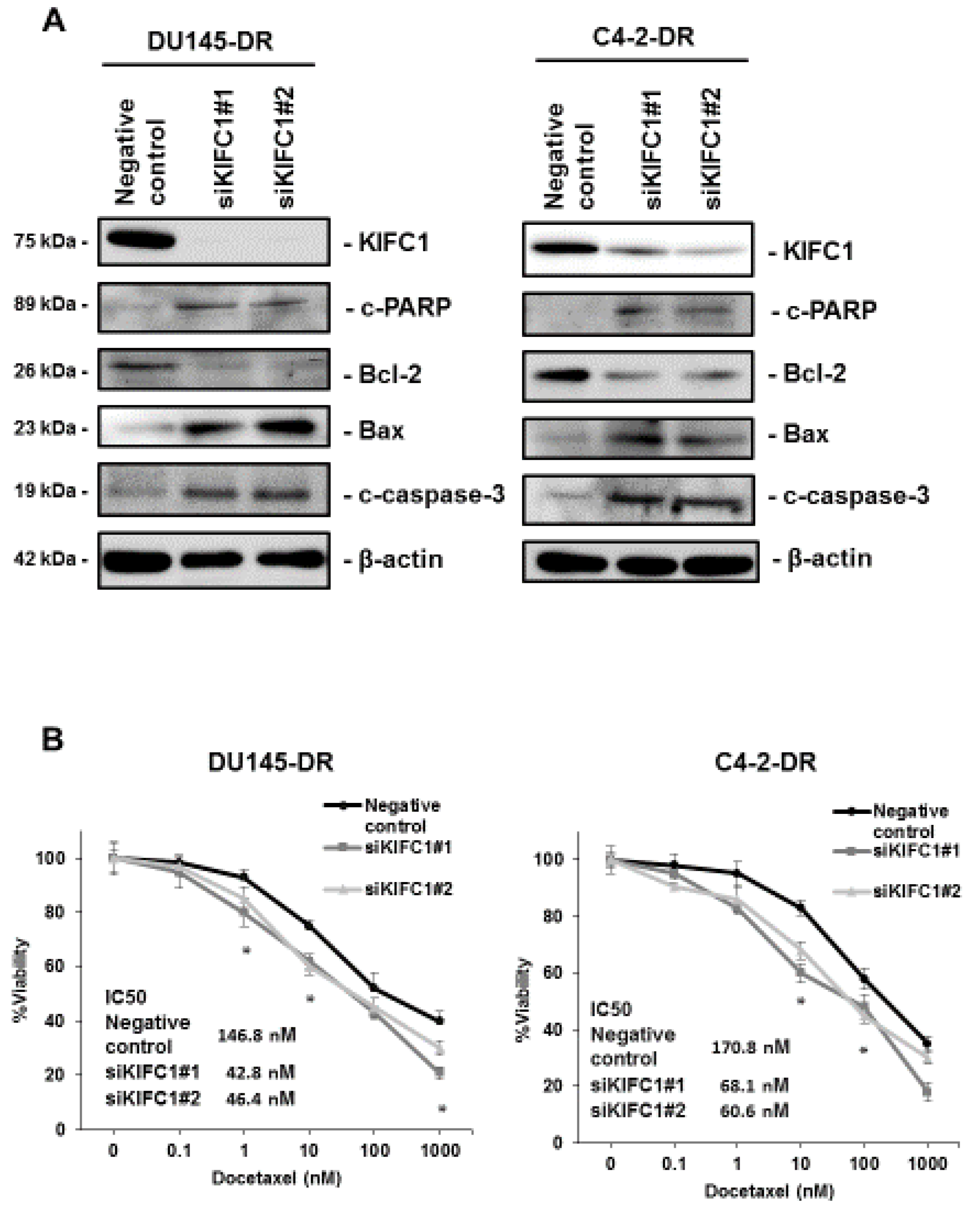
3.3. Inhibition of KIFC1 Induces Apoptosis Pathway and Reverses DTX Resistance In Vitro
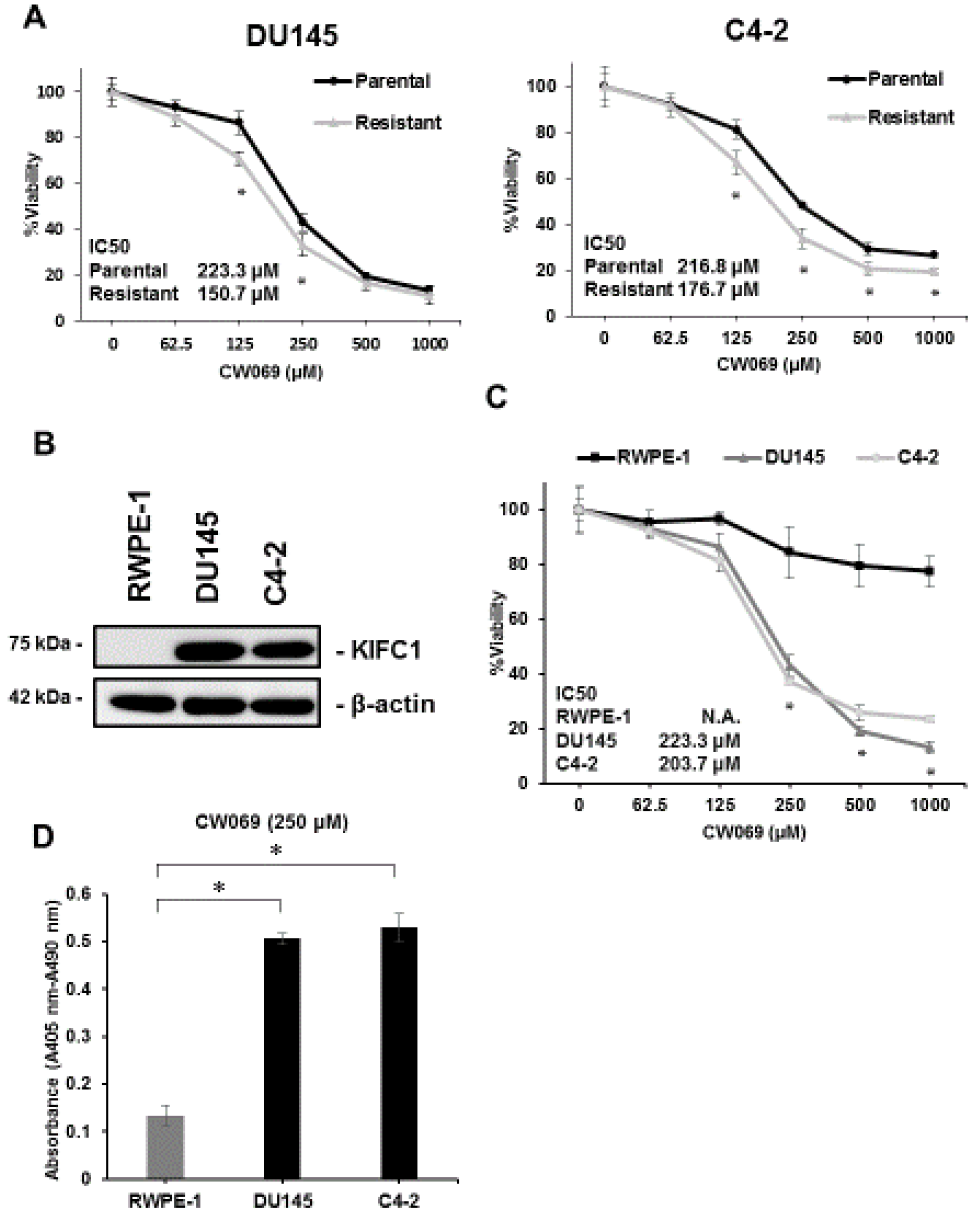
3.4. Effect of KIFC1 Inhibitor CW069 on Cell Viability
3.5. CW069 Re-Sensitizes DTX-Resistant Cell Lines to DTX Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Neste, L.; Herman, J.G.; Otto, G.; Bigley, J.W.; Epstein, J.I.; Van Criekinge, W. The epigenetic promise for prostate cancer diagnosis. Prostate 2012, 72, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Cecchetti, R.; Riuzzi, F.; Peirce, M.J.; Talesa, V.N. Glyoxalase 1 sustains the metastatic phenotype of prostate cancer cells via EMT control. J. Cell Mol. Med. 2018, 22, 2865–2883. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.; Belldegrun, A. Novel therapeutic strategies for castration resistant prostate cancer: Inhibition of persistent androgen production and androgen receptor mediated signaling. J. Urol. 2011, 185, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef]
- Quintyne, N.J.; Reing, J.E.; Hoffelder, D.R.; Gollin, S.M.; Saunders, W.S. Spindle multipolarity is prevented by centrosomal clustering. Science 2005, 307, 127–129. [Google Scholar] [CrossRef]
- Godinho, S.A.; Kwon, M.; Pellman, D. Centrosomes and cancer: How cancer cells divide with too many centrosomes. Cancer Metast. Rev. 2009, 28, 85–98. [Google Scholar] [CrossRef]
- Kwon, M.; Bagonis, M.; Danuser, G.; Pellman, D. Direct microtubule-binding by myosin-10 orients centrosomes toward retraction fibers and subcortical actin clouds. Dev. Cell. 2015, 34, 323–337. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Kleylein-Sohn, J.; Pöllinger, B.; Ohmer, M.; Hofmann, F.; Nigg, E.A.; Hemmings, B.A.; Wartmann, M.; et al. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J. Cell Sci. 2012, 125, 5391–5402. [Google Scholar] [CrossRef]
- Leber, B.; Maier, B.; Fuchs, F.; Chi, J.; Riffel, P.; Anderhub, S.; Wagner, L.; Ho, A.D.; Salisbury, J.L.; Boutros, M.; Krämer, A. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2010, 2, 33ra8. [Google Scholar] [CrossRef] [PubMed]
