Milrinone-Induced Pharmacological Preconditioning in Cardioprotection: Hints for a Role of Mitochondrial Mechanisms

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Surgical Preparation
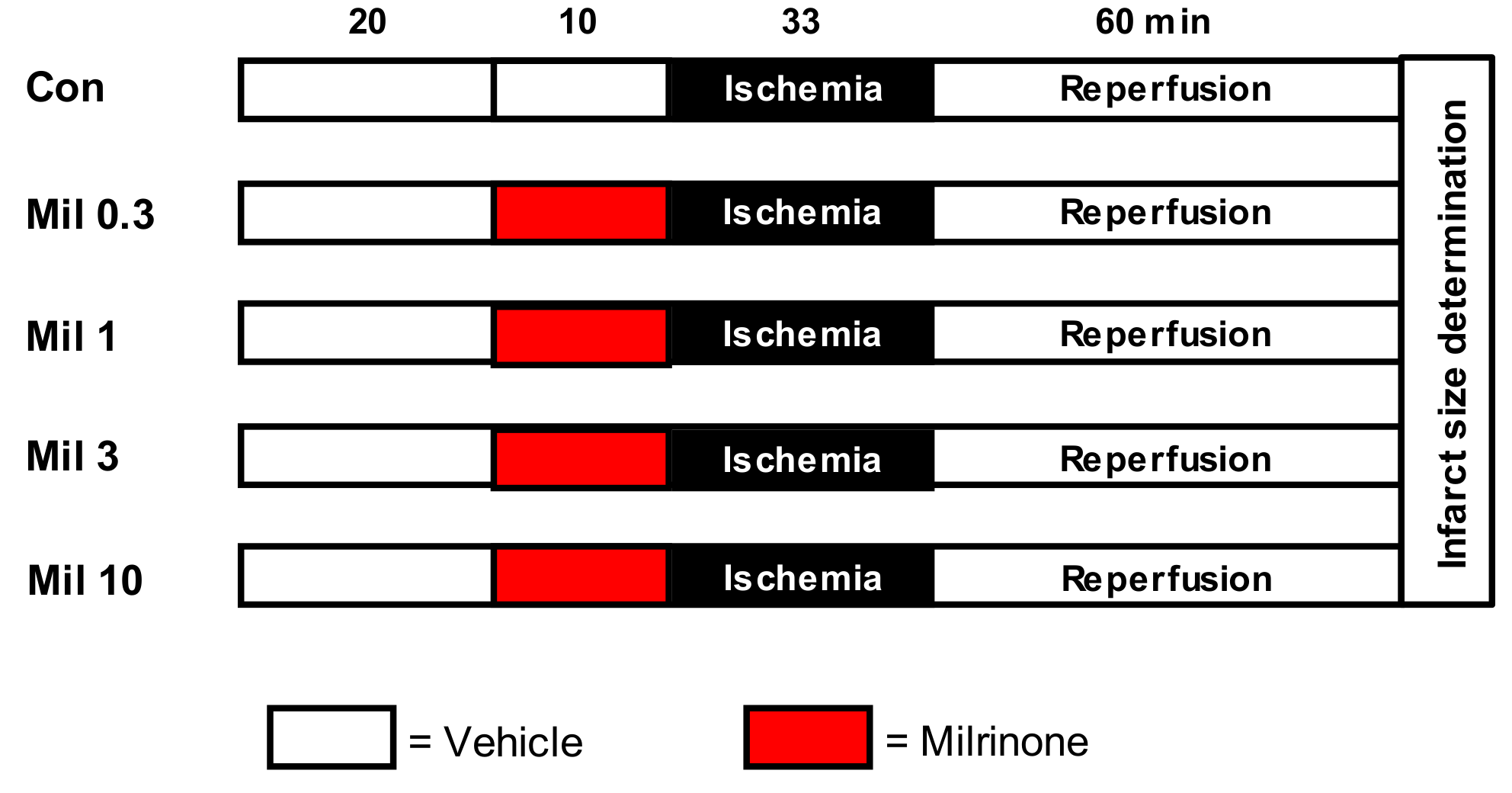
2.2. Experimental Protocol
- Control (Con): Hearts received no further treatment.
