
From Skin Barrier Dysfunction to Systemic Impact of Atopic Dermatitis: Implications for a Precision Approach in Dermocosmetics and Medicine

 ,
,
Abstract
:1. Introduction
2. Evidence That Skin Barrier Dysfunction Is a Key Precursor of AD
2.1. Role of Microbiota in the Inflammation Driven by the Barrier Dysfunction
2.2. Neuroinflammation and Itch
3. Comorbidities of AD
4. Implications for Preventive and Therapeutic Interventions
4.1. Preventive Strategies Based on Skin Barrier Dysfunction: Precision Prevention with Dermocosmetics
4.2. Therapeutic Interventions: Precision Medicine

{kind=link}
| Target | Agent | AD * | Asthma | Other Indications | Remarks for AD and Asthma | ||
|---|---|---|---|---|---|---|---|
| Approv-ed | RCT | Approv-ed | RCT | ||||
| Adaptive immune response | |||||||
| IL-4Rα | dupilumab | + (≥6 y.) | a. | + (≥6 y.) | a. | CRSwNP (add-on), RCT for AR, pruritus, FA, CHP, keloid, HE, AA, AERD, nummular eczema, EE, EG, sinusitis, metastatic non-small cell lung cancer, prostate CA, sleep apnea, cold urticaria, scleredema, Netherton sd, BP, COVID-19, peanut allergy | Severe T2 asthma (eosinophils ≥ 150/µL, FeNO > 25 ppb). Add-on maintenance therapy |
| IL-4Rα | CBP-201 | - | IIb | - | II | RCT for CRSwNP | |
| IL-4Rα | AK 120 | - | Ib | - | II | - | |
| IL-13 | tralokinumab | + (≥18 y.) | a. | - | III | RCT for ulcerative colitis, AA, idiopathic pulmonary fibrosis | ↑ response for subgroups with ↑ levels of periostin, DPP-4, IL-13, inconsistent results for asthma overall |
| IL-13 | lebrikizumab | - | III | - | II | RCT for COPD, idiopathic pulmonary fibrosis | ↑ response for subgroups with ↑ levels of periostin |
| IL-13Rα1 | eblasakimab | - | IIb | - | - | - | |
| IL-5 | mepolizumab | - | - | + (≥6 y.) | a. | CRSwNP(add-on), HES, EPGA RCT for eosinophilia, COPD, eosinophilic fasciitis, esophagitis, angioedema, CSU | Severe eosinophilic asthma (add-on) |
| IL-5Rα | benralizumab | - | II | + (≥18 y.) | a. | RCT for EG, non-cystic fibrosis bonchiectasas, CRwNP, HES, nasal polyps, COPD, skin side effects caused by cancer therapy, eosinophilic chronic rhinosinusitis, cystic fibrosis | Severe eosinophilic asthma |
| IL-5Rα | reslizumab | - | - | + (≥18 y.) | a. | RCT for sinusitis, EE, HES | Severe eosinophilic asthma (add-on) |
| IgE | omalizumab | - | II | + (≥6 y.) | a. | CRSwNP (add-on), urticaria (CSU), RCT for FA, immunotherapy, BP, SLE, AR, Sjogren´s sd, mastocytosis, EE, cholinergic U., solar U., AE anaphylaxis, COPD, CF, HES, Job´s Sd, interstitial cystitis, ASS hypersensitivitiy | Allergic asthma AD: program discontinued |
| IgE | FB825/anti-CεmX | - | IIa | - | II | - | AD: program discontinued |
| Histamine | |||||||
| H4R | adriforant | - | IIb | - | - | - | AD: program discontinued |
| H4R | LEO152020/JW1601 | - | IIb | - | - | RCT for cholinergic urticaria | |
| Other | |||||||
| IL-22 | fezakinumab | - | IIa | - | - | RCT for RA, psoriasis | |
| IL-22R1 | LEO 138559 | - | Ib | - | - | - | |
| IL-17A | secukinumab | - | IIa | - | - | Plaque psoriasis, psoriatic arthritis, axial spondyloarthritis, RCT for HS, psoriasis, discoid LE, necrobiosis lipoidica diabeticorum, pyoderma gangrenosum, autoimmunity | AD: program discontinued |
| IL-23 | risankizumab | - | IIa | - | - | Plaque psoriasis, psoriatic arthritis, RCT for COVID-19, HS, AS, palmoplantar pustulosis, Crohn´s disease, ulcerative colitis, dermatitis | |
| rhIL-2 to Treg cells | Ly3471851 | - | Ib | - | - | RCT for psoriasis, SLE, ulcerative colitis, | |
| OX 40 | GBR 830/ISB 830 | - | IIb | - | - | - | |
| OX 40 | KHK 4083 | - | IIb | - | - | RCT for ulcerative colitis, digestic system diseases | |
| OX 40 | KY1005 | - | IIa | - | - | RCT for immune system diseases | |
| CCR4 | RPT193 | - | IIa | - | - | - | |
| S1PR1,4,5 | etrasimod | - | IIb | - | - | RCT for eosinophilic eophagitis, ulcerative colitis (III), Crohns´s disease, AA, PG | |
| S1PR1 | SCD-044 | - | IIb | - | - | RCT for plaque psoriasis | |
| S1PR1 | BMS-986166 | - | IIb | - | - | RCT for ulcerative colitis | |
| S1PR1 | LC51-0255 | - | I | - | - | RCT for ulcerative colitis | |
| S1PR1 | KT-474 | - | I | - | - | RCT for HS | |
| Innate immune response | |||||||
| TSLP | tezepelumab | - | IIa | + (≥12 y.) | a. | RCT for COPD (IIa), CSU (H2H with omalizumab), CRSwNP | Severe asthma (add-on), RCT for pediatric asthma ≥5–11 y (I), AD: discontinued, ↓ efficacy |
| IL-33 | etokimab | - | IIa | - | - | RCT for CRSwNP | AD: IIa: primary end point not reached |
| IL-33 | Itepekimab # | - | IIa | - | II | RCT for COPD (IIa) | IIa: improved asthma control, QoL, reduction of eosinophils |
| Il-33 | astegolimab | - | IIa | - | - | RCT for COPD, COVID-pneumonia | |
| IL-33 | tozorakimab (MEDI3506) | - | IIa | - | II | RCT for COPD, chronic bronchitis, diabetic kidney disease | |
| IL-1α | bermekimab | - | IIa | - | - | RCT for hidradenitis suppurativa (HS), systemic scleroderma, metastatic colorectal cancer, advanced cancers, type 2 diabetes | |
| IL-36 R | spesolimab | - | IIa | - | - | RCT for Crohn’s disease, HS, generalized pustular psoriasis, palmoplantar pustulosis, ulcerative colitis | AD: program discontinued |
| Microbiome | |||||||
| micro-biome | OM-85 | - | II | - | IV | Prevention of recurrent and lower respiratory tract infections (bronchitis, sinusitis), RCT for COPD, bronchiectasias, sleep, pain, stress, adenoid hypertrophy/ hyperplasia, COVID-19, mucositis, stomatitis, uveitis, head and neck squamous cell cancer, solid tumors, hematologic malignancies, overweight, essential fatty acid defiency, hypercholesterolemia, hypertriglyceridemia, hypertension, Parkinson’s disease, RA, HIV, SLE, psychiatric disorders, metabolic syndrome | |