- Basto, R.; Brunk, K.; Vinadogrova, T.; Peel, N.; Franz, A.; Khodjakov, A.; Raff, J.W. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008, 133, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, F.; Lan, Y.; Wang, J.; Nie, C.; Liang, Y.; Song, R.; Zheng, T.; Pan, S.; Pei, T.; et al. KIFC1 regulated by miR-532-3p promotes epithelial-to-mesenchymal transition and metastasis of hepatocellular carcinoma via gankyrin/AKT signaling. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Cipriano, R.; Jackson, M.W.; Stark, G.R. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res. 2009, 69, 8035–8042. [Google Scholar] [CrossRef] [PubMed]
- Sekino, Y.; Oue, N.; Shigematsu, Y.; Ishikawa, A.; Sakamoto, N.; Sentani, K.; Teishima, J.; Matsubara, A.; Yasui, W. KIFC1 induces resistance to docetaxel and is associated with survival of patients with prostate cancer. Urol. Oncol. 2017, 35, e31.e13–e31.e20. [Google Scholar] [CrossRef] [PubMed]
- Sekino, Y.; Sakamoto, N.; Goto, K.; Honma, R.; Shigematsu, Y.; Sentani, K.; Oue, N.; Teishima, J.; Matsubara, A.; Yasui, W. Transcribed ultraconserved region Uc.63+promotes resistance to docetaxel through regulation of androgen receptor signaling in prostate cancer. Oncotarget 2017, 8, 94259–94270. [Google Scholar] [CrossRef]
- Sekino, Y.; Sakamoto, N.; Goto, K.; Honma, R.; Shigematsu, Y.; Quoc, T.P.; Sentani, K.; Oue, N.; Teishima, J.; Kawakami, F.; et al. Uc.416 + A promotes epithelial-to-mesenchymal transition through miR-153 in renal cell carcinoma. BMC Cancer 2018, 18, 952. [Google Scholar] [CrossRef]
- Sekino, Y.; Oue, N.; Mukai, S.; Shigematsu, Y.; Goto, K.; Sakamoto, N.; Sentani, K.; Hayashi, T.; Teishima, J.; Matsubara, A.; et al. Protocadherin B9 promotes resistance to bicalutamide and is associated with the survival of prostate cancer patients. Prostate 2019, 79, 234–242. [Google Scholar] [CrossRef]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Kashiwagi, E.; Yokomizo, A.; Takeuchi, A.; Dejima, T.; Song, Y.; Tatsugami, K.; Inokuchi, J.; Uchiumi, T.; Naito, S. Interaction between docetaxel resistance and castration resistance in prostate cancer: Implications of Twist1, YB-1, and androgen receptor. Prostate 2013, 73, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Itsumi, M.; Yokomizo, A.; Takeuchi, A.; Imada, K.; Kashiwagi, E.