- Milrinone (Mil): Hearts were perfused with 0.3, 1, 3, and 10 µM Mil for 10 min before ischemia.
- Control (Con): Hearts received no further treatment.
- Milrinone (Mil): Hearts were perfused with 1 µM Mil only for 10 min before ischemia.
- Paxilline + milrinone (Pax + Mil): Hearts were perfused with the mBKCa-channel inhibitor Pax (1 µM) [6] combined with 1 µM Mil for 10 min before ischemia.
- Paxilline (Pax): Hearts were perfused with 1 µM Pax only for 10 min.
- N-2-mercaptopropionylglycine + milrinone (MPG + Mil): Hearts were perfused with the ROS sc#avenger MPG (1 mM) [14] combined with 1 µM Mil for 10 min before ischemia.
- N-2-mercaptopropionylglycine (MPG): Hearts were perfused with 1 mM MPG only for 10 min.
- Milrinone + cyclosporine A (Mil + CsA): Hearts were perfused with 1 µM Mil and 0.2 µM CsA [15] for 10 min before ischemia.
2.3. Statistical Analysis
3. Results
3.1. Animal Characteristics
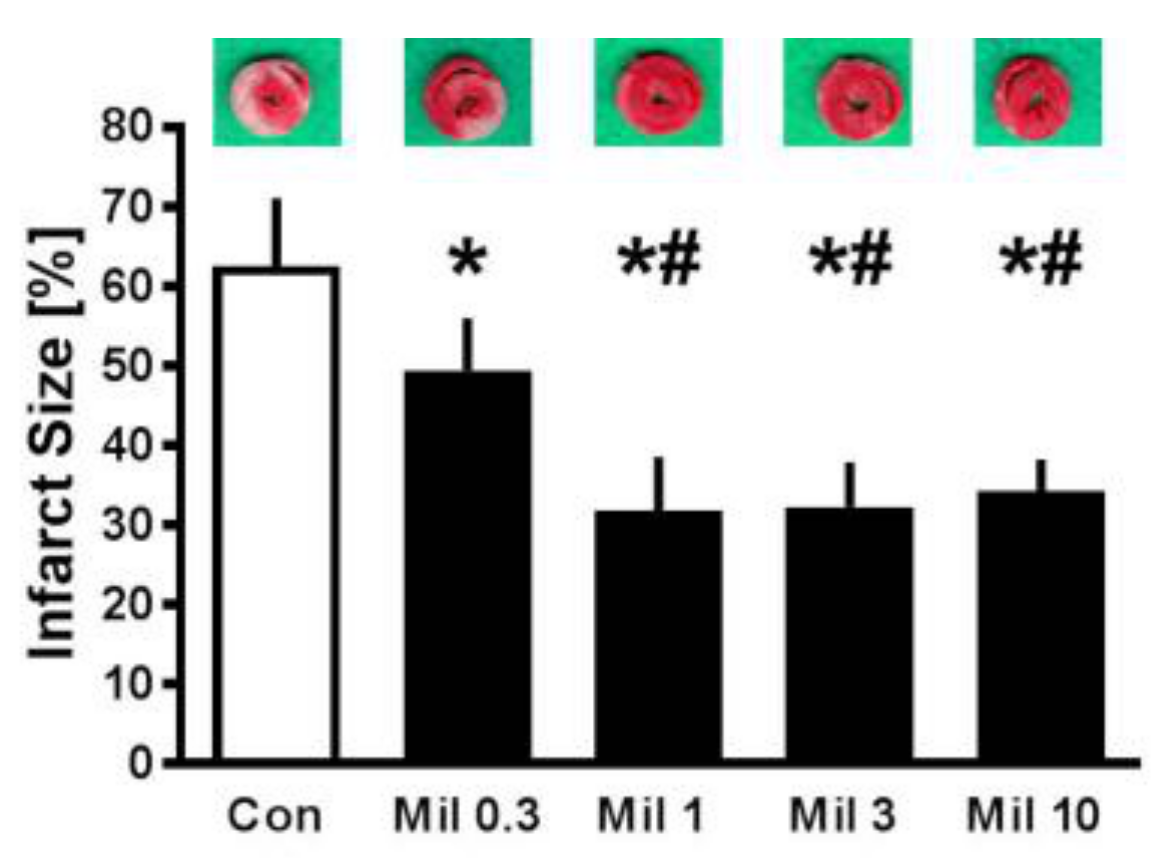
3.2. Infarct Size—Concentration Effect of Milrinone Preconditioning
3.3. Infarct Size—Mechanism of Milrinone Preconditioning
3.4. Cardiac Function
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Rong, L.Q.; Rahouma, M.; Abouarab, A.; di Franco, A.; Calautti, N.M.; Fitzgerald, M.M.; Arisha, M.J.; Ibrahim, D.A.; Girardi, L.N.; Pryor, K.O.; et al. Intravenous and Inhaled Milrinone in Adult Cardiac Surgery Patients: A Pairwise and Network Meta-Analysis. J. Cardiothorac. Vasc. Anesth. 2018. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Endoh, M. Effects of a new cardiotonic agent 1,2-dihydro-6-methyl-2-oxo-5-[imidazo (1,2-a) pyridin-6-yl]-3-pyridine carbonitrile hydrochloride monohydrate (E-1020) on contractile force and cyclic AMP metabolism in canine ventricular muscle. Jpn. J. Pharmacol. 1990, 52, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Sanada, S.; Kitakaze, M.; Papst, P.J.; Asanuma, H.; Node, K.; Takashima, S.; Asakura, M.; Ogita, H.; Liao, Y.; Sakata, Y.; et al. Cardioprotective effect afforded by transient exposure to phosphodiesterase III inhibitors: The role of protein kinase A and p38 mitogen-activated protein kinase. Circulation 2001, 104, 705–710. [Google Scholar] [CrossRef]
