Mitophagy in Cardiovascular Diseases

 ,
,  , ,
, ,
Abstract
:1. Fundamental Aspects of Mitophagy
2. Mitophagy in Cardiovascular Diseases
2.1. Atherosclerosis
2.2. Ischemic Heart Disease
2.3. Cardiomyopathies
2.4. Heart Failure
2.5. Hypertension
2.6. Arrhythmia
2.7. Congenital Heart Disease
2.8. Peripheral Vascular Disease
2.9. Mitophagy in Human Cardiac Surgery
3. Targeting Mitophagy for Cardioprotection
4. Future Perspectives and Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, 146–603. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Vizarra, E.; Enriquez, J.A.; Perez-Martos, A.; Montoya, J.; Fernandez-Silva, P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 2011, 11, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Pedriali, G.; Vezzani, B.; Tarocco, A.; Marchi, S.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Interorganellar calcium signaling in the regulation of cell metabolism: A cancer perspective. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Pinton, P.; Leo, S.; Wieckowski, M.R.; Di Benedetto, G.; Rizzuto, R. Long-term modulation of mitochondrial Ca2+ signals by protein kinase C isozymes. J. Cell Biol. 2004, 165, 223–232. [Google Scholar] [CrossRef]
- Granatiero, V.; De Stefani, D.; Rizzuto, R. Mitochondrial Calcium Handling in Physiology and Disease. Adv. Exp. Med. Biol. 2017, 982, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [Green Version]
- Rius, R.; Cowley, M.J.; Riley, L.; Puttick, C.; Thorburn, D.R.; Christodoulou, J. Biparental inheritance of mitochondrial DNA in humans is not a common phenomenon. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [Green Version]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Morciano, G.; Pedriali, G.; Sbano, L.; Iannitti, T.; Giorgi, C.; Pinton, P. Intersection of mitochondrial fission and fusion machinery with apoptotic pathways: Role of Mcl-1. Biol. Cell 2016, 108, 279–293. [Google Scholar] [CrossRef]
- Patergnani, S.; Pinton, P. Mitophagy and mitochondrial balance. Methods Mol. Biol. 2015, 1241, 181–194. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Um, J.H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissova, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tal, R.; Winter, G.; Ecker, N.; Klionsky, D.J.; Abeliovich, H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J. Biol. Chem. 2007, 282, 5617–5624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarten, M.; Mohrluder, J.; Ma, P.; Stoldt, M.; Thielmann, Y.; Stangler, T.; Hersch, N.; Hoffmann, B.; Merkel, R.; Willbold, D. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy 2009, 5, 690–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ney, P.A. Mechanisms and biology of B-cell leukemia/lymphoma 2/adenovirus E1B interacting protein 3 and Nip-like protein X. Antioxid. Redox Signal. 2011, 14, 1959–1969. [Google Scholar] [CrossRef] [Green Version]
- Chourasia, A.H.; Macleod, K.F. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 2015, 11, 1937–1938. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [Green Version]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Cao, Y.; Tong, T.; Shi, J.; Zhang, Y.; Yang, Y.; Liu, C. Autophagy in atherosclerosis: A phenomenon found in human carotid atherosclerotic plaques. Chin. Med. J. 2015, 128, 69–74. [Google Scholar] [CrossRef]