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Naito, S. Targeting ribosomal S6 kinases/Y-box binding protein-1 signaling improves cellular sensitivity to taxane in prostate cancer. Prostate 2014, 74, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhu, Y.; Zheng, B.; Zou, Y.; Wang, C.; Wu, P.; Wang, J.; Chen, H.; Du, P.; Liang, B.; et al. KIFC1, a novel potential prognostic factor and therapeutic target in hepatocellular carcinoma. Int. J. Oncol. 2018, 52, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Pannu, V.; Rida, P.C.; Ogden, A.; Turaga, R.C.; Donthamsetty, S.; Bowen, N.J.; Rudd, K.; Gupta, M.V.; Reid, M.D.; Cantuaria, G.; et al. HSET overexpression fuels tumor progression via centrosome clustering-independent mechanisms in breast cancer patients. Oncotarget 2015, 6, 6076–6091. [Google Scholar] [CrossRef] [PubMed]
- Watts, C.A.; Richards, F.M.; Bender, A.; Bond, P.J.; Korb, O.; Kern, O.; Riddick, M.; Owen, P.; Myers, R.M.; Raff, J.; et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem. Biol. 2013, 20, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.M.; Bello, D.; Quader, S. Immortalized and tumorigenic adult human prostatic epithelial cell lines: Characteristics and applications. Part 3. Oncogenes, suppressor genes, and applications. Prostate 1997, 30, 136–142. [Google Scholar] [CrossRef]
- Vale, C.L.; Burdett, S.; Rydzewska, L.H.M.; Albiges, L.; Clarke, N.W.; Fisher, D.; Fizazi, K.; Gravis, G.; James, N.D.; Mason, M.D.; et al. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone-sensitive prostate cancer: A systematic review and meta-analyses of aggregate data. Lancet Oncol. 2016, 17, 243–256. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- Kroon, J.; Kooijman, S.; Cho, N.J.; Storm, G.; van der Pluijm, G. Improving taxane-based chemotherapy in castration-resistant prostate cancer. Trends Pharmacol. Sci. 2016, 37, 451–462. [Google Scholar] [CrossRef]
