Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4‒JAK‒STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis


Abstract
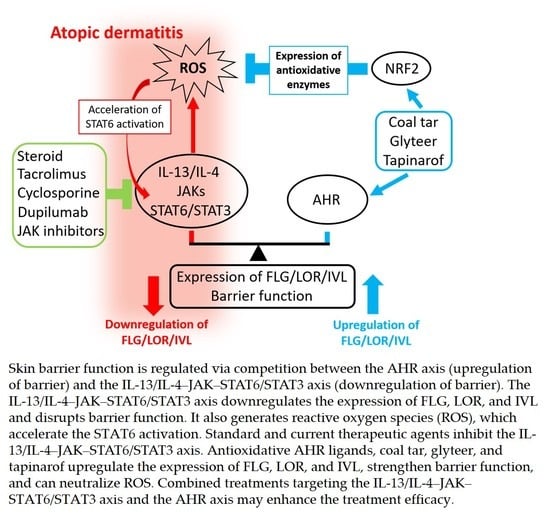
:
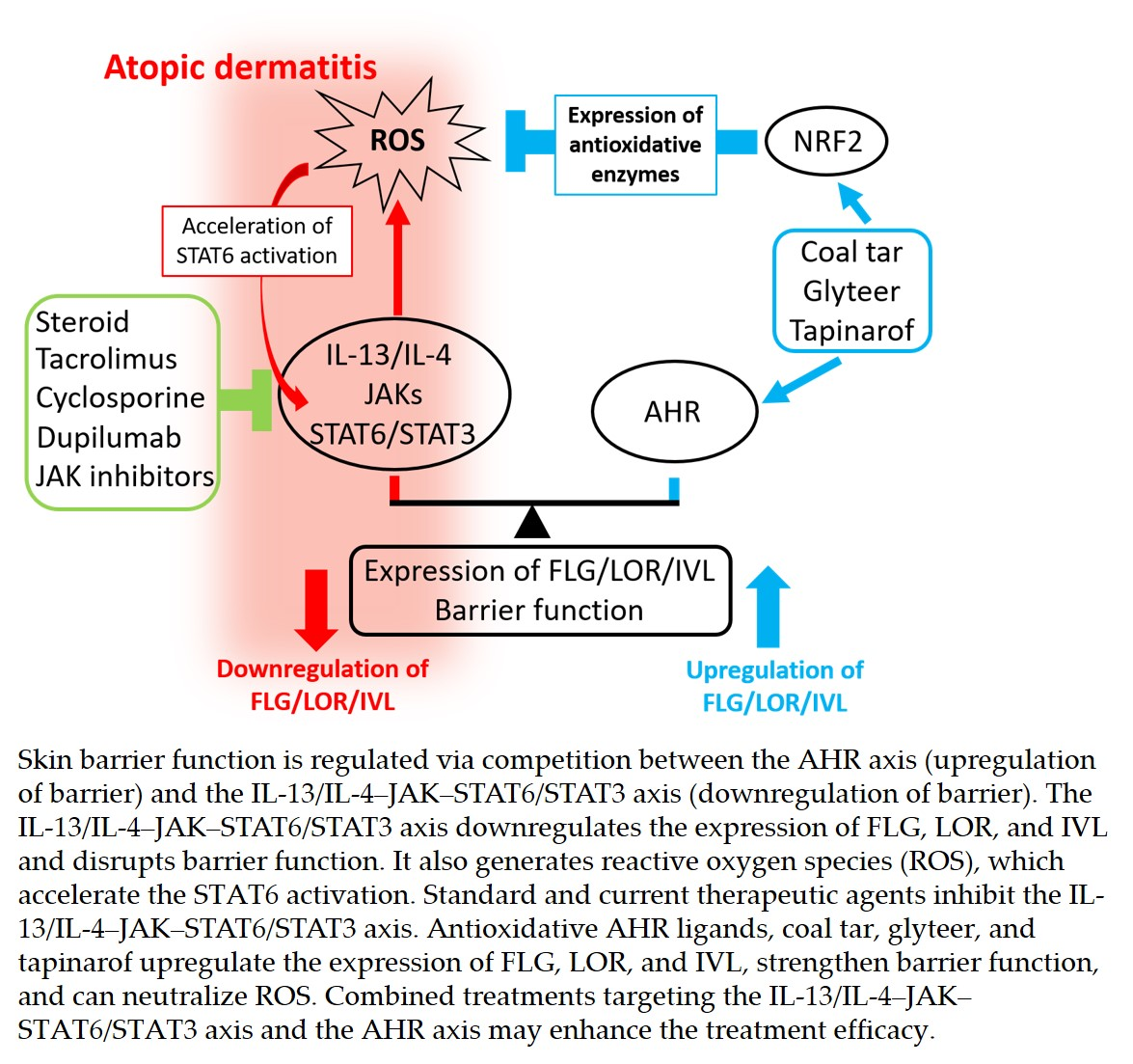{kind=link}
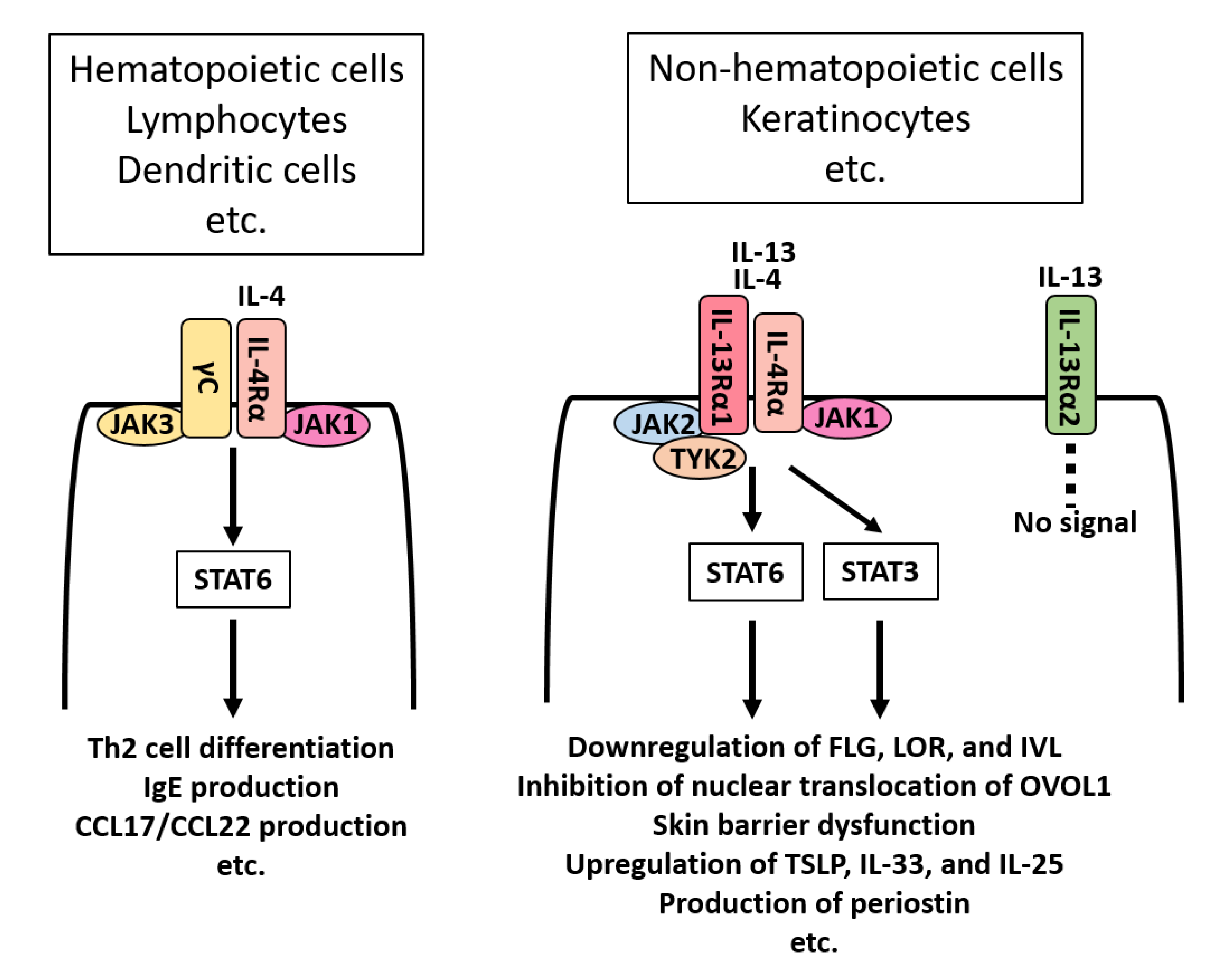{kind=link}
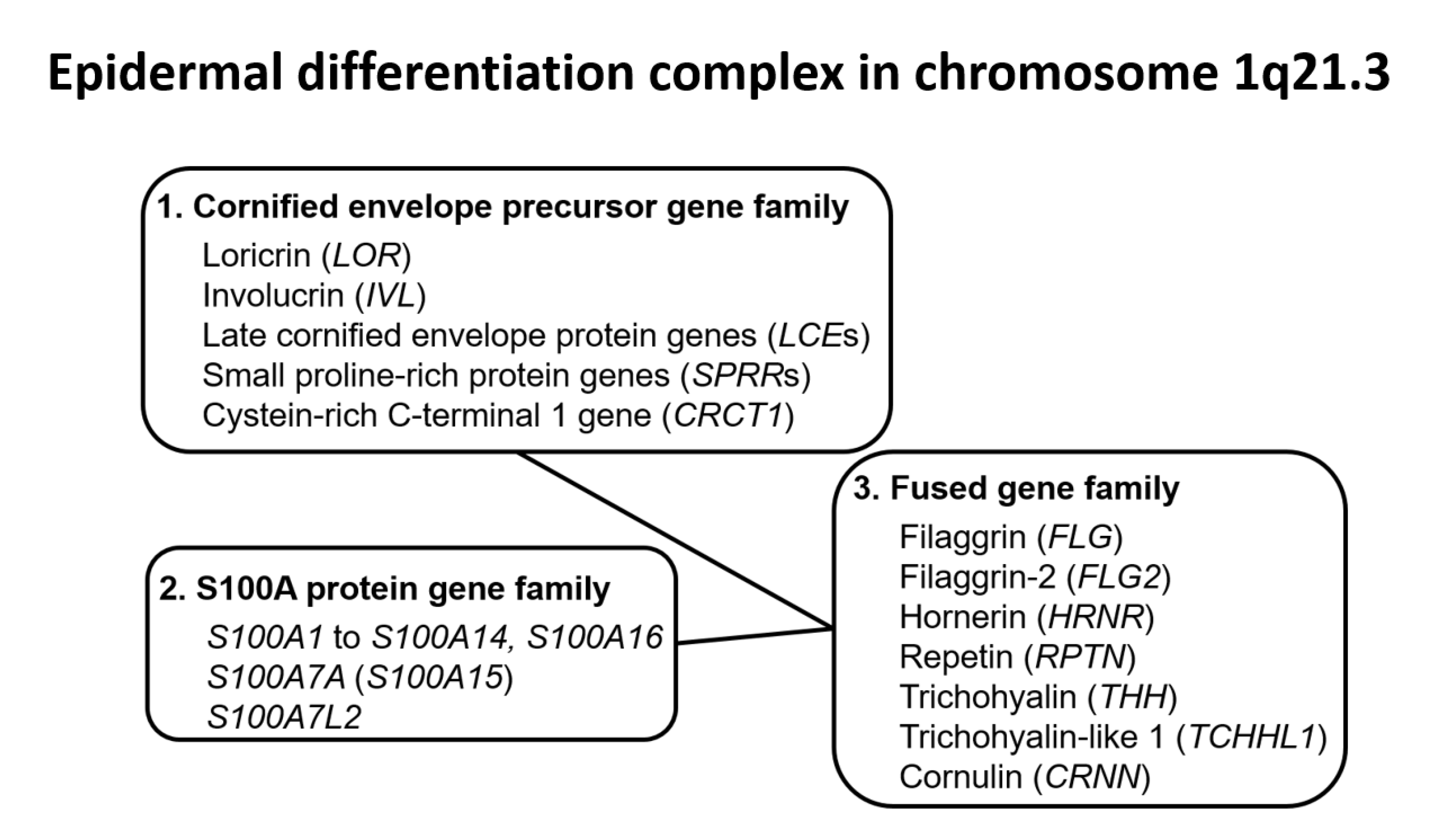{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
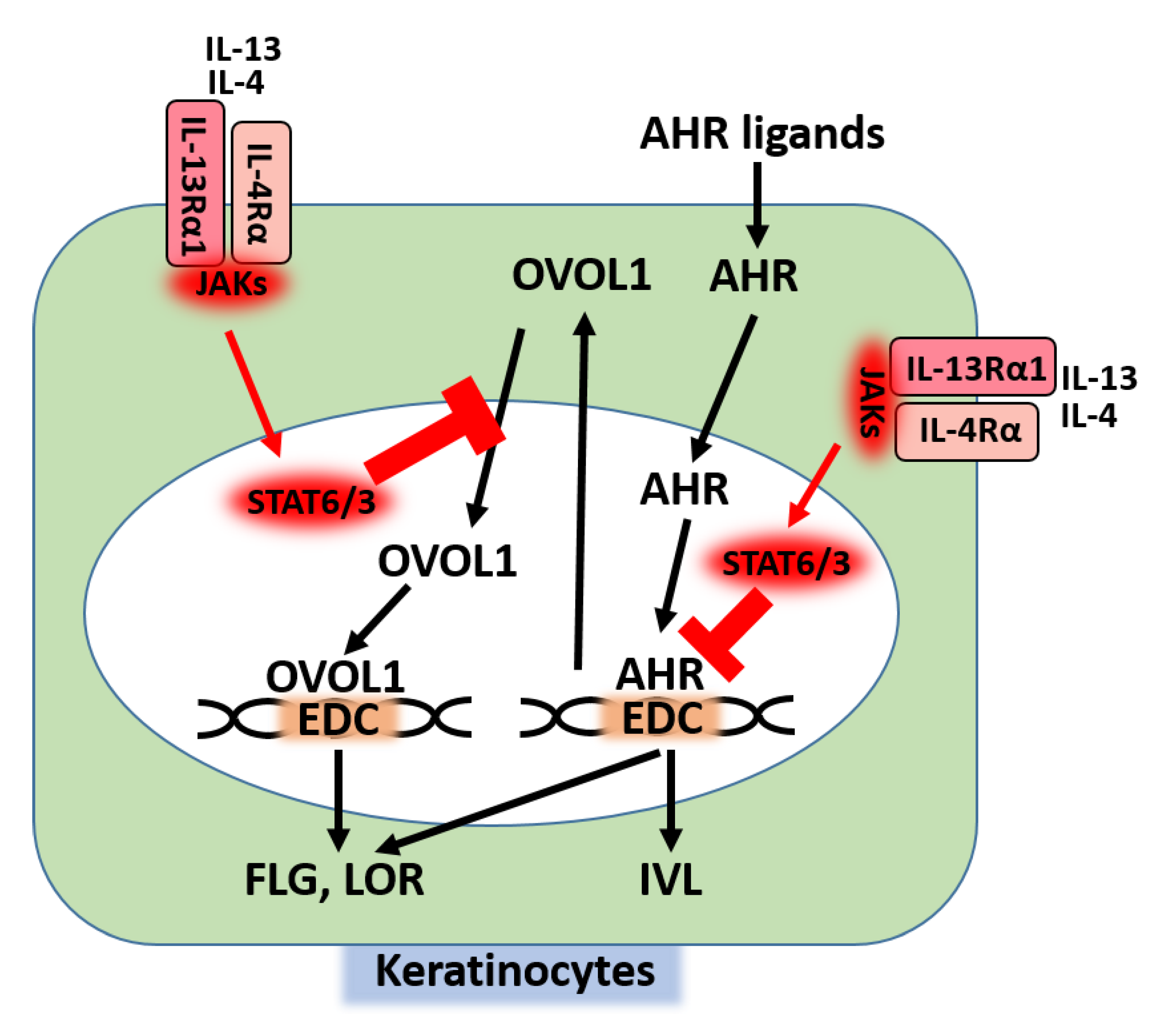
2. IL-13/IL-4 Signaling and AD
3. Role of IL-31 and IL-13/IL-4 in Atopic Pruritus
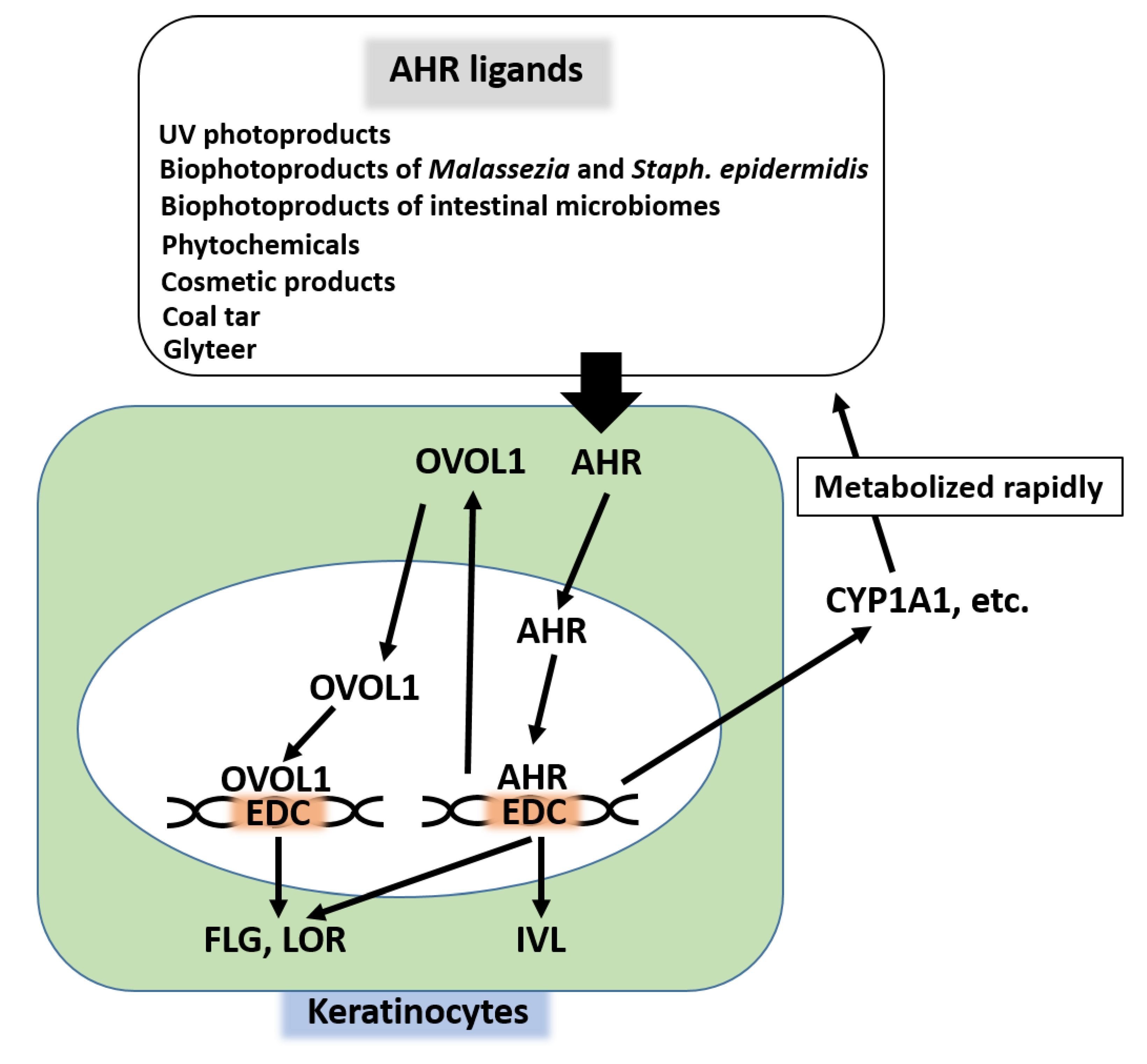
4. Regulation of Skin Barrier Function by Competition between AHR Axis and IL-13/IL-4‒JAK‒STAT6/STAT3 Axis
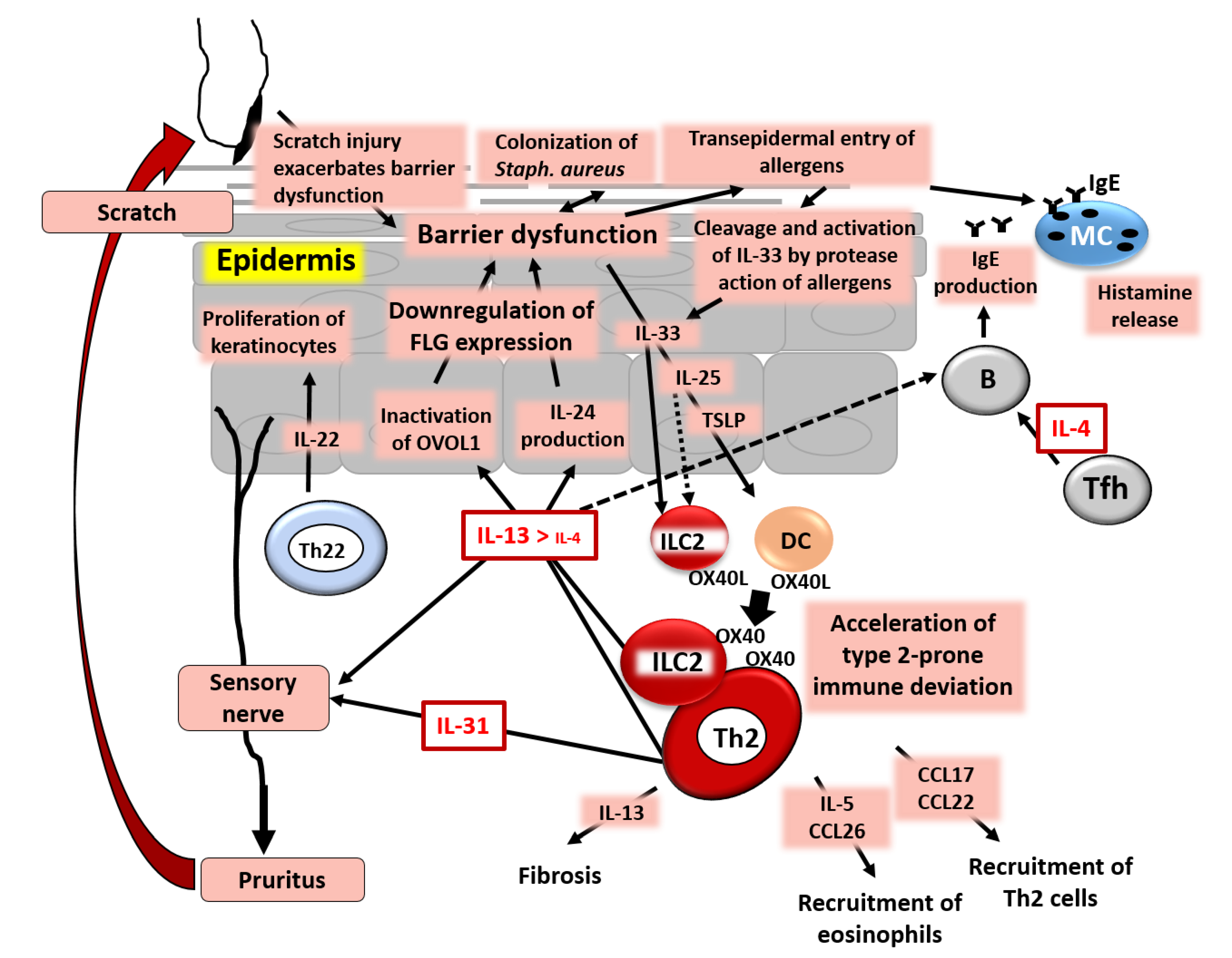
5. Skin Barrier Dysfunction Stimulates Keratinocytes to Produce TSLP, IL-25, and IL-33 and Promotes Type 2 Immune Deviation
6. Th17/Th22 Cells and Chronicity in AD
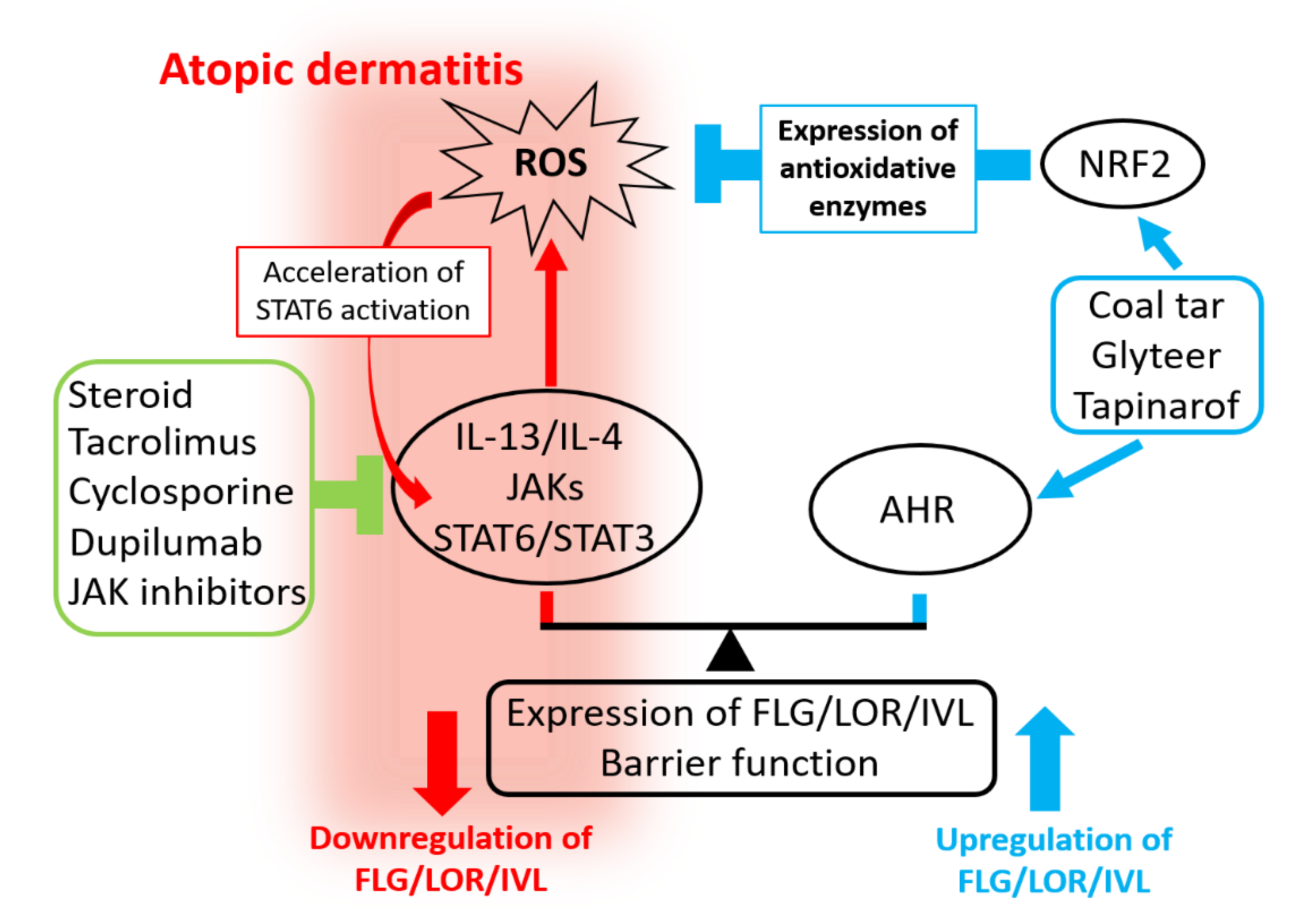
7. IL-13/IL-4–JAK–STAT6/STAT3 Axis and Oxidative Stress
8. Mechanisms of Action of Pharmaceutical Agents in AD
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Furue, M.; Yamazaki, S.; Jimbow, K.; Tsuchida, T.; Amagai, M.; Tanaka, T.; Matsunaga, K.; Muto, M.; Morita, E.; Akiyama, M.; et al. Prevalence of dermatological disorders in Japan: A nationwide, cross-sectional, seasonal, multicenter, hospital-based study. J. Dermatol. 2011, 38, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Stewart, A.; von Mutius, E.; Cookson, W.; Anderson, H.R. Is eczema really on the increase worldwide? J. Allergy Clin. Immunol. 2008, 121, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Chiba, T.; Takeuchi, S. Current status of atopic dermatitis in Japan. Asia Pac. Allergy 2011, 1, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.C.; Strachan, D.P. The natural history of childhood eczema: Observations from the British 1958 birth cohort study. Br. J. Dermatol. 1998, 139, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, L.; Savenije, O.E.M.; Heron, J.; Evans, D.M.; Vonk, J.M.; Brunekreef, B.; Wijga, A.H.; Henderson, A.J.; Koppelman, G.H.; Brown, S.J. Identification of atopic dermatitis subgroups in children from 2 longitudinal birth cohorts. J. Allergy Clin. Immunol. 2018, 141, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef]
- Nakahara, T.; Fujita, H.; Arima, K.; Taguchi, Y.; Motoyama, S.; Furue, M. Treatment satisfaction in atopic dermatitis relates to patient-reported severity: A cross-sectional study. Allergy 2019, 74, 1179–1181. [Google Scholar] [CrossRef]
- Kiebert, G.; Sorensen, S.V.; Revicki, D.; Fagan, S.C.; Doyle, J.J.; Cohen, J.; Fivenson, D. Atopic dermatitis is associated with a decrement in health-related quality of life. Int. J. Dermatol. 2002, 41, 151–158. [Google Scholar] [CrossRef]
- Furue, M.; Onozuka, D.; Takeuchi, S.; Murota, H.; Sugaya, M.; Masuda, K.; Hiragun, T.; Kaneko, S.; Saeki, H.; Shintani, Y.; et al. Poor adherence to oral and topical medication in 3096 dermatological patients as assessed by the Morisky Medication Adherence Scale-8. Br. J. Dermatol. 2015, 172, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Murota, H.; Takeuchi, S.; Sugaya, M.; Tanioka, M.; Onozuka, D.; Hagihara, A.; Saeki, H.; Imafuku, S.; Abe, M.; Shintani, Y.; et al. Characterization of socioeconomic status of Japanese patients with atopic dermatitis showing poor medical adherence and reasons for drug discontinuation. J. Dermatol. Sci. 2015, 79, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Oba, J.; Esaki, H.; Furue, M. Non-corticosteroid adherence and itch severity influence perception of itch in atopic dermatitis. J. Dermatol. 2018, 45, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Iida, K.; Imaji, M.; Nakahara, T. Microbiome analysis of forehead skin in patients with atopic dermatitis and healthy subjects: Implication of Staphylococcus and Corynebacterium. J. Dermatol. 2018, 45, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Moriwaki, M.; Miyake, R.; Hide, M. Staphylococcus aureus in atopic dermatitis: Strain-specific cell wall proteins and skin immunity. Allergol. Int. 2019, 68, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Kadono, T. "Inflammatory skin march" in atopic dermatitis and psoriasis. Inflamm. Res. 2017, 66, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Esaki, H.; Ewald, D.A.; Ungar, B.; Rozenblit, M.; Zheng, X.; Xu, H.; Estrada, Y.D.; Peng, X.; Mitsui, H.; Litman, T.; et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J. Allergy Clin. Immunol. 2015, 135, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Esaki, H.; Brunner, P.M.; Renert-Yuval, Y.; Czarnowicki, T.; Huynh, T.; Tran, G.; Lyon, S.; Rodriguez, G.; Immaneni, S.; Johnson, D.B.; et al. Early-onset pediatric atopic dermatitis is T(H)2 but also T(H)17 polarized in skin. J. Allergy Clin. Immunol. 2016, 138, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Hamid, Q.; Boguniewicz, M.; Leung, D.Y. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J. Clin. Investig. 1994, 94, 870–876. [Google Scholar] [CrossRef]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.P.; et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef]
- Simpson, E.L.; Gadkari, A.; Worm, M.; Soong, W.; Blauvelt, A.; Eckert, L.; Wu, R.; Ardeleanu, M.; Graham, N.M.H.; Pirozzi, G.; et al. Dupilumab therapy provides clinically meaningful improvement in patient-reported outcomes (PROs): A phase IIb, randomized, placebo-controlled, clinical trial in adult patients with moderate to severe atopic dermatitis (AD). J. Am. Acad. Dermatol. 2016, 75, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suárez-Fariñas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, K.; Ito, T.; Tsuji, G.; Ulzii, D.; Vu, Y.H.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology 2019, 158, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, M.; Ulzii, D.; Vu, Y.H.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of atopic dermatitis: Current paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar]