- Perrotta, I. The use of electron microscopy for the detection of autophagy in human atherosclerosis. Micron 2013, 50, 7–13. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; Esen, E.; Stokes, C.J.; Evans, T.D.; Arif, B.; Curci, J.A.; Razani, B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, B.; Goikuria, H.; Vega, R.; Rodriguez-Antiguedad, A.; Lopez Medina, A.; Freijo Mdel, M.; Vandenbroeck, K.; Alloza, I. Autophagic marker MAP1LC3B expression levels are associated with carotid atherosclerosis symptomatology. PLoS ONE 2014, 9, e115176. [Google Scholar] [CrossRef]
- Grootaert, M.O.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; De Meyer, G.R.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef]
- Nahapetyan, H.; Moulis, M.; Grousset, E.; Faccini, J.; Grazide, M.H.; Mucher, E.; Elbaz, M.; Martinet, W.; Vindis, C. Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell Death Dis. 2019, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 28821–28835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, Z.; Zhang, Y.; Zhao, Q.; Wang, X.; Lu, P.; Zhang, H.; Wang, Z.; Dong, H.; Zhang, Z. PEDF protects cardiomyocytes by promoting FUNDC1mediated mitophagy via PEDF-R under hypoxic condition. Int. J. Mol. Med. 2018, 41, 3394–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Siraj, S.; Zhang, R.; Chen, Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 2017, 13, 1080–1081. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Foglio, E.; Puddighinu, G.; Germani, A.; Russo, M.A.; Limana, F. HMGB1 Inhibits Apoptosis Following MI and Induces Autophagy via mTORC1 Inhibition. J. Cell. Physiol. 2017, 232, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Cahill, T.J.; Leo, V.; Kelly, M.; Stockenhuber, A.; Kennedy, N.W.; Bao, L.; Cereghetti, G.M.; Harper, A.R.; Czibik, G.; Liao, C.; et al. Resistance of dynamin-related protein 1 oligomers to disassembly impairs mitophagy, resulting in myocardial inflammation and heart failure. J. Biol. Chem. 2016, 291, 25762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhao, Z.; Feng, X.; Cheng, Z.; Xiong, Z.; Wang, T.; Lin, J.; Zhang, M.; Hu, J.; Fan, Y.; et al. Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J. Cell. Mol. Med. 2018, 22, 5132–5144. [Google Scholar] [CrossRef] [PubMed]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Z.L.; Eirin, A.; Ebrahimi, B.; Pawar, A.S.; Zhu, X.Y.; Lerman, A.; Lerman, L.O. Cardiac metabolic alterations in hypertensive obese pigs. Hypertension 2015, 66, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Yu, B.B.; Zhi, H.; Zhang, X.Y.; Liang, J.W.; He, J.; Su, C.; Xia, W.H.; Zhang, G.X.; Tao, J. Mitochondrial dysfunction-mediated decline in angiogenic capacity of endothelial progenitor cells is associated with capillary rarefaction in patients with hypertension via downregulation of CXCR4/JAK2/SIRT5 signaling. EBioMedicine 2019, 42, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.R.; Baggett, B.; Cooper, L.L.; Lu, Y.J.O.U.; Sedivy, J.M.; Terentyev, D.; Koren, G. Enhancing Autophagy Diminishes Aberrant Ca(2+) Homeostasis and Arrhythmogenesis in Aging Rabbit Hearts. Front. Physiol. 