- Kume, M.; Banafsche, R.; Yamamoto, Y.; Yamaoka, Y.; Nobiling, R.; Gebhard, M.M.; Klar, E. Dynamic changes of post-ischemic hepatic microcirculation improved by a pre-treatment of phosphodiesterase-3 inhibitor, milrinone. J. Surg. Res. 2006, 136, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Saklani, R.; Jaggi, A.; Singh, N. Pharmacological preconditioning by milrinone: Memory preserving and neuroprotective effect in ischemia-reperfusion injury in mice. Arch. Pharm. Res. 2010, 33, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Behmenburg, F.; Trefz, L.; Dorsch, M.; Strothoff, M.; Mathes, A.; Raupach, A.; Heinen, A.; Hollmann, M.W.; Berger, M.M.; Huhn, R. Milrinone-Induced Postconditioning Requires Activation of Mitochondrial Ca(2+)-sensitive Potassium (mBKCa) Channels. J. Cardiothorac. Vasc. Anesth. 2018, 32, 2142–2148. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Menabo, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Cho, S.; Tosaka, S.; Higashijima, U.; Maekawa, T.; Hara, T.; Sumikawa, K. Hyperglycemia raises the threshold of levosimendan- but not milrinone-induced postconditioning in rat hearts. Cardiovasc. Diabetol. 2012, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Schulz, R.; Gres, P.; Korth, H.G.; Heusch, G. Attenuation of ischemic preconditioning in pigs by scavenging of free oxyradicals with ascorbic acid. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H698–H703. [Google Scholar] [CrossRef] [PubMed]
- Bunte, S.; Behmenburg, F.; Bongartz, A.; Stroethoff, M.; Raupach, A.; Heinen, A.; Minol, J.P.; Hollmann, M.W.; Huhn, R.; Sixt, S.U. Preconditioning by Levosimendan is Mediated by Activation of Mitochondrial Ca(2+)-Sensitive Potassium (mBKCa) Channels. Cardiovasc. Drugs Ther. 2018, 32, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Behmenburg, F.; Dorsch, M.; Huhn, R.; Mally, D.; Heinen, A.; Hollmann, M.W.; Berger, M.M. Impact of Mitochondrial Ca2+-Sensitive Potassium (mBKCa) Channels in Sildenafil-Induced Cardioprotection in Rats. PLoS ONE 2015, 10, e0144737. [Google Scholar] [CrossRef] [PubMed]
- Pasdois, P.; Quinlan, C.L.; Rissa, A.; Tariosse, L.; Vinassa, B.; Costa, A.D.; Pierre, S.V.; Dos Santos, P.; Garlid, K.D. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1470–H1478. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, M.; Behmenburg, F.; Raible, M.; Blase, D.; Grievink, H.; Hollmann, M.W.; Heinen, A.; Huhn, R. Morphine-Induced Preconditioning: Involvement of Protein Kinase A and Mitochondrial Permeability Transition Pore. PLoS ONE 2016, 11, e0151025. [Google Scholar] [CrossRef]
- Bailey, J.M.; Levy, J.H.; Kikura, M.; Szlam, F.; Hug, C.C., Jr. Pharmacokinetics of intravenous milrinone in patients undergoing cardiac surgery. Anesthesiology 1994, 81, 616–622. [Google Scholar] [CrossRef]