- Furue, M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic implications in atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. [Google Scholar] [CrossRef]
- Takemura, M.; Nakahara, T.; Hashimoto-Hachiya, A.; Furue, M.; Tsuji, G. Glyteer, soybean tar, impairs IL-4/Stat6 signaling in murine bone marrow-derived dendritic cells: The basis of its therapeutic effect on atopic dermatitis. Int. J. Mol. Sci. 2018, 19, 1169. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Shi, V.Y.; Chan, L.S. IL-4 up-regulates epidermal chemotactic, angiogenic, and pro-inflammatory genes and down-regulates antimicrobial genes in vivo and in vitro: Relevant in the pathogenesis of atopic dermatitis. Cytokine 2013, 61, 419–425. [Google Scholar] [CrossRef]
- Bogiatzi, S.I.; Fernandez, I.; Bichet, J.C.; Marloie-Provost, M.A.; Volpe, E.; Sastre, X.; Soumelis, V. Cutting Edge: Proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J. Immunol. 2007, 178, 3373–3377. [Google Scholar] [CrossRef] [Green Version]
- Ranasinghe, C.; Trivedi, S.; Wijesundara, D.K.; Jackson, R.J. IL-4 and IL-13 receptors: Roles in immunity and powerful vaccine adjuvants. Cytokine Growth Factor Rev. 2014, 25, 437–442. [Google Scholar] [CrossRef]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. Galactomyces fermentation filtrate prevents T helper 2-mediated reduction of filaggrin in an aryl hydrocarbon receptor-dependent manner. Clin. Exp. Dermatol. 2015, 40, 786–793. [Google Scholar] [CrossRef]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Uchi, H.; Takahara, M.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. Antioxidant soybean tar Glyteer rescues T-helper-mediated downregulation of filaggrin expression via aryl hydrocarbon receptor. J. Dermatol. 2015, 42, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.E.; Reinhardt, R.L.; Bando, J.K.; Sullivan, B.M.; Ho, I.C.; Locksley, R.M. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat. Immunol. 2011, 3, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meli, A.P.; Fontés, G.; Leung Soo, C.; King, I.L. T follicular helper cell-derived IL-4 is required for IgE production during intestinal helminth infection. J. Immunol. 2017, 199, 244–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrell, B.P.; Shafiei Jahani, P.; Akbari, O. Social networking of group two innate lymphoid cells in allergy and asthma. Front. Immunol. 2018, 9, 2694. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- Czarnowicki, T.; Esaki, H.; Gonzalez, J.; Malajian, D.; Shemer, A.; Noda, S.; Talasila, S.; Berry, A.; Gray, J.; Becker, L.; et al. Early pediatric atopic dermatitis shows only a cutaneous lymphocyte antigen (CLA) (+) TH2/TH1 cell imbalance, whereas adults acquire CLA(+) TH22/TC22 cell subsets. J. Allergy Clin. Immunol. 2015, 136, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Ohtsuki, M.; Ogata, F.; Ishibashi, Y. Responsiveness to interleukin 4 and interleukin 2 of peripheral blood mononuclear cells in atopic dermatitis. J. Investig. Dermatol. 1991, 96, 468–472. [Google Scholar] [CrossRef] [Green Version]
- Shoda, T.; Futamura, K.; Kobayashi, F.; Saito, H.; Matsumoto, K.; Matsuda, A. Expression of thymus and activation-regulated chemokine (TARC) by human dermal cells, but not epidermal keratinocytes. J. Dermatol. Sci. 2014, 76, 90–95. [Google Scholar] [CrossRef]
- Shinkai, A.; Yoshisue, H.; Koike, M.; Shoji, E.; Nakagawa, S.; Saito, A.; Takeda, T.; Imabeppu, S.; Kato, Y.; Hanai, N.; et al. A novel human CC chemokine, eotaxin-3, which is expressed in IL-4-stimulated vascular endothelial cells, exhibits potent activity toward eosinophils. J. Immunol. 1999, 163, 1602–1610. [Google Scholar]
- Guttman-Yassky, E.; Ungar, B.; Malik, K.; Dickstein, D.; Suprun, M.; Estrada, Y.D.; Xu, H.; Peng, X.; Oliva, M.; Todd, D.; et al. Molecular signatures order the potency of topically applied anti-inflammatory drugs in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2017, 140, 1032–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakinuma, T.; Nakamura, K.; Wakugawa, M.; Mitsui, H.; Tada, Y.; Saeki, H.; Torii, H.; Asahina, A.; Onai, N.; Matsushima, K.; et al. Thymus and activation-regulated chemokine in atopic dermatitis: Serum thymus and activation-regulated chemokine level is closely related with disease activity. J. Allergy Clin. Immunol. 2001, 107, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, T.; Nakamura, K.; Wakugawa, M.; Mitsui, H.; Tada, Y.; Saeki, H.; Torii, H.; Komine, M.; Asahina, A.; Tamaki, K. Serum macrophage-derived chemokine (MDC) levels are closely related with the disease activity of atopic dermatitis. Clin. Exp. Immunol. 2002, 127, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Inagaki, S.; Kawano, T.; Azuma, Y.; Nomura, N.; Noguchi, Y.; Ohta, S.; Kawaguchi, A.; Odajima, H.; Ohya, Y.; et al. SCCA2 is a reliable biomarker for evaluating pediatric atopic dermatitis. J. Allergy Clin. Immunol. 2018, 141, 1934–1936. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Furusyo, N.; Ono, J.; Azuma, Y.; Takemura, M.; Esaki, H.; Yamamura, K.; Mitamura, Y.; Tsuji, G.; Kiyomatsu-Oda, M.; et al. Serum squamous cell carcinoma antigen (SCCA)-2 correlates with clinical severity of pediatric atopic dermatitis in Ishigaki cohort. J. Dermatol. Sci. 2019, 95, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Izuhara, K.; Onozuka, D.; Nunomura, S.; Tamagawa-Mineoka, R.; Masuda, K.; Ichiyama, S.; Saeki, H.; Kabata, Y.; Abe, R.; et al. Exploration of biomarkers to predict clinical improvement of atopic dermatitis in patients treated with dupilumab: A study protocol. Medicine (Baltimore) 2020, 99, e22043. [Google Scholar] [CrossRef]