- Huang, X.; Chau, C.H.; Figg, W.D. Challenges to improved therapeutics for metastatic castrate resistant prostate cancer: From recent successes and failures. J. Hematol. Oncol. 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.M.; Gao, A.C. Drug resistance in castration resistant prostate cancer: Resistance mechanisms and emerging treatment strategies. Am. J. Clin. Exp. Urol. 2015, 3, 64–76. [Google Scholar] [PubMed]
- Liu, C.; Zhu, Y.; Lou, W.; Nadiminty, N.; Chen, X.; Zhou, Q.; Shi, X.B.; deVere White, R.W.; Gao, A.C. Functional p53 determines docetaxel sensitivity in prostate cancer cells. Prostate 2013, 73, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Ploussard, G.; Terry, S.; Maillé, P.; Allory, Y.; Sirab, N.; Kheuang, L.; Soyeux, P.; Nicolaiew, N.; Coppolani, E.; Paule, B.; et al. Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res. 2010, 70, 9253–9264. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Harashima, N.; Hiraki, M.; Arichi, N.; Nishimura, N.; Shiina, H.; Naora, K.; Harada, M. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget 2014, 5, 11399–11412. [Google Scholar] [CrossRef] [PubMed]
- Kroon, J.; Puhr, M.; Buijs, J.T.; van der Horst, G.; Hemmer, DM.; Marijt, K.A.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr. Relat. Cancer 2016, 23, 35–45. [Google Scholar] [CrossRef]
- Choe, M.H.; Kim, J.; Ahn, J.; Hwang, S.G.; Oh, J.S.; Kim, J.S. Centrosome clustering is a tumor-selective target for the improvement of radiotherapy in breast cancer cells. Anticancer Res. 2018, 38, 3393–3400. [Google Scholar] [CrossRef]
- Zhang, W.; Zhai, L.; Wang, Y.; Boohaker, R.J.; Lu, W.; Gupta, V.V.; Padmalayam, I.; Bostwick, R.J.; White, E.L.; Ross, L.J.; et al. Discovery of a novel inhibitor of kinesin-like protein KIFC1. Biochem. J. 2016, 473, 1027–1035. [Google Scholar] [CrossRef]
- Wu, J.; Mikule, K.; Wang, W.; Su, N.; Petteruti, P.; Gharahdaghi, F.; Code, E.; Zhu, X.; Jacques, K.; Lai, Z.; et al. Discovery and mechanistic study of a small molecule inhibitor for motor protein KIFC1. ACS Chem. Biol. 2013, 8, 2201–2208. [Google Scholar] [CrossRef]