- Cox, Z.L.; Calcutt, M.W.; Morrison, T.B.; Akers, W.S.; Davis, M.B.; Lenihan, D.J. Elevation of plasma milrinone concentrations in stage D heart failure associated with renal dysfunction. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.H.; Wu, Y.; Nguyen, V.; Rastogi, S.; McConnell, B.K.; Wijaya, C.; Uretsky, B.F.; Poh, K.K.; Tan, H.C.; Fujise, K. Heart protection by combination therapy with esmolol and milrinone at late-ischemia and early reperfusion. Cardiovasc. Drugs Ther. 2011, 25, 223–232. [Google Scholar] [CrossRef]
- Yu, M.; Liu, S.L.; Sun, P.B.; Pan, H.; Tian, C.L.; Zhang, L.H. Peptide toxins and small-molecule blockers of BK channels. Acta Pharmacol. Sin. 2016, 37, 56–66. [Google Scholar] [CrossRef]
- Sanchez, M.; McManus, O.B. Paxilline inhibition of the alpha-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology 1996, 35, 963–968. [Google Scholar] [CrossRef]
- Zhou, Y.; Lingle, C.J. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J. Gen. Physiol. 2014, 144, 415–440. [Google Scholar] [CrossRef] [PubMed]
- Saleem, F.; Rowe, I.C.; Shipston, M.J. Characterization of BK channel splice variants using membrane potential dyes. Br. J. Pharmacol. 2009, 156, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Bilmen, J.G.; Wootton, L.L.; Michelangeli, F. The mechanism of inhibition of the sarco/endoplasmic reticulum Ca2+ ATPase by paxilline. Arch. Biochem. Biophys. 2002, 406, 55–64. [Google Scholar] [CrossRef]
- Longland, C.L.; Dyer, J.L.; Michelangeli, F. The mycotoxin paxilline inhibits the cerebellar inositol 1,4, 5-trisphosphate receptor. Eur. J. Pharmacol. 2000, 408, 219–225. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Olesen, S.P.; Ronn, L.C.; Grunnet, M. BK channel activators and their therapeutic perspectives. Front. Physiol. 2014, 5, 389. [Google Scholar] [CrossRef]
- Soltysinska, E.; Bentzen, B.H.; Barthmes, M.; Hattel, H.; Thrush, A.B.; Harper, M.E.; Qvortrup, K.; Larsen, F.J.; Schiffer, T.A.; Losa-Reyna, J.; et al. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS ONE 2014, 9, e103402. [Google Scholar] [CrossRef] [PubMed]
- Sausbier, M.; Hu, H.; Arntz, C.; Feil, S.; Kamm, S.; Adelsberger, H.; Sausbier, U.; Sailer, C.A.; Feil, R.; Hofmann, F.; et al. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc. Natl. Acad. Sci. USA 2004, 101, 9474–9478. [Google Scholar] [CrossRef]
- Behmenburg, F.; Holscher, N.; Flogel, U.; Hollmann, M.W.; Heinen, A.; Huhn, R. Opening of calcium-activated potassium channels improves long-term left-ventricular function after coronary artery occlusion in mice. Int. J. Cardiol. 2017, 241, 351–357. [Google Scholar] [CrossRef]