- Szegedi, K.; Lutter, R.; Res, P.C.; Bos, J.D.; Luiten, R.M.; Kezic, S.; Middelkamp-Hup, M.A. Cytokine profiles in interstitial fluid from chronic atopic dermatitis skin. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2136–2144. [Google Scholar] [CrossRef]
- Morita, E.; Takahashi, H.; Niihara, H.; Dekio, I.; Sumikawa, Y.; Murakami, Y.; Matsunaka, H. Stratum corneum TARC level is a new indicator of lesional skin inflammation in atopic dermatitis. Allergy 2010, 65, 1166–1172. [Google Scholar] [CrossRef]
- Hulshof, L.; Hack, D.P.; Hasnoe, Q.C.J.; Dontje, B.; Jakasa, I.; Riethmüller, C.; McLean, W.H.I.; van Aalderen, W.M.C.; Van’t Land, B.; Kezic, S.; et al. Stratum corneum analysis provide a minimal invasive tool to study immune response and skin barrier in atopic dermatitis children. Br. J. Dermatol. 2019, 180, 621–630. [Google Scholar] [CrossRef]
- Oldhoff, J.M.; Darsow, U.; Werfel, T.; Katzer, K.; Wulf, A.; Laifaoui, J.; Hijnen, D.J.; Plötz, S.; Knol, E.F.; Kapp, A.; et al. Anti-IL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy 2005, 60, 693–696. [Google Scholar] [CrossRef]
- Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Tsuji, G.; Furue, K.; Hashimoto-Hachiya, A.; Furue, M. Scratching counteracts IL-13 signaling by upregulating the decoy receptor IL-13Rα2 in keratinocytes. Int. J. Mol. Sci. 2019, 20, 3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, M.; Ulzii, D.; Nakahara, T.; Tsuji, G.; Furue, K.; Hashimoto-Hachiya, A.; Kido-Nakahara, M. Implications of IL-13Rα2 in atopic skin inflammation. Allergol. Int. 2020, 69, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Yasukawa, F.; Furue, M.; Katz, S.I. Collared mice: A model to assess the effects of scratching. J. Dermatol. Sci. 2010, 57, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kido-Nakahara, M.; Furue, M.; Ulzii, D.; Nakahara, T. Itch in atopic dermatitis. Immunol. Allergy Clin. N. Am. 2017, 37, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Hide, M.; Yagami, A.; Togawa, M.; Saito, A.; Furue, M. Efficacy and safety of bilastine in Japanese patients with chronic spontaneous urticaria: A multicenter, randomized, double-blind, placebo-controlled, parallel-group phase II/III study. Allergol. Int. 2017, 66, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Urabe, K.; Moroi, Y.; Morita, K.; Furue, M. Bepotastine besilate rapidly inhibits mite-antigen induced immediate reactions in atopic dermatitis. J. Dermatol. Sci. 2003, 32, 237–238. [Google Scholar] [CrossRef]
- Urabe, K.; Nakahara, T.; Moroi, Y.; Morita, K.; Furue, M. Mite-antigen induced immediate reactions in atopic dermatitis are inhibited by daily administration of fexofenadine. J. Dermatol. 2003, 30, 847–848. [Google Scholar] [CrossRef]
- Matterne, U.; Böhmer, M.M.; Weisshaar, E.; Jupiter, A.; Carter, B.; Apfelbacher, C.J. Oral H1 antihistamines as ‘add-on’ therapy to topical treatment for eczema. Cochrane Database Syst. Rev. 2019, 1, CD012167. [Google Scholar] [CrossRef]
- Furue, M.; Yamamura, K.; Kido-Nakahara, M.; Nakahara, T.; Fukui, Y. Emerging role of interleukin-31 and interleukin-31 receptor in pruritus in atopic dermatitis. Allergy 2018, 73, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, K.; Uruno, T.; Shiraishi, A.; Tanaka, Y.; Ushijima, M.; Nakahara, T.; Watanabe, M.; Kido-Nakahara, M.; Tsuge, I.; Furue, M.; et al. The transcription factor EPAS1 links DOCK8 deficiency to atopic skin inflammation via IL-31 induction. Nat. Commun. 2017, 8, 13946. [Google Scholar] [CrossRef] [Green Version]
- Feld, M.; Garcia, R.; Buddenkotte, J.; Katayama, S.; Lewis, K.; Muirhead, G.; Hevezi, P.; Plesser, K.; Schrumpf, H.; Krjutskov, K.; et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J. Allergy Clin. Immunol. 2016, 138, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kido-Nakahara, M.; Wang, B.; Ohno, F.; Tsuji, G.; Ulzii, D.; Takemura, M.; Furue, M.; Nakahara, T. Inhibition of mite-induced dermatitis, pruritus, and nerve sprouting in mice by the endothelin receptor antagonist bosentan. Allergy 2020. [Google Scholar] [CrossRef]
- Sakata, D.; Uruno, T.; Matsubara, K.; Andoh, T.; Yamamura, K.; Magoshi, Y.; Kunimura, K.; Kamikaseda, Y.; Furue, M.; Fukui, Y. Selective role of neurokinin B in IL-31-induced itch response in mice. J. Allergy Clin. Immunol. 2019, 144, 1130–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiratori-Hayashi, M.; Hasegawa, A.; Toyonaga, H.; Andoh, T.; Nakahara, T.; Kido-Nakahara, M.; Furue, M.; Kuraishi, Y.; Inoue, K.; Dong, X.; et al. Role of P2X3 receptors in scratching behavior in mouse models. J. Allergy Clin. Immunol. 2019, 143, 1252–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiratori-Hayashi, M.; Koga, K.; Tozaki-Saitoh, H.; Kohro, Y.; Toyonaga, H.; Yamaguchi, C.; Hasegawa, A.; Nakahara, T.; Hachisuka, J.; Akira, S.; et al. STAT3-dependent reactive astrogliosis in the spinal dorsal horn underlies chronic itch. Nat. Med. 2015, 21, 927–931. [Google Scholar] [CrossRef]
- Nemoto, O.; Furue, M.; Nakagawa, H.; Shiramoto, M.; Hanada, R.; Matsuki, S.; Imayama, S.; Kato, M.; Hasebe, I.; Taira, K.; et al. The first trial of CIM331, a humanized antihuman interleukin-31 receptor A antibody, in healthy volunteers and patients with atopic dermatitis to evaluate safety, tolerability and pharmacokinetics of a single dose in a randomized, double-blind, placebo-controlled study. Br. J. Dermatol. 2016, 174, 296–304. [Google Scholar]