2019, 10, 1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.H.; McDermott, M.M.; Sufit, R.L.; Kosmac, K.; Bugg, A.W.; Gonzalez-Freire, M.; Ferrucci, L.; Tian, L.; Zhao, L.; Gao, Y.; et al. Walking performance is positively correlated to calf muscle fiber size in peripheral artery disease subjects, but fibers show aberrant mitophagy: An observational study. J. Transl Med. 2016, 14, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andres, A.M.; Tucker, K.C.; Thomas, A.; Taylor, D.J.; Sengstock, D.; Jahania, S.M.; Dabir, R.; Pourpirali, S.; Brown, J.A.; Westbrook, D.G.; et al. Mitophagy and mitochondrial biogenesis in atrial tissue of patients undergoing heart surgery with cardiopulmonary bypass. JCI Insight 2017, 2, e89303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Li, D.; Zhu, P.; Hu, S.; Hu, N.; Ma, S.; Zhang, Y.; Han, T.; Ren, J.; Cao, F.; et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Tonet, E.; Bernucci, D.; Morciano, G.; Campo, G. Pharmacological protection of reperfusion injury in ST-segment elevation myocardial infarction. Gone with the wind? Postepy Kardiol Interwencyjnej 2018, 14, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Campo, G.; Morciano, G.; Pavasini, R.; Bonora, M.; Sbano, L.; Biscaglia, S.; Bovolenta, M.; Pinotti, M.; Punzetti, S.; Rizzo, P.; et al. Fo ATP synthase C subunit serum levels in patients with ST-segment Elevation Myocardial Infarction: Preliminary findings. Int. J. Cardiol. 2016, 221, 993–997. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Decker, R.S.; Wildenthal, K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am. J. Pathol. 1980, 98, 425–444. [Google Scholar] [PubMed]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Gurusamy, N.; Lekli, I.; Gorbunov, N.V.; Gherghiceanu, M.; Popescu, L.M.; Das, D.K. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J. Cell. Mol. Med. 2009, 13, 373–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sybers, H.D.; Ingwall, J.; DeLuca, M. Autophagy in cardiac myocytes. Recent Adv. Stud. Cardiac Struct. Metab. 1976, 12, 453–463. [Google Scholar] [PubMed]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar] [CrossRef]
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001, 15, 2286–2287. [Google Scholar] [CrossRef] [Green Version]
- Teckman, J.H.; An, J.K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G851–G862. [Google Scholar] [CrossRef]
- Carreira, R.S.; Lee, Y.; Ghochani, M.; Gustafsson, A.B.; Gottlieb, R.A. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 2010, 6, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrington, D.D.; Van Vleet, T.R.; Schnellmann, R.G. Calpain 10: A mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am. J. Physiol. Cell Physiol. 2006, 291, C1159–C1171. [Google Scholar] [CrossRef] [PubMed]
- Siddall, H.K.; Yellon, D.M.; Ong, S.B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS ONE 2013, 8, e62400. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubasiak, L.A.; Hernandez, O.M.; Bishopric, N.H.; Webster, K.A. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. USA 2002, 99, 12825–12830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Vallentin, A.; Churchill, E.; Mochly-Rosen, D. deltaPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart. J. Mol. Cell. Cardiol. 2007, 43, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Duffy, A.; O’Mahoney, M.E.; Logue, S.E.; Mylotte, L.A.; O’Brien, T.; Samali, A. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem. Biophys. Res. Commun. 2006, 349, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Terai, K.; Hiramoto, Y.; Masaki, M.; Sugiyama, S.; Kuroda, T.; Hori, M.; Kawase, I.; Hirota, H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol. Cell. Biol. 2005, 25, 9554–9575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severino, A.; Campioni, M.; Straino, S.; Salloum, F.N.; Schmidt, N.; Herbrand, U.; Frede, S.; Toietta, G.; Di Rocco, G.; Bussani, R.; et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 1029–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azfer, A.; Niu, J.; Rogers, L.M.; Adamski, F.M.; Kolattukudy, P.E. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1411–H1420. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Organelle dynamics: ER embraces mitochondria for fission. Curr. Biol. 2011, 21, R922–R924. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Wales, P.; Schuberth, C.E.; Aufschnaiter, R.; Fels, J.; Garcia-Aguilar, I.; Janning, A.; Dlugos, C.P.; Schafer-Herte, M.; Klingner, C.; Walte, M.; et al. Calcium-mediated actin reset (CaAR) mediates acute cell adaptations. Elife 2016, 5. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Ono, K.; Nagao, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2261–H2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitahara, T.; Takeishi, Y.; Harada, M.; Niizeki, T.; Suzuki, S.; Sasaki, T.; Ishino, M.; Bilim, O.; Nakajima, O.; Kubota, I. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc. Res. 2008, 80, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Bei, Y.; Lin, S.; Zhang, H.; Zhou, Y.; Jiang, J.; Chen, P.; Shen, S.; Xiao, J.; Li, X. Exercise Training Protects Against Acute Myocardial Infarction via Improving Myocardial Energy Metabolism and Mitochondrial Biogenesis. Cell. Physiol. Biochem. 2015, 37, 162–175. [Google Scholar] [CrossRef]
- Dandamudi, S.; Slusser, J.; Mahoney, D.W.; Redfield, M.M.; Rodeheffer, R.J.; Chen, H.H. The prevalence of diabetic cardiomyopathy: A population-based study in Olmsted County, Minnesota. J. Card Fail. 2014, 20, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Paolillo, S.; Marsico, F.; Prastaro, M.; Renga, F.; Esposito, L.; De Martino, F.; Di Napoli, P.; Esposito, I.; Ambrosio, A.; Ianniruberto, M.; et al. Diabetic Cardiomyopathy: Definition, Diagnosis, and Therapeutic Implications. Heart Fail. Clin. 2019, 15, 341–347. [Google Scholar] [CrossRef]
- Duncan, J.G.; Finck, B.N. The PPARalpha-PGC-1alpha Axis Controls Cardiac Energy Metabolism in Healthy and Diseased Myocardium. PPAR Res. 2008, 2008, 253817. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Kelly, D.P. Peroxisome proliferator-activated receptor alpha (PPARalpha) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart. J. Mol. Cell. Cardiol. 2002, 34, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.D. The mitochondria in diabetic heart failure: From pathogenesis to therapeutic promise. Antioxid. Redox Signal. 2015, 22, 1515–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Sadoshima, J. Mitochondrial autophagy in cardiomyopathy. Curr. Opin. Genet. Dev. 2016, 38, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Kobayashi, S.; Chen, K.; Timm, D.; Volden, P.; Huang, Y.; Gulick, J.; Yue, Z.; Robbins, J.; Epstein, P.N.; et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J. Biol. Chem. 2013, 288, 18077–18092. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Liang, Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim. Biophys. Acta 2015, 1852, 252–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Brady, C.; Zablocki, D.; Ohishi, M.; Sadoshima, J. Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. J. Mol. Cell. Cardiol. 2015, 78, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Hoke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell. Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhu, H.; Li, H.; Zou, M.H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, L.; Qiao, Y.; Zhou, X.; Wu, G.; Wang, L.; Peng, Y.; Dong, X.; Huang, H.; Si, L.; et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE 2013, 8, e75927. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Riehle, C.; Wende, A.R.; Sena, S.; Pires, K.M.; Pereira, R.O.; Zhu, Y.; Bugger, H.; Frank, D.; Bevins, J.; Chen, D.; et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J. Clin. Investig. 2013, 123, 5319–5333. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Knaapen, M.W.; Davies, M.J.; De Bie, M.; Haven, A.J.; Martinet, W.; Kockx, M.M. Apoptotic versus autophagic cell death in heart failure. Cardiovasc. Res. 2001, 51, 304–312. [Google Scholar] [CrossRef]
- Kostin, S.; Pool, L.; Elsasser, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klovekorn, W.P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Bugger, H.; Schwarzer, M.; Chen, D.; Schrepper, A.; Amorim, P.A.; Schoepe, M.; Nguyen, T.D.; Mohr, F.W.; Khalimonchuk, O.; Weimer, B.C.; et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc. Res. 2010, 85, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, A.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Vatner, D.E.; Kim, S.J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [Green Version]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Pisano, A.; Cerbelli, B.; Perli, E.; Pelullo, M.; Bargelli, V.; Preziuso, C.; Mancini, M.; He, L.; Bates, M.G.; Lucena, J.R.; et al. Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end-stage ischemic heart failure. Cardiovasc. Pathol. 2016, 25, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Arnon, E.; Kostin, S.; Schonburg, M.; Elsasser, A.; Polyakova, V.; Bauer, E.P.; Klovekorn, W.P.; Schaper, J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef]
- Page, I.H. The mosaic theory of arterial hypertension--its interpretation. Perspect. Biol. Med. 1967, 10, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Bazan, I.; Zhang, Y.; Fares, W.H.; Lee, P.J. Mitochondrial dysfunction and pulmonary hypertension: Cause, effect, or both. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L782–L796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, S.; Mannam, P.; Zhang, J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L433–L452. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Albrecht, M.; Fisher, A.J.; Selej, M.; Patel, N.G.; Brown, J.A.; Justice, M.J.; Brown, M.B.; Van Demark, M.; Trulock, K.M.; et al. 17beta-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am. J. Respir. Crit. Care Med. 2012, 185, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Smith, A.; Guo, L.; Alastalo, T.P.; Li, M.; Sawada, H.; Liu, X.; Chen, Z.H.; Ifedigbo, E.; Jin, Y.; et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2011, 183, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, D.K.; Alzoubi, A.; Gupte, R.; Chettimada, S.; Watanabe, M.; Kahn, A.G.; Okada, T.; McMurtry, I.F.; Gupte, S.A. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 2014, 64, 1266–1274. [Google Scholar] [CrossRef] [Green Version]
- Teng, R.J.; Du, J.; Welak, S.; Guan, T.; Eis, A.; Shi, Y.; Konduri, G.G. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L651–L663. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, V.; Marijon, E. Cardiac arrhythmias: Diagnosis and management. Rev. Med. Interne 2016, 37, 608–615. [Google Scholar] [CrossRef]