| micro biome | EDP1815 | - | II | - | - | RCT for COVID-19, psoriasis, | |
| micro-biome | STMC-103 | - | Ib | - | - | RCT for type 1 hypersensitivity, atopic IgE mediated allergic disorder | |
| Pruritus | |||||||
| IL-31R | nemolizumab | - | III | - | - | RCT for PN, systemic sclerosis, chronic kidney disease-associated severe pruritus | Prurigo-form AD best responses, RCT also for pediatric AD (age 2-6 (II), 7-11 (II), 12-17 y (II)) |
| OSMRß | vixarelimab | - | IIb | - | - | RCT for pruritus, PN, chronic idiopathic urticaria, lichen planus, lichen simplex chronicus, plaque psoriasis | |
| NK1R | serlopitant | - | II | - | - | RCT for pruritus, PN, psoriasis, refractory chronic cough, burns, epidermolysis bullosa | AD: program discontinued |
| NK1R | tradipitant | - | II | - | - | RCT for COVID-19, pastorparesis, motion sickness, pruritus | |
| P2X3 | BLU-5937 | - | II | - | - | RCT for pruritus, chronic (refractory) cough | |
| Janus kinases | |||||||
| JAK1/JAK2 | baricitinib | + (≥18 y.) | a. | - | - | RA, RCT for AA, COVID-19, pneumonia, SARS, ACD, vitiligo, lichen planus, pyoderma gangrenosum, wound heal, dermatomyositis, systemic sclerosis, SLE, Sjogren´s syndrome, psoriasis, other skin diseases, polymylagia rheumatic, mypathies, uveitis, chronic graft vs. Host disease, type 1 diabetes, diabetic kidney disease, liver diseases, hepatic insufficiency, arteritis (giant cell), ALS, Alzheimer’s disease, systemic sclerosis, NNS/CANDLE, SAVI, AGS, ankylosing spondylitis, psoriasis arthritis | |
| JAK1 | upadacitinib | + (≥12 y.) | a. | - | - | RA, psoriatic arthritis, ankylosing spondylitis, RCT for SLE, juvenile idipathic arthritis, ulcerative colitis, Crohn´s disease, arteriits (Takayasu, giant cell), non-segmental vitiligo, | |
| JAK1 | abrocitinib | + (≥18 y.) | a. | - | - | -, RCT for FA, PN, pruritus, psoriasis, renal impairment | |
| JAK1 | SHR0302 | - | II | - | - | -, RCT for RA, AS, PsA, AA, GVHD, vitiligo, ulcerative colitis, primary membranous nephopathy | |
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laughter, M.R.; Maymone, M.B.C.; Mashayekhi, S.; Arents, B.W.M.; Karimkhani, C.; Langan, S.M.; Dellavalle, R.; Flohr, C. The global burden of atopic dermatitis: Lessons from the Global Burden of Disease Study 1990–2017. Br. J. Dermatol. 2021, 184, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S. Atopic Dermatitis. N. Engl. J. Med. 2021, 384, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Bieber, T. Atopic dermatitis: An expanding therapeutic pipeline for a complex disease. Nat. Rev. Drug Discov. 2021, 21, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Spergel, J.M. The atopic march: Where we are going? Can we change it? Ann. Allergy Asthma Immunol. 2021, 127, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Tham, E.H.; Leung, D.Y. Mechanisms by Which Atopic Dermatitis Predisposes to Food Allergy and the Atopic March. Allergy Asthma Immunol. Res. 2019, 11, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Deraison, C.; Lacroix, M.; Bonnart, C.; Robin, A.; Besson, C.; Dubus, P.; Hovnanian, A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 2009, 206, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, S.P.; Goh, C.S.M.; Brown, S.J.; Palmer, C.N.A.; Porter, R.M.; Cole, C.; Campbell, L.E.; Gierlinski, M.; Barton, G.J.; Schneider, G.; et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J. Allergy Clin. Immunol. 2013, 132, 1121–1129. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.A.; Spergel, J.M. The atopic march: Critical evidence and clinical relevance. Ann. Allergy Asthma Immunol. 2018, 120, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Ungar, B.; Garcet, S.; Gonzalez, J.; Dhingra, N.; Correa da Rosa, J.; Shemer, A.; Krueger, J.G.; Suarez-Farinas, M.; Guttman-Yassky, E. An Integrated Model of Atopic Dermatitis Biomarkers Highlights the Systemic Nature of the Disease. J. Investig. Dermatol. 2017, 137, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Cork, M.J.; Danby, S.G.; Ogg, G.S. Atopic dermatitis epidemiology and unmet need in the United Kingdom. J. Dermatol. Treat. 2020, 31, 801–809. [Google Scholar] [CrossRef]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.F.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.E.; Kim, J.; Goleva, E.; Berdyshev, E.; Lee, J.; Vang, K.A.; Lee, U.H.; Han, S.; Leung, S.; Hall, C.F.; et al. Particulate matter causes skin barrier dysfunction. JCI Insight 2021, 6, e145185. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat. Rev. Immunol. 2021, 21, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Samuel, M.; van Bever, H.; Tham, E.H. Emollients in infancy to prevent atopic dermatitis: A systematic review and meta-analysis. Allergy 2021, 1–15. [Google Scholar] [CrossRef]
- Lack, G. Update on risk factors for food allergy. J. Allergy Clin. Immunol. 2012, 129, 1187–1197. [Google Scholar] [CrossRef]
- Brough, H.A.; Lanser, B.J.; Sindher, S.B.; Teng, J.M.C.; Leung, D.Y.M.; Venter, C.; Chan, S.M.; Santos, A.F.; Bahnson, H.T.; Guttman-Yassky, E.; et al. Early intervention and prevention of allergic diseases. Allergy 2022, 77, 416–441. [Google Scholar] [CrossRef]
- Mu, Z.; Zhang, J. The Role of Genetics, the Environment, and Epigenetics in Atopic Dermatitis. Adv. Exp. Med. Biol. 2020, 1253, 107–140. [Google Scholar]