- Ruzicka, T.; Hanifin, J.M.; Furue, M.; Pulka, G.; Mlynarczyk, I.; Wollenberg, A.; Galus, R.; Etoh, T.; Mihara, R.; Yoshida, H.; et al. Anti-interleukin-31 receptor A antibody for atopic dermatitis. N. Engl. J. Med. 2017, 376, 826–835. [Google Scholar] [CrossRef]
- Kabashima, K.; Furue, M.; Hanifin, J.M.; Pulka, G.; Wollenberg, A.; Galus, R.; Etoh, T.; Mihara, R.; Nakano, M.; Ruzicka, T. Nemolizumab in patients with moderate-to-severe atopic dermatitis: Randomized, phase II, long-term extension study. J. Allergy Clin. Immunol. 2018, 142, 1121–1130. [Google Scholar] [CrossRef]
- Kabashima, K.; Matsumura, T.; Komazaki, H.; Kawashima, M. Trial of nemolizumab and topical agents for atopic dermatitis with pruritus. N. Engl. J. Med. 2020, 383, 141–150. [Google Scholar] [CrossRef]
- Souza, C.P.; Rosychuk, R.A.W.; Contreras, E.T.; Schissler, J.R.; Simpson, A.C. A retrospective analysis of the use of lokivetmab in the management of allergic pruritus in a referral population of 135 dogs in the western USA. Vet. Dermatol. 2018, 29, 489-e164. [Google Scholar] [CrossRef]
- Oetjen, L.K.; Mack, M.R.; Feng, J.; Whelan, T.M.; Niu, H.; Guo, C.J.; Chen, S.; Trier, A.M.; Xu, A.Z.; Tripathi, S.V.; et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell 2017, 171, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campion, M.; Smith, L.; Gatault, S.; Métais, C.; Buddenkotte, J.; Steinhoff, M. Interleukin-4 and interleukin-13 evoke scratching behaviour in mice. Exp. Dermatol. 2019, 28, 1501–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Drongelen, V.; Haisma, E.M.; Out-Luiting, J.J.; Nibbering, P.H.; El Ghalbzouri, A. Reduced filaggrin expression is accompanied by increased Staphylococcus aureus colonization of epidermal skin models. Clin. Exp. Allergy. 2014, 44, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Ran, L.; Wang, W.; Zhou, R.; Cai, X.; Li, R.; Li, Y.; Zhou, C.; He, W.; Wang, R. Glucocorticoid insensitivity by staphylococcal enterotoxin B in keratinocytes of allergic dermatitis is associated with impaired nuclear translocation of the glucocorticoid receptor α. J. Dermatol. Sci. 2018, 92, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egawa, G.; Kabashima, K. Barrier dysfunction in the skin allergy. Allergol. Int. 2018, 67, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Kypriotou, M.; Huber, M.; Hohl, D. The human epidermal differentiation complex: Cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp. Dermatol. 2012, 21, 643–649. [Google Scholar] [CrossRef]
- Kennedy, L.H.; Sutter, C.H.; Leon Carrion, S.; Tran, Q.T.; Bodreddigari, S.; Kensicki, E.; Mohney, R.P.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol. Sci. 2013, 132, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Van den Bogaard, E.H.; Podolsky, M.A.; Smits, J.P.; Cui, X.; John, C.; Gowda, K.; Desai, D.; Amin, S.G.; Schalkwijk, J.; Perdew, G.H.; et al. Genetic and pharmacological analysis identifies a physiological role for the AHR in epidermal differentiation. J. Investig. Dermatol. 2015, 135, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Tsuji, G.; Mitoma, C.; Nakahara, T.; Chiba, T.; Morino-Koga, S.; Uchi, H. Gene regulation of filaggrin and other skin barrier proteins via aryl hydrocarbon receptor. J. Dermatol. Sci. 2015, 80, 83–88. [Google Scholar] [CrossRef]
- Fritsche, E.; Schäfer, C.; Calles, C.; Bernsmann, T.; Bernshausen, T.; Wurm, M.; Hübenthal, U.; Cline, J.E.; Hajimiragha, H.; Schroeder, P.; et al. Lightening up the UV response by identification of the aryl hydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 8851–8856. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyomatsu-Oda, M.; Uchi, H.; Morino-Koga, S.; Furue, M. Protective role of 6-formylindolo[3,2-b]carbazole (FICZ), an endogenous ligand for arylhydrocarbon receptor, in chronic mite-induced dermatitis. J. Dermatol. Sci. 2018, 90, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Tanaka, Y.; Ito, T.; Tsuji, G. Implications of tryptophan photoproduct FICZ in oxidative stress and terminal differentiation of keratinocytes. G. Ital. Dermatol. Venereol. 2019, 154, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Magiatis, P.; Pappas, P.; Gaitanis, G.; Mexia, N.; Melliou, E.; Galanou, M.; Vlachos, C.; Stathopoulou, K.; Skaltsounis, A.L.; Marselos, M.; et al. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J. Investig. Dermatol. 2013, 133, 2023–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Luo, Y.; Zhu, Z.; Zhou, Y.; Sun, L.; Gao, J.; Sun, J.; Wang, G.; Yao, X.; Li, W. A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J. Allergy Clin. Immunol. 2019, 143, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Nakahara, T.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Uchi, H.; Yan, X.; Hachisuka, J.; Chiba, T.; Esaki, H.; et al. Antioxidant Opuntia ficus-indica extract activates AHR-NRF2 signaling and upregulates filaggrin and loricrin expression in human keratinocytes. J. Med. Food 2015, 18, 1143–1149. [Google Scholar] [CrossRef]
- Doi, K.; Mitoma, C.; Nakahara, T.; Uchi, H.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, M.; Furue, M. Antioxidant Houttuynia cordata extract upregulates filaggrin expression in an aryl hydrocarbon-dependent manner. Fukuoka Igaku Zasshi 2014, 105, 205–213. [Google Scholar]
- Hirano, A.; Goto, M.; Mitsui, T.; Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M. Antioxidant Artemisia princeps extract enhances the expression of filaggrin and loricrin via the AHR/OVOL1 pathway. Int. J. Mol. Sci. 2017, 18, E1948. [Google Scholar] [CrossRef]