- Heinen, A.; Strothoff, M.; Schmidt, A.; Stracke, N.; Behmenburg, F.; Bauer, I.; Hollmann, M.W.; Huhn, R. Pharmacological options to protect the aged heart from ischemia and reperfusion injury by targeting the PKA-BK(Ca) signaling pathway. Exp. Gerontol. 2014, 56, 99–105. [Google Scholar] [CrossRef]
- Shim, Y.H. Cardioprotection and ageing. Korean J. Anesthesiol. 2010, 58, 223–230. [Google Scholar] [CrossRef]
- Heinen, A.; Aldakkak, M.; Stowe, D.F.; Rhodes, S.S.; Riess, M.L.; Varadarajan, S.G.; Camara, A.K. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1400–H1407. [Google Scholar] [CrossRef]
- Baines, C.P.; Goto, M.; Downey, J.M. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J. Mol. Cell. Cardiol. 1997, 29, 207–216. [Google Scholar] [CrossRef]
- Toufektsian, M.C.; Morel, S.; Tanguy, S.; Jeunet, A.; de Leiris, J.; Boucher, F. Involvement of reactive oxygen species in cardiac preconditioning in rats. Antioxid. Redox Signal. 2003, 5, 115–122. [Google Scholar] [CrossRef]
- Vigneron, F.; Dos Santos, P.; Lemoine, S.; Bonnet, M.; Tariosse, L.; Couffinhal, T.; Duplaá, C.; Jaspard-Vinassa, B. GSK-3β at the crossroads in the signalling of heart preconditioning: Implication of mTOR and Wnt pathways. Cardiovasc. Res. 2011, 49–56. [Google Scholar] [CrossRef]
- Tanonaka, K.; Iwai, T.; Motegi, K.; Takeo, S. Effects of N-(2-mercaptopropionyl)-glycine on mitochondrial function in ischemic-reperfused heart. Cardiovasc. Res. 2003, 57, 416–425. [Google Scholar] [CrossRef]
- Stowe, D.F.; Aldakkak, M.; Camara, A.K.; Riess, M.L.; Heinen, A.; Varadarajan, S.G.; Jiang, M.T. Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H434–H440. [Google Scholar] [CrossRef]
- Behmenburg, F.; van Caster, P.; Bunte, S.; Brandenburger, T.; Heinen, A.; Hollmann, M.W.; Huhn, R. Impact of Anesthetic Regimen on Remote Ischemic Preconditioning in the Rat Heart In Vivo. Anesth. Analg. 2018, 126, 1377–1380. [Google Scholar] [CrossRef]
- Skrzypiec-Spring, M.; Grotthus, B.; Szelag, A.; Schulz, R. Isolated heart perfusion according to Langendorff—Still viable in the new millennium. J. Pharmacol. Toxicol. Methods 2007, 55, 113–126. [Google Scholar] [CrossRef]
- Dai, H.; Wang, M.; Patel, P.N.; Kalogeris, T.; Liu, Y.; Durante, W.; Korthuis, R.J. Preconditioning with the BKCa channel activator NS-1619 prevents ischemia-reperfusion-induced inflammation and mucosal barrier dysfunction: Roles for ROS and heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H988–H999. [Google Scholar] [CrossRef]