- Yang, K.C.; Bonini, M.G.; Dudley, S.C., Jr. Mitochondria and arrhythmias. Free Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Burkhalter, M.D.; Sridhar, A.; Sampaio, P.; Jacinto, R.; Burczyk, M.S.; Donow, C.; Angenendt, M.; Competence Network for Congenital Heart Defects, I.; Hempel, M.; Walther, P.; et al. Imbalanced mitochondrial function provokes heterotaxy via aberrant ciliogenesis. J. Clin. Investig. 2019, 129, 2841–2855. [Google Scholar] [CrossRef]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Pampliega, O.; Cuervo, A.M. Autophagy and primary cilia: Dual interplay. Curr. Opin. Cell Biol. 2016, 39, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Livingston, M.J.; Su, Y.; Dong, Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 2015, 11, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, J.; Liu, L.; McKeehan, W.L.; Wang, F. The fibroblast growth factor signaling axis controls cardiac stem cell differentiation through regulating autophagy. Autophagy 2012, 8, 690–691. [Google Scholar] [CrossRef] [Green Version]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salabei, J.K.; Hill, B.G. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free Radic. Biol. Med. 2013, 65, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Kong, L.S.; Chen, H.; Li, W.D.; Qian, A.M.; Wang, X.Y.; Du, X.L.; Li, C.L.; Yu, X.B.; Li, X.Q. Autophagy protein 5 enhances the function of rat EPCs and promotes EPCs homing and thrombus recanalization via activating AKT. Thromb. Res. 2015, 136, 642–651. [Google Scholar] [CrossRef]
- Sarafian, V.; Dikov, D.; Karaivanov, M.; Belovejdov, V.; Stefanova, P. Differential expression of ABH histo-blood group antigens and LAMPs in infantile hemangioma. J. Mol. Histol. 2005, 36, 455–460. [Google Scholar] [CrossRef]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef]
- Jahania, S.M.; Sengstock, D.; Vaitkevicius, P.; Andres, A.; Ito, B.R.; Gottlieb, R.A.; Mentzer, R.M., Jr. Activation of the homeostatic intracellular repair response during cardiac surgery. J. Am. Coll. Surg. 2013, 216, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andres, A.M.; Hernandez, G.; Lee, P.; Huang, C.; Ratliff, E.P.; Sin, J.; Thornton, C.A.; Damasco, M.V.; Gottlieb, R.A. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal. 2014, 21, 1960–1973. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell Longev. 2014, 2014, 345105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, J.; Xu, J.; Lu, Y.; Jiang, J.; Wang, L.; Shen, H.M.; Xia, D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget 2016, 7, 75659–75671. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, X.; Yang, X. Possible antioxidant mechanism of melanoidins extract from Shanxi aged vinegar in mitophagy-dependent and mitophagy-independent pathways. J. Agric. Food Chem. 2014, 62, 8616–8622. [Google Scholar] [CrossRef]
- Ma, L.; Zhu, J.; Gao, Q.; Rebecchi, M.J.; Wang, Q.; Liu, L. Restoring Pharmacologic Preconditioning in the Aging Heart: Role of Mitophagy/Autophagy. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Thangaraj, A.; Periyasamy, P.; Guo, M.L.; Chivero, E.T.; Callen, S.; Buch, S. Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy 2019, 1–24. [Google Scholar] [CrossRef]