- Palmer, C.N.A.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.D.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef]
- Dumortier, A.; Durham, A.-D.; Di Piazza, M.; Vauclair, S.; Koch, U.; Ferrand, G.; Ferrero, I.; Demehri, S.; Song, L.L.; Farr, A.G.; et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PLoS ONE 2010, 5, e9258. [Google Scholar] [CrossRef]
- Gupta, J.; Grube, E.; Ericksen, M.B.; Stevenson, M.D.; Lucky, A.W.; Sheth, A.P.; Assa’Ad, A.H.; Hershey, G.K.K. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J. Allergy Clin. Immunol. 2008, 121, 725–730.e2. [Google Scholar] [CrossRef]
- Strugar, T.L.; Kuo, A.; Seité, S.; Lin, M.; Lio, P. Connecting the Dots: From Skin Barrier Dysfunction to Allergic Sensitization, and the Role of Moisturizers in Repairing the Skin Barrier. J. Drugs Dermatol. 2019, 18, 581. [Google Scholar] [PubMed]
- Elias, P.M. Primary role of barrier dysfunction in the pathogenesis of atopic dermatitis. Exp. Dermatol. 2018, 27, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakok, T.; Woolf, R.; Smith, C.H.; Weidinger, S.; Flohr, C. Atopic dermatitis: The skin barrier and beyond. Br. J. Dermatol. 2019, 180, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity 2015, 43, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; DeBenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y.M. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2009, 124, R7–R12. [Google Scholar] [CrossRef]
- Liu, F.-T.; Goodarzi, H.; Chen, H.-Y. IgE, mast cells, and eosinophils in atopic dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 298–310. [Google Scholar] [CrossRef]
- He, H.; Del Duca, E.; Diaz, A.; Kim, H.J.; Gay-Mimbrera, J.; Zhang, N.; Wu, J.; Beaziz, J.; Estrada, Y.; Krueger, J.G.; et al. Mild atopic dermatitis lacks systemic inflammation and shows reduced nonlesional skin abnormalities. J. Allergy Clin. Immunol. 2021, 147, 1369–1380. [Google Scholar] [CrossRef]
- Dyjack, N.; Goleva, E.; Rios, C.; Kim, B.E.; Bin, L.; Taylor, P.; Bronchick, C.; Hall, C.F.; Richers, B.N.; Seibold, M.A.; et al. Minimally invasive skin tape strip RNA sequencing identifies novel characteristics of the type 2-high atopic dermatitis disease endotype. J. Allergy Clin. Immunol. 2018, 141, 1298–1309. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, C.; Torres, T. More than skin deep: The systemic nature of atopic dermatitis. Eur. J. Dermatol. 2019, 29, 250–258. [Google Scholar]
- He, J.-Q.; Chan-Yeung, M.; Becker, A.B.; Dimich-Ward, H.; Ferguson, A.C.; Manfreda, J.; Watson, W.T.A.; Sandford, A.J. Genetic variants of the IL13 and IL4 genes and atopic diseases in at-risk children. Genes Immun. 2003, 4, 385–389. [Google Scholar] [CrossRef] [Green Version]
- Namkung, J.-H.; Lee, J.-E.; Kim, E.; Kim, H.-J.; Seo, E.-Y.; Jang, H.-Y.; Shin, E.-S.; Cho, E.-Y.; Yang, J.-M. Association of polymorphisms in genes encoding IL-4, IL-13 and their receptors with atopic dermatitis in a Korean population. Exp. Dermatol. 2011, 20, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Zhang, H.; Chan, L.S. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAK-STAT 2013, 2, e24137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renert-Yuval, Y.; Thyssen, J.P.; Bissonnette, R.; Bieber, T.; Kabashima, K.; Hijnen, D.; Guttman-Yassky, E. Biomarkers in atopic dermatitis—A review on behalf of the International Eczema Council. J. Allergy Clin. Immunol. 2021, 147, 1174–1190.e1. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Emerson, R.O.; Tipton, C.; Garcet, S.; Khattri, S.; Coats, I.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin shares similar T-cell clones with lesional tissues. Allergy 2017, 72, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Fariñas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964.e4. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Altunbulakli, C.; Reiger, M.; Neumann, A.U.; Garzorz-Stark, N.; Fleming, M.; Huelpuesch, C.; Castro-Giner, F.; Eyerich, K.; Akdis, C.A.; Traidl-Hoffmann, C. Relations between epidermal barrier dysregulation and Staphylococcus species-dominated microbiome dysbiosis in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2018, 142, 1643–1647.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnowicki, T.; Krueger, J.G.; Guttman-Yassky, E. Skin barrier and immune dysregulation in atopic dermatitis: An evolving story with important clinical implications. J. Allergy Clin. Immunol. Pract. 2014, 2, 371–379. [Google Scholar] [CrossRef]
- Le Nguyen, H.T.; Trujillo-Paez, J.V.; Umehara, Y.; Yue, H.; Peng, G.; Kiatsurayanon, C.; Chieosilapatham, P.; Song, P.; Okumura, K.; Ogawa, H.; et al. Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals with Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 7607. [Google Scholar] [CrossRef]
- Forbes-Blom, E.; Camberis, M.; Prout, M.; Tang, S.-C.; Le Gros, G. Staphylococcal-derived superantigen enhances peanut induced Th2 responses in the skin. Clin. Exp. Allergy 2012, 42, 305–314. [Google Scholar] [CrossRef]