- Hashimoto-Hachiya, A.; Tsuji, G.; Murai, M.; Yan, X.; Furue, M. Upregulation of FLG, LOR, and IVL expression by Rhodiola crenulata root extract via aryl hydrocarbon receptor: Differential involvement of OVOL1. Int. J. Mol. Sci. 2018, 19, 1654. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Antioxidative phytochemicals accelerate epidermal terminal differentiation via the AHR-OVOL1 pathway: Implications for atopic dermatitis. Acta Derm. Venereol. 2018, 98, 918–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, M.; Matsumoto, T.; Yamamoto, T.; Takeuchi, S.; Esaki, H.; Chiba, T.; Yamaguchi, H. Correlation between serum thymus and activation-regulated chemokine levels and stratum corneum barrier function in healthy individuals and patients with mild atopic dermatitis. J. Dermatol. Sci. 2012, 66, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Takahashi, A.; Kubo, M.; Tsunoda, T.; Tomita, K.; Sakashita, M.; Yamada, T.; Fujieda, S.; Tanaka, S.; Doi, S.; et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 2012, 44, 1222–1226. [Google Scholar] [CrossRef]
- Tamari, M.; Hirota, T. Genome-wide association studies of atopic dermatitis. J. Dermatol. 2014, 41, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Paternoster, L.; Standl, M.; Waage, J.; Baurecht, H.; Hotze, M.; Strachan, D.P.; Curtin, J.A.; Bønnelykke, K.; Tian, C.; Takahashi, A.; et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015, 47, 1449–1456. [Google Scholar]
- Cascella, R.; Foti Cuzzola, V.; Lepre, T.; Galli, E.; Moschese, V.; Chini, L.; Mazzanti, C.; Fortugno, P.; Novelli, G.; Giardina, E. Full sequencing of the FLG gene in Italian patients with atopic eczema: Evidence of new mutations, but lack of an association. J. Investig. Dermatol. 2011, 131, 982–984. [Google Scholar] [CrossRef]
- Thawer-Esmail, F.; Jakasa, I.; Todd, G.; Wen, Y.; Brown, S.J.; Kroboth, K.; Campbell, L.E.; O’Regan, G.M.; McLean, W.H.; Irvine, A.D.; et al. South African amaXhosa patients with atopic dermatitis have decreased levels of filaggrin breakdown products but no loss-of-function mutations in filaggrin. J. Allergy Clin. Immunol. 2014, 133, 280–282. [Google Scholar] [CrossRef] [Green Version]
- Winge, M.C.; Bilcha, K.D.; Liedén, A.; Shibeshi, D.; Sandilands, A.; Wahlgren, C.F.; McLean, W.H.; Nordenskjöld, M.; Bradley, M. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. Br. J. Dermatol. 2011, 165, 1074–1080. [Google Scholar] [CrossRef]
- Fukiwake, N.; Furusyo, N.; Kubo, N.; Takeoka, H.; Toyoda, K.; Morita, K.; Shibata, S.; Nakahara, T.; Kido, M.; Hayashida, S.; et al. Incidence of atopic dermatitis in nursery school children—A follow-up study from 2001 to 2004, Kyushu University Ishigaki Atopic Dermatitis Study (KIDS). Eur. J. Dermatol. 2006, 16, 416–419. [Google Scholar]
- Sasaki, T.; Furusyo, N.; Shiohama, A.; Takeuchi, S.; Nakahara, T.; Uchi, H.; Hirota, T.; Tamari, M.; Shimizu, N.; Ebihara, T.; et al. Filaggrin loss-of-function mutations are not a predisposing factor for atopic dermatitis in an Ishigaki Island under subtropical climate. J. Dermatol. Sci. 2014, 76, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Ito, T.; Chiba, T.; Mitoma, C.; Nakahara, T.; Uchi, H.; Furue, M. The role of the OVOL1-OVOL2 axis in normal and diseased human skin. J. Dermatol. Sci. 2018, 90, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurakic Toncic, R.; Kezic, S.; Jakasa, I.; Ljubojevic Hadzavdic, S.; Balic, A.; Petkovic, M.; Pavicic, B.; Zuzul, K.; Marinovic, B. Filaggrin loss-of-function mutations and levels of filaggrin degradation products in adult patients with atopic dermatitis in Croatia. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Ogawa, M.; Yoshihara, T.; Masuoka, M.; Tsuji, G.; Nakahara, T.; Hashimoto-Hachiya, A.; Conway, S.J.; et al. The IL-13/periostin/IL-24 pathway causes epidermal barrier dysfunction in allergic skin inflammation. Allergy 2018, 73, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, Y.; Nunomura, S.; Furue, M.; Izuhara, K. IL-24: A new player in the pathogenesis of pro-inflammatory and allergic skin diseases. Allergol. Int. 2020, 69, 405–411. [Google Scholar] [CrossRef]
- Kobayashi, J.; Inai, T.; Morita, K.; Moroi, Y.; Urabe, K.; Shibata, Y.; Furue, M. Reciprocal regulation of permeability through a cultured keratinocyte sheet by IFN-gamma and IL-4. Cytokine 2004, 28, 186–189. [Google Scholar] [CrossRef]
- Fujii-Maeda, S.; Kajiwara, K.; Ikizawa, K.; Shinazawa, M.; Yu, B.; Koga, T.; Furue, M.; Yanagihara, Y. Reciprocal regulation of thymus and activation-regulated chemokine/macrophage-derived chemokine production by interleukin (IL)-4/IL-13 and interferon-gamma in HaCaT keratinocytes is mediated by alternations in E-cadherin distribution. J. Investig. Dermatol. 2004, 122, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sakai, T.; Matsuda-Hirose, H.; Goto, M.; Yamate, T.; Hatano, Y. Cutaneous permeability barrier function in signal transducer and activator of transcription 6-deficient mice is superior to that in wild-type mice. J. Dermatol. Sci. 2018, 92, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Taylor, S.; Ogg, G.S. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol. 2011, 165, 492–498. [Google Scholar] [CrossRef]
- Cornelissen, C.; Marquardt, Y.; Czaja, K.; Wenzel, J.; Frank, J.; Lüscher-Firzlaff, J.; Lüscher, B.; Baron, J.M. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J. Allergy Clin. Immunol. 2012, 129, 426–433. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; Ogg, G.S. Cytokine regulation of the epidermal barrier. Clin. Exp. Allergy 2013, 43, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Hvid, M.; Vestergaard, C.; Kemp, K.; Christensen, G.B.; Deleuran, B.; Deleuran, M. IL-25 in atopic dermatitis: A possible link between inflammation and skin barrier dysfunction? J. Investig. Dermatol. 2011, 131, 150–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seltmann, J.; Roesner, L.M.; von Hesler, F.W.; Wittmann, M.; Werfel, T. IL-33 impacts on the skin barrier by downregulating the expression of filaggrin. J. Allergy Clin. Immunol. 2015, 135, 1659–1661. [Google Scholar] [CrossRef]