- Yukawa, M.; Yamauchi, T.; Kurisawa, N.; Ahmed, S.; Kimura, K.I.; Toda, T. Fission yeast cells overproducing HSET/KIFC1 provides a useful tool for identification and evaluation of human kinesin-14 inhibitors. Fungal Genet. Biol. 2018, 116, 33–41. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; Cucchiara, V.; Gu, X.; Lou, W.; Evans, C.P.; Gao, A.C. ABCB1 Mediates Cabazitaxel–Docetaxel Cross-Resistance in Advanced Prostate Cancer. Mol. Cancer Ther. 2017, 16, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, R.J.; De Morrée, E.S.; Kweldam, C.F.; De Ridder, C.M.; Wiemer, E.A.; Mathijssen, R.H.; De Wit, R.; Van Weerden, W.M. Targeting the Androgen Receptor Confers In Vivo Cross-resistance Between Enzalutamide and Docetaxel, But Not Cabazitaxel, in Castration-resistant Prostate Cancer. Eur. Urol. 2015, 67, 981–985. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DU145-DR | Docetaxel (nM) | |||
| 5 | 10 | 20 | ||
| CW069 (μM) | 50 | 0.55 | 0.45 | 0.45 |
| 100 | 0.73 | 0.48 | 0.54 | |
| 200 | 0.72 | 0.71 | 0.47 | |
| C4-2-DR | Docetaxel (nM) | |||
| 5 | 10 | 20 | ||
| CW069 (μM) | 50 | 0.66 | 0.51 | 0.62 |
| 100 | 0.77 | 0.46 | 0.53 | |
| 200 | 0.66 | 0.67 | 0.42 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekino, Y.; Oue, N.; Koike, Y.; Shigematsu, Y.; Sakamoto, N.; Sentani, K.; Teishima, J.; Shiota, M.; Matsubara, A.; Yasui, W. KIFC1 Inhibitor CW069 Induces Apoptosis and Reverses Resistance to Docetaxel in Prostate Cancer. J. Clin. Med. 2019, 8, 225. https://doi.org/10.3390/jcm8020225
Sekino Y, Oue N, Koike Y, Shigematsu Y, Sakamoto N, Sentani K, Teishima J, Shiota M, Matsubara A, Yasui W. KIFC1 Inhibitor CW069 Induces Apoptosis and Reverses Resistance to Docetaxel in Prostate Cancer. Journal of Clinical Medicine. 2019; 8(2):225. https://doi.org/10.3390/jcm8020225
Chicago/Turabian StyleSekino, Yohei, Naohide Oue, Yuki Koike, Yoshinori Shigematsu, Naoya Sakamoto, Kazuhiro Sentani, Jun Teishima, Masaki Shiota, Akio Matsubara, and Wataru Yasui. 2019. "KIFC1 Inhibitor CW069 Induces Apoptosis and Reverses Resistance to Docetaxel in Prostate Cancer" Journal of Clinical Medicine 8, no. 2: 225. https://doi.org/10.3390/jcm8020225