- Chen, Y.E.; Fischbach, M.A.; Belkaid, Y. Skin microbiota-host interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Glatz, M.; Horiuchi, K.; Kawasaki, H.; Akiyama, H.; Kaplan, D.H.; Kong, H.H.; Amagai, M.; Nagao, K. Dysbiosis and Staphylococcus aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity 2015, 42, 756–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, K.; Nümm, T.J.; Koch, S.; Herrmann, N.; Leib, N.; Bieber, T. Langerhans and inflammatory dendritic epidermal cells in atopic dermatitis are tolerized toward TLR2 activation. Allergy 2018, 73, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S.; Yosipovitch, G.; Bushmakin, A.G.; Cappelleri, J.C.; Luger, T.; Tom, W.L.; Ports, W.; Zielinski, M.; Tallman, A.; Tan, H.; et al. Examining the association between pruritus and quality of life in patients with atopic dermatitis treated with crisaborole. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1742–1746. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S.; Simpson, E.L.; Guttman-Yassky, E.; Thyssen, J.P.; Kabashima, K.; Ball, S.G.; Rueda, M.J.; DeLozier, A.M.; Silverberg, J.I. Clinical Relevance of Skin Pain in Atopic Dermatitis. J. Drugs Dermatol. 2020, 19, 921–926. [Google Scholar] [CrossRef]
- Lerner, E.A. Pathophysiology of Itch. Dermatol. Clin. 2018, 36, 175–177. [Google Scholar] [CrossRef]
- Oetjen, L.K.; Mack, M.R.; Feng, J.; Whelan, T.M.; Niu, H.; Guo, C.J.; Chen, S.; Trier, A.M.; Xu, A.Z.; Tripathi, S.V.; et al. Sensory Neurons Co-opt Classical Immune Signaling Pathways to Mediate Chronic Itch. Cell 2017, 171, 217–228.e13. [Google Scholar] [CrossRef] [Green Version]
- Cevikbas, F.; Lerner, E.A. Physiology and Pathophysiology of Itch. Physiol. Rev. 2020, 100, 945–982. [Google Scholar] [CrossRef]
- Werfel, T.; Layton, G.; Yeadon, M.; Whitlock, L.; Osterloh, I.; Jimenez, P.; Liu, W.; Lynch, V.; Asher, A.; Tsianakas, A.; et al. Efficacy and safety of the histamine H4 receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 1830–1837.e4. [Google Scholar] [CrossRef]
- Kabashima, K.; Matsumura, T.; Komazaki, H.; Kawashima, M. Trial of Nemolizumab and Topical Agents for Atopic Dermatitis with Pruritus. N. Engl. J. Med. 2020, 383, 141–150. [Google Scholar] [CrossRef]
- Kabashima, K.; Matsumura, T.; Komazaki, H.; Kawashima, M. Nemolizumab plus topical agents in patients with atopic dermatitis (AD) and moderate-to-severe pruritus provide improvement in pruritus and signs of AD for up to 68 weeks: Results from two phase III; long-term studies. Br. J. Dermatol. 2022, 186, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Feld, M.; Garcia, R.; Buddenkotte, J.; Katayama, S.; Lewis, K.; Muirhead, G.; Hevezi, P.; Plesser, K.; Schrumpf, H.; Krjutskov, K.; et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J. Allergy Clin. Immunol. 2016, 138, 500–508.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, L.-S.; Yen, Y.-T.; Lee, C.-H. The Implications of Pruritogens in the Pathogenesis of Atopic Dermatitis. Int. J. Mol. Sci. 2021, 22, 7227. [Google Scholar] [CrossRef]
- Simpson, E.L.; Guttman-Yassky, E.; Margolis, D.J.; Feldman, S.R.; Qureshi, A.; Hata, T.; Mastey, V.; Wei, W.; Eckert, L.; Chao, J.; et al. Association of Inadequately Controlled Disease and Disease Severity with Patient-Reported Disease Burden in Adults with Atopic Dermatitis. JAMA Dermatol. 2018, 154, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Thijs, J.L.; Strickland, I.; Bruijnzeel-Koomen, C.A.F.M.; Nierkens, S.; Giovannone, B.; Knol, E.F.; Csomor, E.; Sellman, B.R.; Mustelin, T.; Sleeman, M.A.; et al. Serum biomarker profiles suggest that atopic dermatitis is a systemic disease. J. Allergy Clin. Immunol. 2018, 141, 1523–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.H.; Patel, K.R.; Singam, V.; Rastogi, S.; Silverberg, J.I. A systematic review and meta-analysis of the prevalence and phenotype of adult-onset atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 1526–1532.e7. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I. Adult-Onset Atopic Dermatitis. J. Allergy Clin. Immunol. Pract. 2019, 7, 28–33. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Vakharia, P.P.; Chopra, R.; Sacotte, R.; Patel, N.; Immaneni, S.; White, T.; Kantor, R.; Hsu, D.Y. Phenotypical Differences of Childhood- and Adult-Onset Atopic Dermatitis. J. Allergy Clin. Immunol. Pract. 2018, 6, 1306–1312. [Google Scholar] [CrossRef]
- Barker, J.N.W.N.; Palmer, C.N.A.; Zhao, Y.; Liao, H.; Hull, P.R.; Lee, S.P.; Allen, M.H.; Meggitt, S.J.; Reynolds, N.J.; Trembath, R.C.; et al. Null mutations in the filaggrin gene (FLG) determine major susceptibility to early-onset atopic dermatitis that persists into adulthood. J. Investig. Dermatol. 2007, 127, 564–567. [Google Scholar] [CrossRef] [Green Version]
- Mortz, C.G.; Andersen, K.E.; Dellgren, C.; Barington, T.; Bindslev-Jensen, C. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: Prevalence, persistence and comorbidities. Allergy 2015, 70, 836–845. [Google Scholar] [CrossRef]
- Silverberg, J.I. Persistence of childhood eczema into adulthood. JAMA Dermatol. 2014, 150, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Abuabara, K.; Yu, A.M.; Okhovat, J.-P.; Allen, I.E.; Langan, S.M. The prevalence of atopic dermatitis beyond childhood: A systematic review and meta-analysis of longitudinal studies. Allergy 2018, 73, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Abuabara, K.; Langan, S.M.; Yu, A.M. Conclusions about atopic dermatitis persistence might be premature. J. Am. Acad. Dermatol. 2017, 76, e177–e178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuabara, K.; Ye, M.; McCulloch, C.E.; Sullivan, A.; Margolis, D.J.; Strachan, D.P.; Paternoster, L.; Yew, Y.W.; Williams, H.C.; Langan, S.M. Clinical onset of atopic eczema: Results from 2 nationally representative British birth cohorts followed through midlife. J. Allergy Clin. Immunol. 