- Andrienko, T.; Pasdois, P.; Rossbach, A.; Halestrap, A.P. Real-Time Fluorescence Measurements of ROS and [Ca2+] in Ischemic/Reperfused Rat Hearts: Detectable Increases Occur only after Mitochondrial Pore Opening and Are Attenuated by Ischemic Preconditioning. PLoS ONE 2016, 11, e0167300. [Google Scholar] [CrossRef]
- Zhou, T.; Prather, E.R.; Garrison, D.E.; Zuo, L. Interplay between ROS and Antioxidants during Ischemia-Reperfusion Injuries in Cardiac and Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 417. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Body Weight (g) | Heart Weight Dry (g) | Heart Weight Wet (g) | Time of Max. Ischemic Contracture (min) | Level of Max. Ischemic Contracture (mmHg) | |
|---|---|---|---|---|---|---|
| Con | 9 | 284 ± 22 | 0.12 ± 0.02 | 1.27 ± 0.13 | 16 ± 2 | 69 ± 25 |
| Mil 0.3 | 9 | 287 ± 14 | 0.13 ± 0.01 | 1.37 ± 0.07 | 16 ± 2 | 58 ± 12 |
| Mil 1 | 7 | 285 ± 20 | 0.13 ± 0.02 | 1.33 ± 0.11 | 17 ± 2 | 55 ± 12 |
| Mil 3 | 7 | 278 ± 18 | 0.13 ± 0.02 | 1.36 ± 0.19 | 18 ± 4 | 74 ± 22 |
| Mil 10 | 7 | 286 ± 28 | 0.14 ± 0.01 | 1.33 ± 0.20 | 17 ± 3 | 60 ± 18 |
| Con | 9 | 299 ± 17 | 0.14 ± 0.02 | 1.35 ± 0.08 | 17 ± 2 | 61 ± 9 |
| Mil | 9 | 287 ± 14 | 0.13 ± 0.01 | 1.35 ± 0.09 | 16 ± 2 | 58 ± 17 |
| Pax + Mil | 7 | 284 ± 19 | 0.13 ± 0.01 | 1.33 ± 0.08 | 17 ± 2 | 63 ± 14 |
| Pax | 9 | 297 ± 13 | 0.14 ± 0.02 | 1.29 ± 0.11 | 17 ± 1 | 62 ± 15 |
| MPG + Mil | 9 | 297 ± 14 | 0.14 ± 0.01 | 1.35 ± 0.06 | 16 ± 1 | 59 ± 15 |
| MPG | 9 | 293 ± 15 | 0.15 ± 0.02 | 1.33 ± 0.07 | 16 ± 1 | 70 ± 12 |
| MPG + Mil + CsA | 9 | 293 ± 18 | 0.13 ± 0.01 | 1.27 ± 0.10 | 16 ± 2 | 63 ± 23 |
| Mil + CsA | 9 | 299 ± 19 | 0.13 ± 0.01 | 1.32 ± 0.11 | 16 ± 2 | 59 ± 18 |
| Baseline | PC | Reperfusion | ||
|---|---|---|---|---|
| 30 | 60 | |||
| Heart Rate (bpm) | ||||
| Con | 311 ± 37 | 302 ± 30 | 244 ± 46 * | 248 ± 34 * |
| Mil 0.3 | 313 ± 27 | 316 ± 32 | 262 ± 54 | 285 ± 47 |
| Mil 1 | 301 ± 21 | 282 ± 36 | 252 ± 36 | 232 ± 68 |
| Mil 3 | 315 ± 32 | 344 ± 38 | 231 ± 78 | 275 ± 72 |
| Mil 10 | 325 ± 61 | 328 ± 45 | 258 ± 43 | 263 ± 40 |
| Phasic LVP (mmHg) | ||||
| Con | 117 ± 20 | 125 ± 23 | 26 ± 12 * | 27 ± 12 * |
| Mil 0.3 | 123 ± 16 | 123 ± 15 | 24 ± 11 * | 22 ± 7 * |
| Mil 1 | 137 ± 9 | 138 ± 12 | 17 ± 5 * | 29 ± 5 * |
| Mil 3 | 129 ± 21 | 131 ± 19 | 31 ± 11 * | 36 ± 8 * |
| Mil 10 | 118 ± 21 | 130 ± 28 | 22 ± 14 * | 25 ± 12 * |
| Coronary flow (ml * min−1) | ||||
| Con | 13 ± 3 | 13 ± 2 | 7 ± 2 * | 6 ± 1 * |
| Mil 0.3 | 17 ± 2 # | 17 ± 2 # | 9 ± 2 * | 7 ± 2 * |
| Mil 1 | 14 ± 2 | 14 ± 2 | 8 ± 1 * | 6 ± 1 * |