- Bian, X.; Teng, T.; Zhao, H.; Qin, J.; Qiao, Z.; Sun, Y.; Liun, Z.; Xu, Z. Zinc prevents mitochondrial superoxide generation by inducing mitophagy in the setting of hypoxia/reoxygenation in cardiac cells. Free Radic. Res. 2018, 52, 80–91. [Google Scholar] [CrossRef]
- Qiao, H.; Ren, H.; Du, H.; Zhang, M.; Xiong, X.; Lv, R. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol. Med. Rep. 2018, 17, 3722–3734. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [Green Version]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a master regulator of cell death and inflammation: Molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carloni, S.; Favrais, G.; Saliba, E.; Albertini, M.C.; Chalon, S.; Longini, M.; Gressens, P.; Buonocore, G.; Balduini, W. Melatonin modulates neonatal brain inflammation through endoplasmic reticulum stress, autophagy, and miR-34a/silent information regulator 1 pathway. J. Pineal Res. 2016, 61, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Riparini, G.; Buonocore, G.; Balduini, W. Rapid modulation of the silent information regulator 1 by melatonin after hypoxia-ischemia in the neonatal rat brain. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Hu, S.; Shi, C.; Zhu, P.; Ma, Q.; Jin, Q.; Cao, F.; Tian, F.; Chen, Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Gabisonia, K.; Prosdocimo, G.; Aquaro, G.D.; Carlucci, L.; Zentilin, L.; Secco, I.; Ali, H.; Braga, L.; Gorgodze, N.; Bernini, F.; et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 2019, 569, 418–422. [Google Scholar] [CrossRef]
- Yang, F.; Li, T.; Dong, Z.; Mi, R. MicroRNA-410 is involved in mitophagy after cardiac ischemia/reperfusion injury by targeting high-mobility group box 1 protein. J. Cell Biochem. 2018, 119, 2427–2439. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J. Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef] [Green Version]



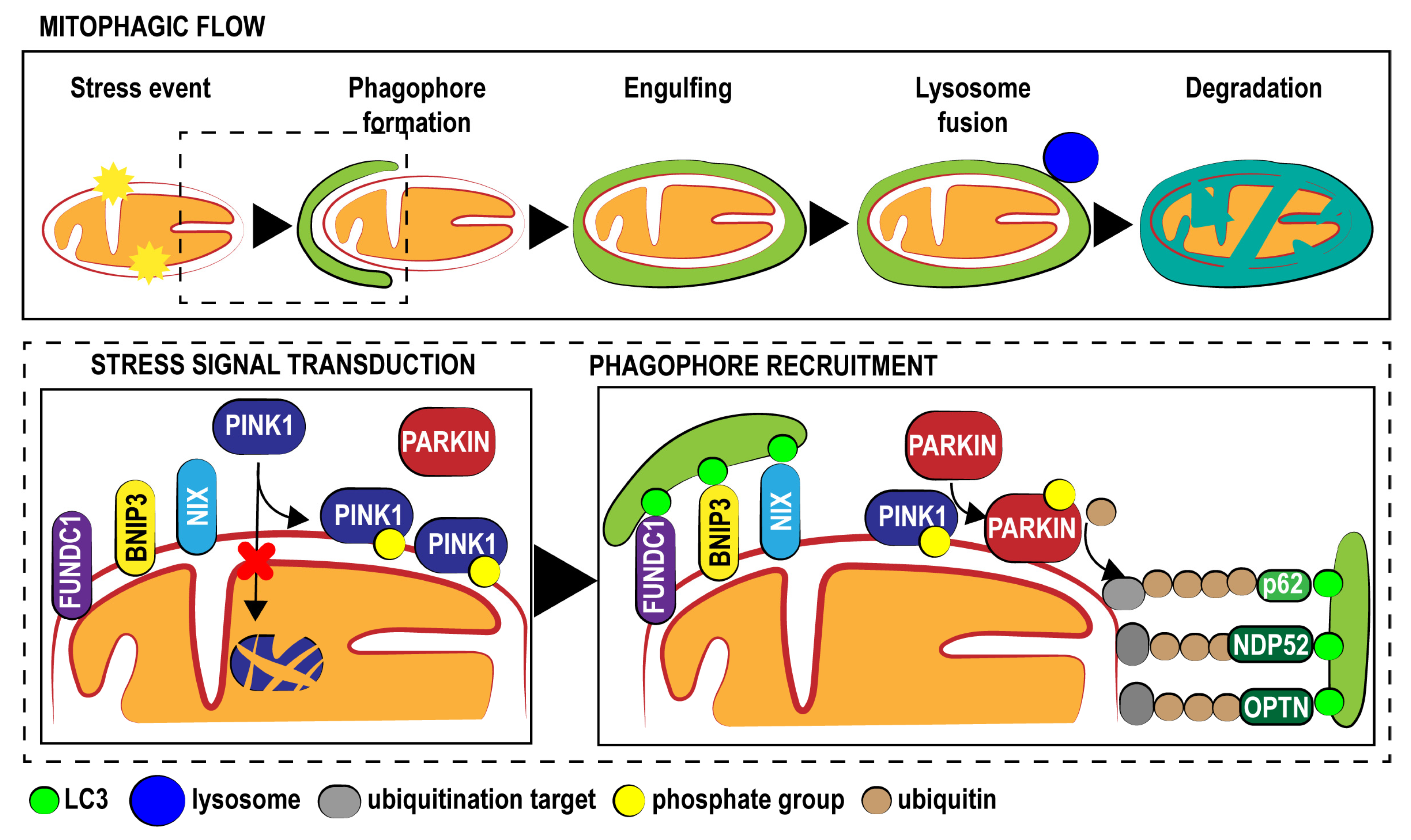{kind=link}
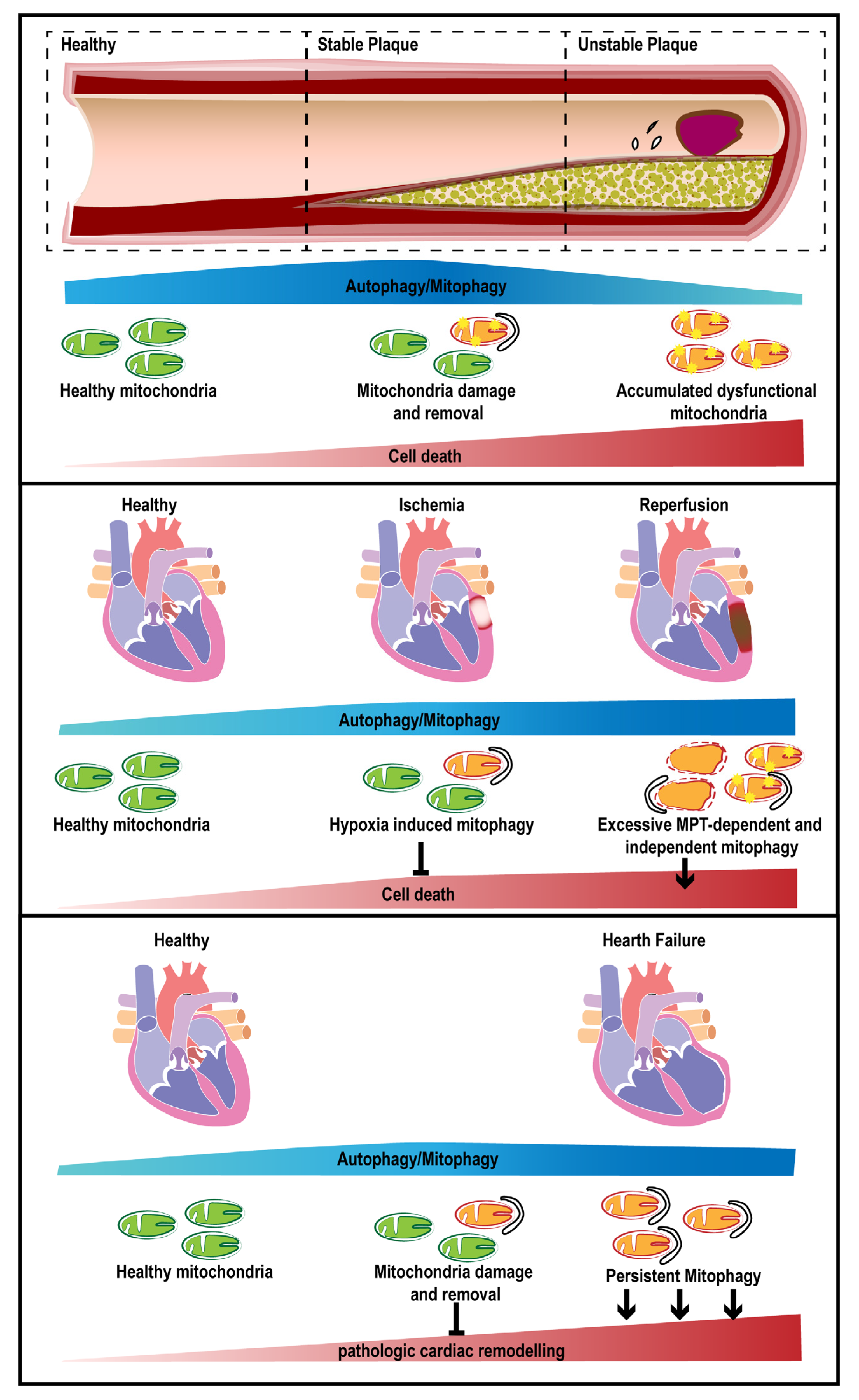{kind=link}
| Pathology | Mechanism of Action | References |
|---|---|---|
| Atherosclerosis | Atherosclerotic plaque stabilization mediated by ox-LDL. | [52] |
| NLRP3 inflammasome activation by the SIRT3/FOXO3a/Parkin signaling pathway. | [53] | |
| Ischemic heart disease | Mitophagy is an adaptive metabolic response to hypoxia mediated by BNIP3, Beclin-1 and ATG5. | [54] |
| Pigment epithelium-derived factor (PEDF) protects hypoxic cardiomyocytes in a rat model through the PEDF/PEDF-R/PA/DAG/PKC-α/ULK1/FUNDC1 pathway. | [55] | |
| Fundc1-knockout (KO) platelets present impaired mitochondria, which cause more I/R heart injury. | [56] | |
| FUNDC1 loss of function through CK2α-mediated phosphorylation leads to the development of cardiac I/R injury in mice. | [57] | |
| HMGB1 treatment in a murine model inhibits apoptosis via mTORC1 inhibition. | [58] | |
| Cardiomyopathies and heart failure | Inhibition of the dynamic process through the expression of dominant-negative Drp1 (Drp1K38A) results in disruption of mitophagy. C452F Drp1 mutation causes spontaneous development of monogenic dilated CM. | [59] |
| Mice lacking transferrin receptor (Tfrc) develop lethal CM and drastically ineffective mitophagy due to altered expression of proteins involved in mitophagy. | [60] | |
| Enhancement of mitophagy by TAT-Beclin1 attenuates the development of DCM. | [61] | |
| Melatonin inhibits Mst1 phosphorylation, increases LC3-II levels and enhances Parkin activity in mice with CM. | [62] | |