2019, 144, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Margolis, J.S.; Abuabara, K.; Bilker, W.; Hoffstad, O.; Margolis, D.J. Persistence of mild to moderate atopic dermatitis. JAMA Dermatol. 2014, 150, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Brunner, P.M.; Silverberg, J.I.; Guttman-Yassky, E.; Paller, A.S.; Kabashima, K.; Amagai, M.; Luger, T.A.; Deleuran, M.; Werfel, T.; Eyerich, K.; et al. Increasing Comorbidities Suggest that Atopic Dermatitis Is a Systemic Disorder. J. Investig. Dermatol. 2017, 137, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Marenholz, I.; Esparza-Gordillo, J.; Rüschendorf, F.; Bauerfeind, A.; Strachan, D.P.; Spycher, B.D.; Baurecht, H.; Margaritte-Jeannin, P.; Sääf, A.; Kerkhof, M.; et al. Meta-Analysis Identifies Seven Susceptibility Loci Involved in the Atopic March; Universität Würzburg: Würzburg, Germany, 2016. [Google Scholar]
- Thyssen, J.P.; Tang, L.; Husemoen, L.L.N.; Stender, S.; Szecsi, P.B.; Menné, T.; Johansen, J.D.; Linneberg, A. Filaggrin gene mutations are not associated with food and aeroallergen sensitization without concomitant atopic dermatitis in adults. J. Allergy Clin. Immunol. 2015, 135, 1375–1378.e1. [Google Scholar] [CrossRef]
- Rogers, A.J.; Celedón, J.C.; Lasky-Su, J.A.; Weiss, S.T.; Raby, B.A. Filaggrin mutations confer susceptibility to atopic dermatitis but not to asthma. J. Allergy Clin. Immunol. 2007, 120, 1332–1337. [Google Scholar] [CrossRef]
- Tsuge, M.; Ikeda, M.; Matsumoto, N.; Yorifuji, T.; Tsukahara, H. Current Insights into Atopic March. Children 2021, 8, 1067. [Google Scholar] [CrossRef]
- Pasha, M.A.; Patel, G.; Hopp, R.; Yang, Q. Role of innate lymphoid cells in allergic diseases. Allergy Asthma Proc. 2019, 40, 138–145. [Google Scholar] [CrossRef]
- Roediger, B.; Kyle, R.; Yip, K.H.; Sumaria, N.; Guy, T.V.; Kim, B.S.; Mitchell, A.J.; Tay, S.S.; Jain, R.; Forbes-Blom, E.; et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 564–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnowicki, T.; Krueger, J.G.; Guttman-Yassky, E. Novel concepts of prevention and treatment of atopic dermatitis through barrier and immune manipulations with implications for the atopic march. J. Allergy Clin. Immunol. 2017, 139, 1723–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
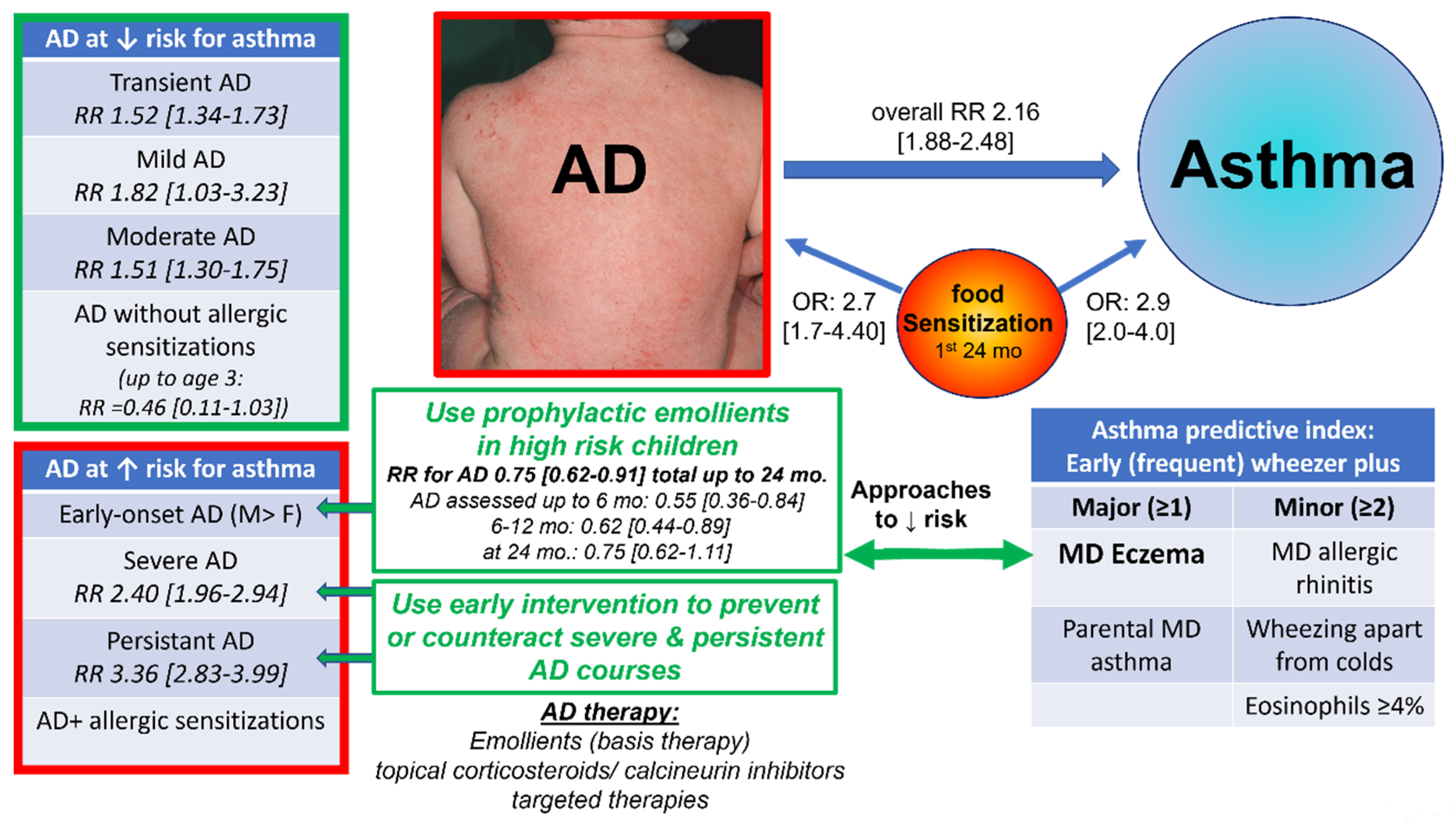
- Li, H.; Dai, T.; Liu, C.; Liu, Q.; Tan, C. Phenotypes of atopic dermatitis and the risk for subsequent asthma: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2022, 86, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodriguez, J.A.; Forno, E.; Padilla, O.; Casanello, P.; Krause, B.J.; Borzutzky, A. The asthma predictive index as a surrogate diagnostic tool in preschoolers: Analysis of a longitudinal birth cohort. Pediatric Pulmonol. 2021, 56, 3183–3188. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodríguez, J.A.; Holberg, C.J.; Wright, A.L.; Martinez, F.D. A clinical index to define risk of asthma in young children with recurrent wheezing. Am. J. Respir. Crit. Care Med. 2000, 162, 1403–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akar-Ghibril, N.; Casale, T.; Custovic, A.; Phipatanakul, W. Allergic Endotypes and Phenotypes of Asthma. J. Allergy Clin. Immunol. Pract. 2020, 8, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.M.; Lefebvre, D.L.; Dharma, C.; Dai, D.; Lou, W.Y.W.; Subbarao, P.; Becker, A.B.; Mandhane, P.J.; Turvey, S.E.; Sears, M.R.; et al. Predicting the atopic march: Results from the Canadian Healthy Infant Longitudinal Development Study. J. Allergy Clin. Immunol. 2018, 141, 601–607.e8. [Google Scholar] [CrossRef] [Green Version]
- Alduraywish, S.A.; Lodge, C.J.; Campbell, B.; Allen, K.J.; Erbas, B.; Lowe, A.J.; Dharmage, S.C. The march from early life food sensitization to allergic disease: A systematic review and meta-analyses of birth cohort studies. Allergy 2016, 71, 77–89. [Google Scholar] [CrossRef]
- Alduraywish, S.A.; Standl, M.; Lodge, C.J.; Abramson, M.J.; Allen, K.J.; Erbas, B.; von Berg, A.; Heinrich, J.; Lowe, A.J.; Dharmage, S.C. Is there a march from early food sensitization to later childhood allergic airway disease? Results from two prospective birth cohort studies. Pediatric Allergy Immunol. 2017, 28, 30–37. [Google Scholar] [CrossRef]
- Paternoster, L.; Savenije, O.E.M.; Heron, J.; Evans, D.M.; Vonk, J.M.; Brunekreef, B.; Wijga, A.H.; Henderson, A.J.; Koppelman, G.; Brown, S.J. Identification of atopic dermatitis subgroups in children from 2 longitudinal birth cohorts. J. Allergy Clin. Immunol. 2018, 141, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.M.R.; Drucker, A.M.; Alikhan, A.; Bercovitch, L.; Cohen, D.E.; Darr, J.M.; Eichenfield, L.F.; Frazer-Green, L.; Paller, A.S.; Silverberg, J.I.; et al. AAD Guidelines: Awareness of comorbidities associated with atopic dermatitis in adults. J. Am. Acad. Dermatol. 2022, 86, 1335–1336.e18. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Suárez-Fariñas, M.; He, H.; Malik, K.; Wen, H.-C.; Gonzalez, J.; Chan, T.C.-C.; Estrada, Y.; Zheng, X.; Khattri, S.; et al. The atopic dermatitis blood signature is characterized by increases in inflammatory and cardiovascular risk proteins. Sci. Rep. 2017, 7, 8707. [Google Scholar] [CrossRef] [PubMed]