- Kondo, H.; Ichikawa, Y.; Imokawa, G. Percutaneous sensitization with allergens through barrier-disrupted skin elicits a Th2-dominant cytokine response. Eur. J. Immunol. 1998, 28, 769–779. [Google Scholar] [CrossRef]
- Onoue, A.; Kabashima, K.; Kobayashi, M.; Mori, T.; Tokura, Y. Induction of eosinophil- and Th2-attracting epidermal chemokines and cutaneous late-phase reaction in tape-stripped skin. Exp. Dermatol. 2009, 18, 1036–1043. [Google Scholar] [CrossRef]
- Ashida, Y.; Denda, M. Dry environment increases mast cell number and histamine content in dermis in hairless mice. Br. J. Dermatol. 2003, 149, 240–247. [Google Scholar] [CrossRef]
- Dainichi, T.; Kitoh, A.; Otsuka, A.; Nakajima, S.; Nomura, T.; Kaplan, D.H.; Kabashima, K. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol. 2018, 19, 1286–1298. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. Barrier epithelial cells and the control of type 2 immunity. Immunity 2015, 43, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Aktar, M.K.; Kido-Nakahara, M.; Furue, M.; Nakahara, T. Mutual upregulation of endothelin-1 and IL-25 in atopic dermatitis. Allergy 2015, 70, 846–854. [Google Scholar] [CrossRef]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Oyoshi, M.K.; Larson, R.P.; Ziegler, S.F.; Geha, R.S. Mechanical injury polarizes skin dendritic cells to elicit a T(H)2 response by inducing cutaneous thymic stromal lymphopoietin expression. J. Allergy Clin. Immunol. 2010, 126, 976–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilliet, M.; Soumelis, V.; Watanabe, N.; Hanabuchi, S.; Antonenko, S.; de Waal-Malefyt, R.; Liu, Y.J. Human dendritic cells activated by TSLP and CD40L induce proallergic cytotoxic T cells. J. Exp. Med. 2003, 197, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.H.; Malefyt Rde, W.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef]
- Halim, T.Y.F.; Rana, B.M.J.; Walker, J.A.; Kerscher, B.; Knolle, M.D.; Jolin, H.E.; Serrao, E.M.; Haim-Vilmovsky, L.; Teichmann, S.A.; Rodewald, H.R.; et al. Tissue-restricted adaptive Type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity 2018, 48, 1195–1207. [Google Scholar] [CrossRef] [Green Version]
- Kolls, J.K.; Lindén, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; French, D.; Mao, W.; Maruoka, M.; Risser, P.; Lee, J.; Foster, J.; Aggarwal, S.; Nicholes, K.; Guillet, S.; et al. Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. J. Immunol. 2001, 167, 6559–6567. [Google Scholar] [CrossRef] [Green Version]
- Hurst, S.D.; Muchamuel, T.; Gorman, D.M.; Gilbert, J.M.; Clifford, T.; Kwan, S.; Menon, S.; Seymour, B.; Jackson, C.; Kung, T.T.; et al. New IL-17 family members promote Th1 or Th2 responses in the lung: In vivo function of the novel cytokine IL-25. J. Immunol. 2002, 169, 443–453. [Google Scholar] [CrossRef]
- Zheng, R.; Chen, F.H.; Gao, W.X.; Wang, D.; Yang, Q.T.; Wang, K.; Lai, Y.Y.; Deng, J.; Jiang, L.J.; Sun, Y.Q.; et al. The T(H)2-polarizing function of atopic interleukin 17 receptor B-positive dendritic cells up-regulated by lipopolysaccharide. Ann. Allergy Asthma Immunol. 2017, 118, 474–482. [Google Scholar] [CrossRef]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet-Schiltz, O.; Gonzalez-de-Peredo, A.; Girard, J.P. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef]
- Dickel, H.; Gambichler, T.; Kamphowe, J.; Altmeyer, P.; Skrygan, M. Standardized tape stripping prior to patch testing induces upregulation of Hsp90, Hsp70, IL-33, TNF-α and IL-8/CXCL8 mRNA: New insights into the involvement of ‘alarmins’. Contact Dermat. 2010, 63, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Komine, M.; Tsuda, H.; Oshio, T.; Ohtsuki, M. Interleukin-33 is expressed in the lesional epidermis in herpes virus infection but not in verruca vulgaris. J. Dermatol. 2018, 45, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Choi, J.K.; Jin, M.; Choi, Y.A.; Ryoo, Z.Y.; Lee, H.S.; Park, P.H.; Kim, S.U.; Kwon, T.K.; Jang, M.H.; et al. House dust mite increases pro-Th2 cytokines IL-25 and IL-33 via the activation of TLR1/6 signaling. J. Investig. Dermatol. 2017, 137, 2354–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, N.W.; Rosenstein, R.K.; Medzhitov, R. Allergic host defences. Nature 2012, 484, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Sokol, C.L.; Barton, G.M.; Farr, A.G.; Medzhitov, R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat. Immunol. 2008, 9, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechama, M.; Kwon, J.; Wei, S.; Kyi, A.T.; Welner, R.S.; Ben-Dov, I.Z.; Arredouani, M.S.; Asara, J.M.; Chen, C.H.; Tsai, C.Y.; et al. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity in IL-33-induced allergic airway inflammation. Nat. Commun. 2018, 9, 1603. [Google Scholar] [CrossRef] [Green Version]
- Vannella, K.M.; Ramalingam, T.R.; Borthwick, L.A.; Barron, L.; Hart, K.M.; Thompson, R.W.; Kindrachuk, K.N.; Cheever, A.W.; White, S.; Budelsky, A.L.; et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl. Med. 2016, 8, 337ra65. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.fiercebiotech.com/biotech/after-asthma-success-astrazeneca-and-amgen-s-tezepelumab-misses-atopic-dermatitis?utm_source=internal&utm_medium=rss (accessed on 20 November 2020).