| Mil 3 | 13 ± 4 | 15 ± 3 | 8 ± 1 * | 7 ± 1 * |
| Mil 10 | 14 ± 2 | 16 ± 2 # | 8 ± 1 * | 7 ± 1 * |
| Baseline | PC | Reperfusion | ||
|---|---|---|---|---|
| 30 | 60 | |||
| Heart Rate (bpm) | ||||
| Con | 275 ± 28 | 264 ± 13 | 237 ± 65 | 234 ± 45 |
| Mil | 282 ± 34 | 289 ± 37 | 260 ± 54 | 235 ± 46 |
| Pax + Mil | 322 ± 29 | 314 ± 22 | 280 ± 70 | 249 ± 40 |
| Pax | 298 ± 30 | 260 ± 36 | 230 ± 41 | 233 ± 47 |
| MPG + Mil | 301 ± 35 | 294 ± 39 | 274 ± 65 | 227 ± 44 |
| MPG | 294 ± 41 | 284 ± 40 | 218 ± 71 | 229 ± 82 |
| MPG + Mil + CsA | 314 ± 34 | 288 ± 42 | 253 ± 53 | 222 ± 72 |
| Mil + CsA | 299 ± 30 | 295 ± 26 | 251 ± 69 | 204 ± 51 |
| Phasic LVP (mmHg) | ||||
| Con | 146 ± 19 | 151 ± 22 | 18 ± 15 * | 21 ± 8 * |
| Mil | 148 ± 23 | 144 ± 20 | 23 ± 11 * | 32 ± 14 * |
| Pax + Mil | 131 ± 10 | 122 ± 24 # | 25 ± 9 * | 35 ± 12 * |
| Pax | 144 ± 24 | 126 ± 25 | 30 ± 10 * | 35 ± 12 * |
| MPG + Mil | 141 ± 15 | 157 ± 24 | 23 ± 15 * | 26 ± 16 * |
| MPG | 134 ± 18 | 148 ± 18 | 18 ± 7 * | 24 ± 8 * |
| MPG + Mil + CsA | 132 ± 33 | 125 ± 38 # | 22 ± 13 * | 22 ± 10 * |
| Mil + CsA | 133 ± 22 | 139 ± 36 | 26 ± 7 * | 33 ± 11 * |
| Coronary flow (ml * min−1) | ||||
| Con | 16 ± 2 | 16 ± 3 | 8 ± 2 * | 7 ± 2 * |
| Mil | 16 ± 2 | 17 ± 2 | 8 ± 2 * | 7 ± 2 * |
| Pax + Mil | 16 ± 4 | 14 ± 2 | 9 ± 3 * | 8 ± 4 * |
| Pax | 16 ± 2 | 13 ± 3 | 8 ± 2 * | 7 ± 2 * |
| MPG + Mil | 17 ± 3 | 19 ± 2 | 7 ± 2 * | 6 ± 2 * |
| MPG | 15 ± 2 | 17 ± 2 | 7 ± 3 * | 6 ± 3 * |
| MPG + Mil + CsA | 15 ± 3 | 17 ± 5 | 6 ± 2 * | 5 ± 3 * |
| Mil + CsA | 15 ± 4 | 16 ± 4 | 7 ± 2 * | 6 ± 2 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raupach, A.; Reinle, J.; Stroethoff, M.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Bunte, S. Milrinone-Induced Pharmacological Preconditioning in Cardioprotection: Hints for a Role of Mitochondrial Mechanisms. J. Clin. Med. 2019, 8, 507. https://doi.org/10.3390/jcm8040507
Raupach A, Reinle J, Stroethoff M, Mathes A, Heinen A, Hollmann MW, Huhn R, Bunte S. Milrinone-Induced Pharmacological Preconditioning in Cardioprotection: Hints for a Role of Mitochondrial Mechanisms. Journal of Clinical Medicine. 2019; 8(4):507. https://doi.org/10.3390/jcm8040507
Chicago/Turabian StyleRaupach, Annika, Julia Reinle, Martin Stroethoff, Alexander Mathes, André Heinen, Markus W. Hollmann, Ragnar Huhn, and Sebastian Bunte. 2019. "Milrinone-Induced Pharmacological Preconditioning in Cardioprotection: Hints for a Role of Mitochondrial Mechanisms" Journal of Clinical Medicine 8, no. 4: 507. https://doi.org/10.3390/jcm8040507