- Silverwood, R.J.; Forbes, H.J.; Abuabara, K.; Ascott, A.; Schmidt, M.; Schmidt, S.A.J.; Smeeth, L.; Langan, S.M. Severe and predominantly active atopic eczema in adulthood and long term risk of cardiovascular disease: Population based cohort study. BMJ 2018, 361, k1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, W.F.; Leung, D.Y.M.; Beck, L.A.; Berin, C.M.; Boguniewicz, M.; Busse, W.W.; Chatila, T.A.; Geha, R.S.; Gern, J.E.; Guttman-Yassky, E.; et al. Report from the National Institute of Allergy and Infectious Diseases Workshop on “Atopic Dermatitis and the Atopic March. Mechanisms and Interventions”. J. Allergy Clin. Immunol. 2019, 143, 894–913. [Google Scholar] [CrossRef]
- Nomura, T.; Kabashima, K. Advances in atopic dermatitis in 2015. J. Allergy Clin. Immunol. 2016, 138, 1548–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazanfar, M.N.; Kibsgaard, L.; Thomsen, S.F.; Vestergaard, C. Risk of comorbidities in patients diagnosed with chronic urticaria: A nationwide registry-study. World Allergy Organ. J. 2020, 13, 100097. [Google Scholar] [CrossRef] [Green Version]
- Andersen, Y.M.F.; Egeberg, A.; Gislason, G.H.; Skov, L.; Thyssen, J.P. Autoimmune diseases in adults with atopic dermatitis. J. Am. Acad. Dermatol. 2017, 76, 274–280.e1. [Google Scholar] [CrossRef]
- Mohan, G.C.; Silverberg, J.I. Association of Vitiligo and Alopecia Areata with Atopic Dermatitis: A Systematic Review and Meta-analysis. JAMA Dermatol. 2015, 151, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Maintz, L.; Welchowski, T.; Herrmann, N.; Brauer, J.; Kläschen, A.S.; Fimmers, R.; Schmid, M.; Bieber, T.; the CK-CARE Study Group. Machine Learning-Based Deep Phenotyping of Atopic Dermatitis: Severity-Associated Factors in Adolescent and Adult Patients. JAMA Dermatol. 2021, 157, 1414–1424. [Google Scholar] [CrossRef]
- Liezmann, C.; Klapp, B.; Peters, E.M. Stress, atopy and allergy: A re-evaluation from a psychoneuroimmunologic persepective. Derm. Endocrinol. 2011, 3, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Katayama, I.; Murota, H.; Satoh, T. (Eds.) Evolution of Atopic Dermatitis in the 21st Century, 1st ed.; Springer: Singapore, 2018. [Google Scholar]
- Elenkov, I.J. Glucocorticoids and the Th1/Th2 balance. Ann. N. Y. Acad. Sci. 2004, 1024, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Bieber, T.; Cork, M.; Reitamo, S. Atopic dermatitis: A candidate for disease-modifying strategy. Allergy 2012, 67, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; He, H.; Canter, T.; Han, J.; Lefferdink, R.; Erickson, T.; Rangel, S.; Kameyama, N.; Kim, H.J.; Pavel, A.B.; et al. Evolution of pathologic T-cell subsets in patients with atopic dermatitis from infancy to adulthood. J. Allergy Clin. Immunol. 2020, 145, 215–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paller, A.S.; Spergel, J.M.; Mina-Osorio, P.; Irvine, A.D. The atopic march and atopic multimorbidity: Many trajectories, many pathways. J. Allergy Clin. Immunol. 2019, 143, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.M.; Berdyshev, E.; Goleva, E. Cutaneous barrier dysfunction in allergic diseases. J. Allergy Clin. Immunol. 2020, 145, 1485–1497. [Google Scholar] [CrossRef]
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: Part I. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 657–682. [Google Scholar] [CrossRef] [Green Version]
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: Part II. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 850–878. [Google Scholar] [CrossRef] [Green Version]
- Blume-Peytavi, U.; Bagot, M.; Tennstedt, D.; Saint Aroman, M.; Stockfleth, E.; Zlotogorski, A.; Mengeaud, V.; Schmitt, A.M.; Paul, C.; Lim, H.W.; et al. Dermatology today and tomorrow: From symptom control to targeted therapy. J. Eur. Acad. Dermatol. Venereol. 2019, 33 (Suppl. S1), 3–36. [Google Scholar] [CrossRef] [Green Version]
- Bieber, T.; Traidl-Hoffmann, C.; Schäppi, G.; Lauener, R.; Akdis, C.; Schmid-Grendlmeier, P. Unraveling the Complexity of Atopic Dermatitis: The CK-CARE Approach toward Precision Medicine; Universität Augsburg: Augsburg, Germany; Wiley: Hoboken, NJ, USA, 2020. [Google Scholar]
- Bieber, T.; Akdis, C.; Lauener, R.; Traidl-Hoffmann, C.; Schmid-Grendelmeier, P.; Schäppi, G.; Allam, J.-P.; Apfelbacher, C.; Augustin, M.; Beck, L.; et al. Global Allergy Forum and 3rd Davos Declaration 2015, Atopic dermatitis/Eczema: Challenges and opportunities toward precision medicine. Allergy 2016, 71, 588–592. [Google Scholar] [CrossRef]
- Agache, I.; Akdis, C.A. Precision medicine and phenotypes, endotypes, genotypes, regiotypes, and theratypes of allergic diseases. J. Clin. Investig. 2019, 129, 1493–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieber, T.; D’Erme, A.M.; Akdis, C.A.; Traidl-Hoffmann, C.; Lauener, R.; Schäppi, G.; Schmid-Grendelmeier, P. Clinical phenotypes and endophenotypes of atopic dermatitis. Where are we, and where should we go? J. Allergy Clin. Immunol. 2017, 139, S58–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavel, A.B.; Renert-Yuval, Y.; Wu, J.; Del Duca, E.; Diaz, A.; Lefferdink, R.; Fang, M.M.; Canter, T.; Rangel, S.M.; Zhang, N.; et al. Tape strips from early-onset pediatric atopic dermatitis highlight disease abnormalities in nonlesional skin. Allergy 2021, 76, 314–325. [Google Scholar] [CrossRef]
- Bakker, D.S.; de Graaf, M.; Nierkens, S.; Delemarre, E.M.; Knol, E.; van Wijk, F.; de Bruin-Weller, M.S.; Drylewicz, J.; Thijs, J.L. Unraveling heterogeneity in pediatric atopic dermatitis: Identification of serum biomarker based patient clusters. J. Allergy Clin. Immunol. 2022, 149, 125–134. [Google Scholar] [CrossRef]