- Chen, Y.L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11, eaax2945. [Google Scholar] [CrossRef]
- Koga, C.; Kabashima, K.; Shiraishi, N.; Kobayashi, M.; Tokura, Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J. Investig. Dermatol. 2008, 128, 2625–2630. [Google Scholar] [CrossRef] [Green Version]
- Nomura, T.; Honda, T.; Kabashima, K. Multipolarity of cytokine axes in the pathogenesis of atopic dermatitis in terms of age, race, species, disease stage and biomarkers. Int. Immunol. 2018, 30, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Guttman-Yassky, E.; Krueger, J.G. Atopic dermatitis and psoriasis: Two different immune diseases or one spectrum? Curr. Opin. Immunol. 2017, 48, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, S.; Uchi, H.; Takeuchi, S.; Esaki, H.; Moroi, Y.; Furue, M. Significant correlation of serum IL-22 levels with CCL17 levels in atopic dermatitis. J. Dermatol. Sci. 2011, 61, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Raychaudhuri, S.K.; Raychaudhuri, S.P. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine 2012, 60, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Brunner, P.M.; Neumann, A.U.; Khattri, S.; Pavel, A.B.; Malik, K.; Singer, G.K.; Baum, D.; Gilleaudeau, P.; Sullivan-Whalen, M.; et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol. 2018, 78, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Brunner, P.M.; Pavel, A.B.; Khattri, S.; Leonard, A.; Malik, K.; Rose, S.; Jim On, S.; Vekaria, A.S.; Traidl-Hoffmann, C.; Singer, G.K.; et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J. Allergy Clin. Immunol. 2019, 143, 142–154. [Google Scholar] [CrossRef]
- Nakahara, T.; Morimoto, H.; Murakami, N.; Furue, M. Mechanistic insights into topical tacrolimus for the treatment of atopic dermatitis. Pediatr. Allergy Immunol. 2018, 29, 233–238. [Google Scholar] [CrossRef]
- Ohtsuki, M.; Morimoto, H.; Nakagawa, H. Tacrolimus ointment for the treatment of adult and pediatric atopic dermatitis: Review on safety and benefits. J. Dermatol. 2018, 45, 936–942. [Google Scholar] [CrossRef]
- Napolitano, M.; Caiazzo, G.; Fabbrocini, G.; Balato, A.; Di Caprio, R.; Scala, E.; Scalvenzi, M.; Patruno, C. Increased expression of IL-23A in lesional skin of atopic dermatitis patients with psoriasiform reaction during dupilumab treatment. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Tsuji, G.; Nakahara, T.; Furue, M. The CCL20 and CCR6 axis in psoriasis. Scand. J. Immunol. 2020, 91, e12846. [Google Scholar] [CrossRef] [Green Version]
- Furue, K.; Furue, M. A new perspective on IL=17 producing cell infiltration in atopic dermatitis: Role of CCL20 production observed in an in vitro scratched keratinocyte model. Jap. J. Dermatol 2020, 130, 1645–1652. [Google Scholar]
- Furue, K.; Ito, T.; Tanaka, Y.; Yumine, A.; Hashimoto-Hachiya, A.; Takemura, M.; Murata, M.; Yamamura, K.; Tsuji, G.; Furue, M. Cyto/chemokine profile of in vitro scratched keratinocyte model: Implications of significant upregulation of CCL20, CXCL8 and IL36G in Koebner phenomenon. J. Dermatol. Sci. 2019, 94, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, K.; Ito, T.; Tanaka, Y.; Hashimoto-Hachiya, A.; Takemura, M.; Murata, M.; Kido-Nakahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. The EGFR-ERK/JNK-CCL20 pathway in scratched keratinocytes may underpin koebnerization in psoriasis patients. Int. J. Mol. Sci. 2020, 21, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, K.; Ito, T.; Tsuji, G.; Esaki, H.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. Does mechanical scratching cause the recruitment of T-helper 17 cells in atopic dermatitis? J. Dermatol. 2019, 46, e436–e437. [Google Scholar] [CrossRef]
- Furue, K.; Ulzii, D.; Tanaka, Y.; Ito, T.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. Pathogenic implication of epidermal scratch injury in psoriasis and atopic dermatitis. J. Dermatol. 2020, 47, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Furue, K.; Tsuji, G.; Nakahara, T. Interleukin-17A and keratinocytes in psoriasis. Int. J. Mol. Sci. 2020, 21, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Huang, J.; Zhao, X.; Lu, H.; Wang, W.; Yang, X.O.; Shi, Y.; Wang, X.; Lai, Y.; Dong, C. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci. Immunol. 2019, 4, eaau9657. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.H.; Choi, D.; Chun, Y.J.; Noh, M. Keratinocyte-derived IL-24 plays a role in the positive feedback regulation of epidermal inflammation in response to environmental and endogenous toxic stressors. Toxicol. Appl. Pharmacol. 2014, 280, 199–206. [Google Scholar] [CrossRef]
- Lai, X.; Li, X.; Chang, L.; Chen, X.; Huang, Z.; Bao, H.; Huang, J.; Yang, L.; Wu, X.; Wang, Z.; et al. IL-19 up-regulates mucin 5AC production in patients with chronic rhinosinusitis via STAT3 pathway. Front. Immunol. 2019, 10, 1682. [Google Scholar] [CrossRef] [Green Version]
- Chiricozzi, A.; Nograles, K.E.; Johnson-Huang, L.M.; Fuentes-Duculan, J.; Cardinale, I.; Bonifacio, K.M.; Gulati, N.; Mitsui, H.; Guttman-Yassky, E.; Suárez-Fariñas, M.; et al. IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE 2014, 9, e90284. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/results/NCT02594098?term=atopic&cond=secukinumab&draw=2&rank=1 (accessed on 20 November 2020).
- Yang, X.; Zheng, S.G. Interleukin-22: A likely target for treatment of autoimmune diseases. Autoimmun. Rev. 2014, 13, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Boniface, K.; Bernard, F.X.; Garcia, M.; Gurney, A.L.; Lecron, J.C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 2005, 174, 3695–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitabile, S.; Odorisio, T.; Madonna, S.; Eyerich, S.; Guerra, L.; Eyerich, K.; Zambruno, G.; Cavani, A.; Cianfarani, F. Interleukin-22 promotes wound repair in diabetes by improving keratinocyte pro-healing functions. J. Investig. Dermatol. 2015, 135, 2862–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, M.; Yeo, H.; Ko, J.; Kim, H.K.; Lee, C.H. MAP17 is associated with the T-helper cell cytokine-induced down-regulation of filaggrin transcription in human keratinocytes. Exp. Dermatol. 2010, 19, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and non-canonical aspects of JAK-STAT signaling: Lessons from interferons for cytokine responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Di, Z.H.; Ma, L.; Qi, R.Q.; Sun, X.D.; Huo, W.; Zhang, L.; Lyu, Y.N.; Hong, Y.X.; Chen, H.D.; Gao, X.H. T helper 1 and T helper 2 cytokines differentially modulate expression of filaggrin and its processing proteases in human keratinocytes. Chin. Med. J. (Engl.) 2016, 129, 295–303. [Google Scholar] [CrossRef]
- Nishida, J.; Li, Y.; Zhuang, Y.; Huang, Z.; Huang, H. IFN-γ suppresses permissive chromatin remodeling in the regulatory region of the Il4 gene. Cytokine 2013, 62, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Nakagome, K.; Okunishi, K.; Imamura, M.; Harada, H.; Matsumoto, T.; Tanaka, R.; Miyazaki, J.; Yamamoto, K.; Dohi, M. IFN-gamma attenuates antigen-induced overall immune response in the airway as a Th1-type immune regulatory cytokine. J. Immunol. 2009, 183, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Moroi, Y.; Takahara, M.; Tsuji, G.; Oba, J.; Hayashida, S.; Takeuchi, S.; Shan, B.; Uchi, H.; Furue, M. CD10 expressed by fibroblasts and melanoma cells degrades endothelin-1 secreted by human keratinocytes. Eur. J. Dermatol. 2011, 21, 505–509. [Google Scholar] [CrossRef]
- Eto, A.; Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Acrosyringeal endothelin-1 expression: Potential for fostering melanocytes in volar sites. J. Dermatol. 2020, 47, 924–925. [Google Scholar] [CrossRef]
- Nakahara, T.; Kido-Nakahara, M.; Ohno, F.; Ulzii, D.; Chiba, T.; Tsuji, G.; Furue, M. The pruritogenic mediator endothelin-1 shifts the dendritic cell-T-cell response toward Th17/Th1 polarization. Allergy 2018, 73, 511–515. [Google Scholar] [CrossRef]
- Kido-Nakahara, M.; Buddenkotte, J.; Kempkes, C.; Ikoma, A.; Cevikbas, F.; Akiyama, T.; Nunes, F.; Seeliger, S.; Hasdemir, B.; Mess, C.; et al. Neural peptidase endothelin-converting enzyme 1 regulates endothelin 1-induced pruritus. J. Clin. Investig. 2014, 124, 2683–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, T.; Kido-Nakahara, M.; Furue, M. Potential role of endothelin-1 in atopic dermatitis. Curr. Treat. Opt. Allergy 2019, 6, 156–163. [Google Scholar] [CrossRef]
- Nakahara, T.; Kido-Nakahara, M.; Ulzii, D.; Miake, S.; Fujishima, K.; Sakai, S.; Chiba, T.; Tsuji, G.; Furue, M. Topical application of endothelin receptor a antagonist attenuates imiquimod-induced psoriasiform skin inflammation. Sci. Rep. 2020, 10, 9510. [Google Scholar] [CrossRef]
- Ji, H.; Li, X.K. Oxidative stress in atopic dermatitis. Oxid. Med. Cell. Longev. 2016, 2016, 2721469. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, S.; Miyazaki, Y.; Yoshii, C.; Nakaya, M.; Ozaki, N.; Toda, S.; Kuroda, E.; Ishibashi, K.; Yasuda, T.; Natsuaki, Y.; et al. An ITAM-Syk-CARD9 signalling axis triggers contact hypersensitivity by stimulating IL-1 production in dendritic cells. Nat. Commun. 2014, 5, 3755. [Google Scholar] [CrossRef] [Green Version]
- Eto, H.; Tsuji, G.; Chiba, T.; Furue, M.; Hyodo, F. Non-invasive evaluation of atopic dermatitis based on redox status using in vivo dynamic nuclear polarization magnetic resonance imaging. Free Radic. Biol. Med. 2017, 103, 209–215. [Google Scholar] [CrossRef]
- Hirakawa, S.; Saito, R.; Ohara, H.; Okuyama, R.; Aiba, S. Dual oxidase 1 induced by Th2 cytokines promotes STAT6 phosphorylation via oxidative inactivation of protein tyrosine phosphatase 1B in human epidermal keratinocytes. J. Immunol. 2011, 186, 4762–4770. [Google Scholar] [CrossRef]