| Parkin-deficient mice are more sensitive to MI, developing larger infarcts and exhibiting reduced survival. | [63] | |
| An increase in BNIP3 expression is detected in adult rat hearts with chronic HF. | [64] | |
| Tamoxifen-inducible cardiac-specific Drp1-KO mice present cardiac dysfunction and increased susceptibility to I/R linked to an accumulation of damaged mitochondria (due to mitophagy inhibition). | [65] | |
| Phosphorylation of Ser495 in PINK1 by AMPKα2 prevents the progression of HF. | [66] | |
| Cytosolic p53 binding to Parkin prevents its translocation to damaged mitochondria, modulating cardiac dysfunction in HF. | [67] | |
| Hypertension | The coexistence of obesity and HT aggravates mitochondrial dysfunction; RAAS activation inhibits mitochondrial biogenesis. | [68] |
| Spermidine assumption leads to a delayed onset of HT in wild-type animals by stimulating mitophagy in cardiomyocytes, but these effects are abolished in ATG5-KO mice. | [69] | |
| EPCs in HT patients present mitochondrial dysfunctions linked to impairment of the CXCR4/JAK2/SIRT5 signaling pathway and failure of angiogenic capacity. | [70] | |
| Arrhythmia | Decrease in mitophagy leads to pro-arrhythmic spontaneous Ca2+ release via oxidized RyR2s by mito-ROS. | [71] |
| Peripheral vascular disease | PVD fibers present an accumulation of LC3 that is not completely colocalized with LAMP2. | [72] |
| Mitophagy in cardiac surgery | Post-cardiopulmonary bypass samples display decreased levels of mitophagy adapters NDP52 and OPTN, decreased expression of the long form of Opa1, and translocation of Parkin to the mitochondrial fraction. | [73] |
| Reperfusion after CABG surgery induces FUNDC1 dephosphorylation and mitophagy activation, causing platelet aggregation and increased risk of thrombosis. | [74] |
| Compound | Target/Mechanism of Action | Cardiovascular Effect |
|---|---|---|
| TEMPOL | SOD mimetic/reduces ROS and counteracts age-related mitophagy | Improves preconditioning after hypoxia/reoxygenation in aged cultured cardiomyocytes |
| Simvastatin | mTOR-dependent activation of mitophagy through Parkin and p62/SQSTM1 | Improves cardioprotection during left ventricular artery occlusion and reperfusion |
| Zinc | Favors SOD activity and activates PINK1/Beclin1-dependent mitophagy | Improves the recovery of cultured cardiomyocytes after hypoxia/reoxygenation |
| Liraglutide | Agonist of GLP1, stimulates SIRT1 and promotes Parkin-mediated mitophagy | Promotes recovery after left ventricular artery ligation-induced ischemia |
| Melatonin | Inhibits Mst1 phosphorylation, increases LC3-II levels and enhances Parkin-mediated mitophagy | Counteracts diabetic CM |
| Metformin | Activates autophagy by disrupting the Bcl2-Beclin1 complex | Inhibits cardiomyocyte apoptosis in diabetic heart |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. https://doi.org/10.3390/jcm9030892
Morciano G, Patergnani S, Bonora M, Pedriali G, Tarocco A, Bouhamida E, Marchi S, Ancora G, Anania G, Wieckowski MR, et al. Mitophagy in Cardiovascular Diseases. Journal of Clinical Medicine. 2020; 9(3):892. https://doi.org/10.3390/jcm9030892
Chicago/Turabian StyleMorciano, Giampaolo, Simone Patergnani, Massimo Bonora, Gaia Pedriali, Anna Tarocco, Esmaa Bouhamida, Saverio Marchi, Gina Ancora, Gabriele Anania, Mariusz R. Wieckowski, and et al. 2020. "Mitophagy in Cardiovascular Diseases" Journal of Clinical Medicine 9, no. 3: 892. https://doi.org/10.3390/jcm9030892