- Breiteneder, H.; Peng, Y.-Q.; Agache, I.; Diamant, Z.; Eiwegger, T.; Fokkens, W.J.; Traidl-Hoffmann, C.; Nadeau, K.; O’Hehir, R.E.; O’Mahony, L.; et al. Biomarkers for diagnosis and prediction of therapy responses in allergic diseases and asthma. Allergy 2020, 75, 3039–3068. [Google Scholar] [CrossRef] [PubMed]
- Bakker, D.S.; Nierkens, S.; Knol, E.F.; Giovannone, B.; Delemarre, E.M.; van der Schaft, J.; Van Wijk, F.; De Bruin-Weller, M.S.; Drylewicz, J.; Thijs, J.L. Confirmation of multiple endotypes in atopic dermatitis based on serum biomarkers. J. Allergy Clin. Immunol. 2021, 147, 189–198. [Google Scholar] [CrossRef]
- Rindler, K.; Krausgruber, T.; Thaler, F.M.; Alkon, N.; Bangert, C.; Kurz, H.; Fortelny, N.; Rojahn, T.B.; Jonak, C.; Griss, J. Spontaneously Resolved Atopic Dermatitis Shows Melanocyte and Immune Cell Activation Distinct from Healthy Control Skin. Front. Immunol. 2021, 12, 630892. [Google Scholar] [CrossRef]
- McLoughlin, I.J.; Wright, E.M.; Tagg, J.R.; Jain, R.; Hale, J.D.F. Skin Microbiome—The Next Frontier for Probiotic Intervention. Probiotics Antimicrob. Proteins 2021. [Google Scholar] [CrossRef]
- Van Zuuren, E.J.; Fedorowicz, Z.; Arents, B.W.M. Emollients and moisturizers for eczema: Abridged Cochrane systematic review including GRADE assessments. Br. J. Dermatol. 2017, 177, 1256–1271. [Google Scholar] [CrossRef]
- Van Zuuren, E.J.; Fedorowicz, Z.; Christensen, R.; Lavrijsen, A.; Arents, B.W.M. Emollients and moisturisers for eczema. Cochrane Database Syst. Rev. 2017, 2, CD012119. [Google Scholar]
- Kelleher, M.M.; Cro, S.; van Vogt, E.; Cornelius, V.; Lodrup Carlsen, K.C.; Ove Skjerven, H.; Rehbinder, E.M.; Lowe, A.; Dissanayake, E.; Shimojo, N.; et al. Skincare interventions in infants for preventing eczema and food allergy: A cochrane systematic review and individual participant data meta-analysis. Clin. Exp. Allergy 2021, 51, 402–418. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Chalmers, J.R.; Hanifin, J.M.; Thomas, K.S.; Cork, M.J.; McLean, W.H.I.; Brown, S.J.; Chen, Z.; Chen, Y.; Williams, H.C. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J. Allergy Clin. Immunol. 2014, 134, 818–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horimukai, K.; Morita, K.; Narita, M.; Kondo, M.; Kitazawa, H.; Nozaki, M.; Shigematsu, Y.; Yoshida, K.; Niizeki, H.; Motomura, K. Application of moisturizer to neonates prevents development of atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 824–830.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, A.; Su, J.; Tang, M.; Lodge, C.J.; Matheson, M.; Allen, K.J.; Varigos, G.; Sasi, A.; Cranswick, N.; Hamilton, S.; et al. PEBBLES study protocol: A randomised controlled trial to prevent atopic dermatitis, food allergy and sensitisation in infants with a family history of allergic disease using a skin barrier improvement strategy. BMJ Open 2019, 9, e024594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skjerven, H.O.; Rehbinder, E.M.; Vettukattil, R.; LeBlanc, M.; Granum, B.; Haugen, G.; Hedlin, G.; Landrø, L.; Marsland, B.J.; Rudi, K. Skin emollient and early complementary feeding to prevent infant atopic dermatitis (PreventADALL): A factorial, multicentre, cluster-randomised trial. Lancet 2020, 395, 951–961. [Google Scholar] [CrossRef]
- Chalmers, J.R.; Haines, R.H.; Bradshaw, L.E.; Montgomery, A.A.; Thomas, K.S.; Brown, S.J.; Ridd, M.J.; Lawton, S.; Simpson, f.E.L.; Cork, M.J.; et al. Daily emollient during infancy for prevention of eczema: The BEEP randomised controlled trial. Lancet 2020, 395, 962–972. [Google Scholar] [CrossRef]
- Wollenberg, A.; Christen-Zäch, S.; Taieb, A.; Paul, C.; Thyssen, J.P.; de Bruin-Weller, M.; Vestergaard, C.; Seneschal, J.; Werfel, T.; Cork, M.; et al. ETFAD/EADV Eczema task force 2020 position paper on diagnosis and treatment of atopic dermatitis in adults and children. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2717–2744. [Google Scholar] [CrossRef]
- Eichenfield, L.F.; Tom, W.L.; Chamlin, S.L.; Feldman, S.R.; Hanifin, J.M.; Simpson, E.L.; Berger, T.G.; Bergman, J.N.; Cohen, D.E.; Cooper, K.D.; et al. Guidelines of care for the management of atopic dermatitis: Section 1. Diagnosis and assessment of atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Van Halewijn, K.F.; Lahnstein, T.; Bohnen, A.M.; van den Berg, P.J.; Gma Pasmans, S.; Je Bindels, P.; Elshout, G. Recommendations for emollients, bathing and topical corticosteroids for the treatment of atopic dermatitis: A systematic review of guidelines. Eur. J. Dermatol. 2022, 32, 113–123. [Google Scholar] [CrossRef]
- Sweeney, A.; Sampath, V.; Nadeau, K.C. Early intervention of atopic dermatitis as a preventive strategy for progression of food allergy. Allergy Asthma Clin. Immunol. 2021, 17, 30. [Google Scholar] [CrossRef]
- Seite, S.; Bieber, T. Barrier function and microbiotic dysbiosis in atopic dermatitis. Clin. Cosmet. Investig. Dermatol. 2015, 8, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Seité, S.; Zelenkova, H.; Martin, R. Clinical efficacy of emollients in atopic dermatitis patients—Relationship with the skin microbiota modification. Clin. Cosmet. Investig. Dermatol. 2017, 10, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, E.L.; Bruin-Weller, M.; Flohr, C.; Ardern-Jones, M.R.; Barbarot, S.; Deleuran, M.; Bieber, T.; Vestergaard, C.; Brown, S.J.; Cork, M.J.; et al. When does atopic dermatitis warrant systemic therapy? Recommendations from an expert panel of the International Eczema Council. J. Am. Acad. Dermatol. 2017, 77, 623–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyle, M.; Cevikbas, F.; Harden, J.L.; Guttman-Yassky, E. Understanding the immune landscape in atopic dermatitis: The era of biologics and emerging therapeutic approaches. Exp. Dermatol. 2019, 28, 756–768. [Google Scholar] [CrossRef] [Green Version]