- Lu, X.; Malumbres, R.; Shields, B.; Jiang, X.; Sarosiek, K.A.; Natkunam, Y.; Tiganis, T.; Lossos, I.S. PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood 2008, 112, 4098–4108. [Google Scholar] [CrossRef]
- Sharma, P.; Chakraborty, R.; Wang, L.; Min, B.; Tremblay, M.L.; Kawahara, T.; Lambeth, J.D.; Haque, S.J. Redox regulation of interleukin-4 signaling. Immunity 2008, 29, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Fuyuno, Y.; Uchi, H.; Yasumatsu, M.; Morino-Koga, S.; Tanaka, Y.; Mitoma, C.; Furue, M. Perillaldehyde inhibits AHR signaling and activates NRF2 antioxidant pathway in human keratinocytes. Oxid. Med. Cell Longev. 2018, 2018, 9524657. [Google Scholar] [CrossRef]
- Tanaka, Y.; Uchi, H.; Furue, M. Antioxidant cinnamaldehyde attenuates UVB-induced photoaging. J. Dermatol. Sci. 2019, 96, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Uchi, H.; Yasumatsu, M.; Morino-Koga, S.; Mitoma, C.; Furue, M. Inhibition of aryl hydrocarbon receptor signaling and induction of NRF2-mediated antioxidant activity by cinnamaldehyde in human keratinocytes. J. Dermatol. Sci. 2017, 85, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, Y.; Murai, M.; Mitoma, C.; Furue, M. NRF2 activation inhibits both TGF-β1- and IL-13-mediated periostin expression in fibroblasts: Benefit of cinnamaldehyde for antifibrotic treatment. Oxid. Med. Cell Longev. 2018, 2018, 2475047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Ito, T.; Tsuji, G.; Furue, M. Baicalein inhibits benzo[a]pyrene-induced toxic response by downregulating Src phosphorylation and by upregulating NRF2-HMOX1 system. Antioxidants 2020, 9, 507. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M. Antioxidants cinnamaldehyde and Galactomyces fermentation filtrate downregulate senescence marker CDKN2A/p16INK4A via NRF2 activation in keratinocytes. J. Dermatol. Sci. 2019, 96, 53–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, X.; Zhang, L.; Yan, T.; Wu, B.; Xu, F.; Jia, Y. Silychristin A activates Nrf2-HO-1/SOD2 pathway to reduce apoptosis and improve GLP-1 production through upregulation of estrogen receptor α in GLUTag cells. Eur. J. Pharmacol. 2020, 881, 173236. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Chiba, T.; Ito, T.; Nakahara, T.; Tsuji, G. Antioxidants for healthy skin: The emerging role of aryl hydrocarbon receptors and nuclear factor-erythroid 2-related factor-2. Nutrients 2017, 9, 223. [Google Scholar] [CrossRef]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Arima, K.; Yoshihara, T.; Komiya, K.; Fukuda, S.; Takatori, H.; Nakajima, H.; Furue, M.; et al. Hierarchical control of interleukin 13 (IL-13) signals in lung fibroblasts by STAT6 and SOX11. J. Biol. Chem. 2018, 293, 14646–14658. [Google Scholar] [CrossRef] [Green Version]
- Lumsden, R.V.; Worrell, J.C.; Boylan, D.; Walsh, S.M.; Cramton, J.; Counihan, I.; O’Beirne, S.; Medina, M.F.; Gauldie, J.; Fabre, A.; et al. Modulation of pulmonary fibrosis by IL-13Rα2. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L710–L718. [Google Scholar] [CrossRef]
- Kolodsick, J.E.; Toews, G.B.; Jakubzick, C.; Hogaboam, C.; Moore, T.A.; McKenzie, A.; Wilke, C.A.; Chrisman, C.J.; Moore, B.B. Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J. Immunol. 2004, 172, 4068–4076. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.W.; Lee, H.B.; Chung, Y.H.; Choi, Y. The effect of disodium cromoglycate, budesonide, and cyclosporin A on interleukin-4, interleukin-5, and interleukin-13 secretions in Der p I-stimulated T cells from house dust mite-sensitive atopic and nonatopic individuals. Allergy Asthma Proc. 2002, 23, 109–115. [Google Scholar] [PubMed]
- Liu, Z.; Yuan, X.; Luo, Y.; He, Y.; Jiang, Y.; Chen, Z.K.; Sun, E. Evaluating the effects of immunosuppressants on human immunity using cytokine profiles of whole blood. Cytokine 2009, 45, 141–147. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Luo, Y.; Lao, X.; Tan, L.; Sun, E. Cytokine signatures of human whole blood for monitoring immunosuppression. Cent. Eur. J. Immunol. 2014, 39, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamatsu, Y.; Hasegawa, M.; Sato, S.; Takehara, K. IL-13 production by peripheral blood mononuclear cells from patients with atopic dermatitis. Dermatology 1998, 196, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Pacocha, S.E.; Oriente, A.; Huang, S.K.; Essayan, D.M. Regulation of antigen-induced human T-lymphocyte responses by calcineurin antagonists. J. Allergy Clin. Immunol. 1999, 104, 828–835. [Google Scholar] [CrossRef]
- Katagiri, K.; Itami, S.; Hatano, Y.; Yamaguchi, T.; Takayasu, S. In vivo expression of IL-4, IL-5, IL-13 and IFN-gamma mRNAs in peripheral blood mononuclear cells and effect of cyclosporin A in a patient with Kimura’s disease. Br. J. Dermatol. 1997, 137, 972–977. [Google Scholar] [CrossRef]
- Liu, S.; Verma, M.; Michalec, L.; Liu, W.; Sripada, A.; Rollins, D.; Good, J.; Ito, Y.; Chu, H.; Gorska, M.M.; et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2018, 141, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, T.; Fukuoka, A.; Matsushita, K.; Yasuda, K.; Iwasaki, N.; Akasaki, S.; Fujieda, S.; Yoshimoto, T. Activation of group 2 innate lymphoid cells exacerbates and confers corticosteroid resistance to mouse nasal type 2 inflammation. Int. Immunol. 2017, 29, 221–233. [Google Scholar] [CrossRef]
- Kurgonaite, K.; Gandhi, H.; Kurth, T.; Pautot, S.; Schwille, P.; Weidemann, T.; Bökel, C. Essential role of endocytosis for interleukin-4-receptor-mediated JAK/STAT signalling. J. Cell Sci. 2015, 128, 3781–3795. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Nemoto, O.; Yamada, H.; Nagata, T.; Ninomiya, N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE-052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J. Dermatol. 2018, 45, 701–709. [Google Scholar] [CrossRef]
- Amano, W.; Nakajima, S.; Kunugi, H.; Numata, Y.; Kitoh, A.; Egawa, G.; Dainichi, T.; Honda, T.; Otsuka, A.; Kimoto, Y.; et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J. Allergy Clin. Immunol. 2015, 136, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Nemoto, O.; Igarashi, A.; Saeki, H.; Kaino, H.; Nagata, T. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J. Am. Acad. Dermatol. 2020, 82, 823–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Otsuka, A.; Nakashima, C.; Ishida, Y.; Honda, T.; Egawa, G.; Amano, W.; Usui, K.; Hamada, Y.; Wada, M.; et al. Janus kinase inhibitor delgocitinib suppresses pruritus and nerve elongation in an atopic dermatitis murine model. J. Dermatol. Sci. 2020, 97, 161–164. [Google Scholar] [CrossRef]
- Li, H.; Min, J.; Mao, X.; Wang, X.; Yang, Y.; Chen, Y. Edaravone ameliorates experimental autoimmune thyroiditis in rats through HO-1-dependent STAT3/PI3K/Akt pathway. Am. J. Transl. Res. 2018, 10, 2037–2046. [Google Scholar] [PubMed]
- Park, B.; Lim, J.W.; Kim, H. Lycopene treatment inhibits activation of Jak1/Stat3 and Wnt/β-catenin signaling and attenuates hyperproliferation in gastric epithelial cells. Nutr. Res. 2019, 70, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, R.; Kosai, H.; Masuo, M.; Izumiyama, K.; Noshikawaji, T.; Morimoto, M.; Kumaki, S.; Miyazaki, Y.; Motohashi, H.; Yamamoto, M.; et al. Nrf2 suppresses allergic lung Inflammation by attenuating the type 2 innate lymphoid cell response. J. Immunol. 2019, 202, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, Y.N.; Jiang, D.L.; Cai, L.; Chen, X.; Wang, Q.; Xie, Z.W.; Liu, Y.; Zhang, C.Y.; Jing, S.; Chen, G.H.; et al. Use of a dose-response model to guide future clinical trial of Benvitimod cream to treat mild and moderate psoriasis. Int. J. Clin. Pharmacol. Ther. 2016, 54, 87–95. [Google Scholar] [CrossRef]
- Bissonnette, R.; Vasist, L.S.; Bullman, J.N.; Collingwood, T.; Chen, G.; Maeda-Chubachi, T. Systemic pharmacokinetics, safety, and preliminary efficacy of topical AhR agonist Tapinarof: Results of a phase 1 study. Clin. Pharmacol. Drug Dev. 2018, 7, 524–531. [Google Scholar] [CrossRef]
- Bissonnette, R.; Poulin, Y.; Zhou, Y.; Tan, J.; Hong, H.C.; Webster, J.; Ip, W.; Tang, L.; Lyle, M. Efficacy and safety of topical WBI-1001 in patients with mild to severe atopic dermatitis: Results from a 12-week, multicentre, randomized, placebo-controlled double-blind trial. Br. J. Dermatol. 2012, 166, 853–856. [Google Scholar] [CrossRef]
- Peppers, J.; Paller, A.S.; Maeda-Chubachi, T.; Wu, S.; Robbins, K.; Gallagher, K.; Kraus, J.E. A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Miake, S.; Tsuji, G.; Takemura, M.; Hashimoto-Hachiya, A.; Vu, Y.H.; Furue, M.; Nakahara, T. IL-4 augments IL-31/IL-31 receptor alpha interaction leading to enhanced Ccl17 and Ccl22 production in dendritic cells: Implications for atopic dermatitis. Int. J. Mol. Sci. 2019, 20, 4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puar, N.; Chovatiya, R.; Paller, A.S. New treatments in atopic dermatitis. Ann. Allergy Asthma Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furue, M. Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4‒JAK‒STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis. J. Clin. Med. 2020, 9, 3741. https://doi.org/10.3390/jcm9113741
Furue M. Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4‒JAK‒STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis. Journal of Clinical Medicine. 2020; 9(11):3741. https://doi.org/10.3390/jcm9113741
Chicago/Turabian StyleFurue, Masutaka. 2020. "Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4‒JAK‒STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis" Journal of Clinical Medicine 9, no. 11: 3741. https://doi.org/10.3390/jcm9113741