- Werfel, T.; Heratizadeh, A.; Aberer, W.; Ahrens, F.; Augustin, M.; Biedermann, T.; Diepgen, T.; Fölster-Holst, R.; Gieler, U.; Kahle, J.; et al. S2k guideline on diagnosis and treatment of atopic dermatitis—Short version. Allergo J. Int. 2016, 25, 82–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratchataswan, T.; Banzon, T.M.; Thyssen, J.P.; Weidinger, S.; Guttman-Yassky, E.; Phipatanakul, W. Biologics for Treatment of Atopic Dermatitis: Current Status and Future Prospect. J. Allergy Clin. Immunol. Pract. 2021, 9, 1053–1065. [Google Scholar] [CrossRef]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.; et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef]
- Edris, A.; de Feyter, S.; Maes, T.; Joos, G.; Lahousse, L. Monoclonal antibodies in type 2 asthma: A systematic review and network meta-analysis. Respir. Res. 2019, 20, 179. [Google Scholar] [CrossRef] [Green Version]
- Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; Naghavi, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar]
- Wechsler, M.E.; Ruddy, M.K.; Pavord, I.D.; Israel, E.; Rabe, K.F.; Ford, L.B.; Maspero, J.F.; Abdulai, R.M.; Hu, C.-C.; Martincova, R.; et al. Efficacy and Safety of Itepekimab in Patients with Moderate-to-Severe Asthma. N. Engl. J. Med. 2021, 385, 1656–1668. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suárez-Fariñas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callewaert, C.; Nakatsuji, T.; Knight, R.; Kosciolek, T.; Vrbanac, A.; Kotol, P.; Ardeleanu, M.; Hultsch, T.; Guttman-Yassky, E.; Bissonnette, R.; et al. IL-4Rα Blockade by Dupilumab Decreases Staphylococcus aureus Colonization and Increases Microbial Diversity in Atopic Dermatitis. J. Investig. Dermatol. 2020, 140, 191–202.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panettieri, R.A.; Sjöbring, U.; Péterffy, A.; Wessman, P.; Bowen, K.; Piper, E.; Colice, G.; Brightling, C.E. Tralokinumab for severe, uncontrolled asthma (STRATOS 1 and STRATOS 2): Two randomised, double-blind, placebo-controlled, phase 3 clinical trials. Lancet 2018, 6, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, A.; Edwards, M.; Nair, P.; Drew, S.; Vyas, A.; Sharma, R.; Marsden, P.A.; Evans, D.J.W. Anti-interleukin-13 and anti-interleukin-4 agents versus placebo, anti-interleukin-5 or anti-immunoglobulin-E agents, for people with asthma. Cochrane Database Syst. Rev. 2021, 10, CD012929. [Google Scholar]
- Emson, C.; Pham, T.-H.; Manetz, S.; Newbold, P. Periostin and Dipeptidyl Peptidase-4: Potential Biomarkers of Interleukin 13 Pathway Activation in Asthma and Allergy. Immunol. Allergy Clin. N. Am. 2018, 38, 611–628. [Google Scholar] [CrossRef]
- De Yang, H.Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Ghezzi, M.; Pozzi, E.; Abbattista, L.; Lonoce, L.; Zuccotti, G.V.; D’Auria, E. Barrier Impairment and Type 2 Inflammation in Allergic Diseases: The Pediatric Perspective. Children 2021, 8, 1165. [Google Scholar] [CrossRef]
- Saikumar Jayalatha, A.K.; Hesse, L.; Ketelaar, M.E.; Koppelman, G.H.; Nawijn, M.C. The central role of IL-33/IL-1RL1 pathway in asthma: From pathogenesis to intervention. Pharmacol. Ther. 2021, 225, 107847. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11, eaax2945. [Google Scholar] [CrossRef]
- Cardinale, F.; Lombardi, E.; Rossi, O.; Bagnasco, D.; Bellocchi, A.; Menzella, F. Epithelial dysfunction, respiratory infections and asthma: The importance of immunomodulation. A focus on OM-85. Expert Rev. Respir. Med. 2020, 14, 1019–1026. [Google Scholar] [CrossRef]
- Pivniouk, V.; Gimenes, J.A., Jr.; Ezeh, P.; Michael, A.; Pivniouk, O.; Hahn, S.; VanLinden, S.R.; Malone, S.P.; Abidov, A.; Anderson, D.; et al. Airway administration of OM-85, a bacterial lysate, blocks experimental asthma by targeting dendritic cells and the epithelium/IL-33/ILC2 axis. J. Allergy Clin. Immunol. 2022, 149, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Bodemer, C.; Guillet, G.; Cambazard, F.; Boralevi, F.; Ballarini, S.; Milliet, C.; Bertuccio, P.; la Vecchia, C.; Bach, J.; de Prost, Y. Adjuvant treatment with the bacterial lysate (OM-85) improves management of atopic dermatitis: A randomized study. PLoS ONE 2017, 12, e0161555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, M.A.; Torres, T. JAK/STAT inhibitors for the treatment of atopic dermatitis. J. Dermatol. Treat. 2019, 31, 33–40. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maintz, L.; Bieber, T.; Simpson, H.D.; Demessant-Flavigny, A.-L. From Skin Barrier Dysfunction to Systemic Impact of Atopic Dermatitis: Implications for a Precision Approach in Dermocosmetics and Medicine. J. Pers. Med. 2022, 12, 893. https://doi.org/10.3390/jpm12060893
Maintz L, Bieber T, Simpson HD, Demessant-Flavigny A-L. From Skin Barrier Dysfunction to Systemic Impact of Atopic Dermatitis: Implications for a Precision Approach in Dermocosmetics and Medicine. Journal of Personalized Medicine. 2022; 12(6):893. https://doi.org/10.3390/jpm12060893
Chicago/Turabian StyleMaintz, Laura, Thomas Bieber, Helen D. Simpson, and Anne-Laure Demessant-Flavigny. 2022. "From Skin Barrier Dysfunction to Systemic Impact of Atopic Dermatitis: Implications for a Precision Approach in Dermocosmetics and Medicine" Journal of Personalized Medicine 12, no. 6: 893. https://doi.org/10.3390/jpm12060893