
Synthesis, Molecular Modelling and Biological Evaluation of Novel Heterodimeric, Multiple Ligands Targeting Cholinesterases and Amyloid Beta

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
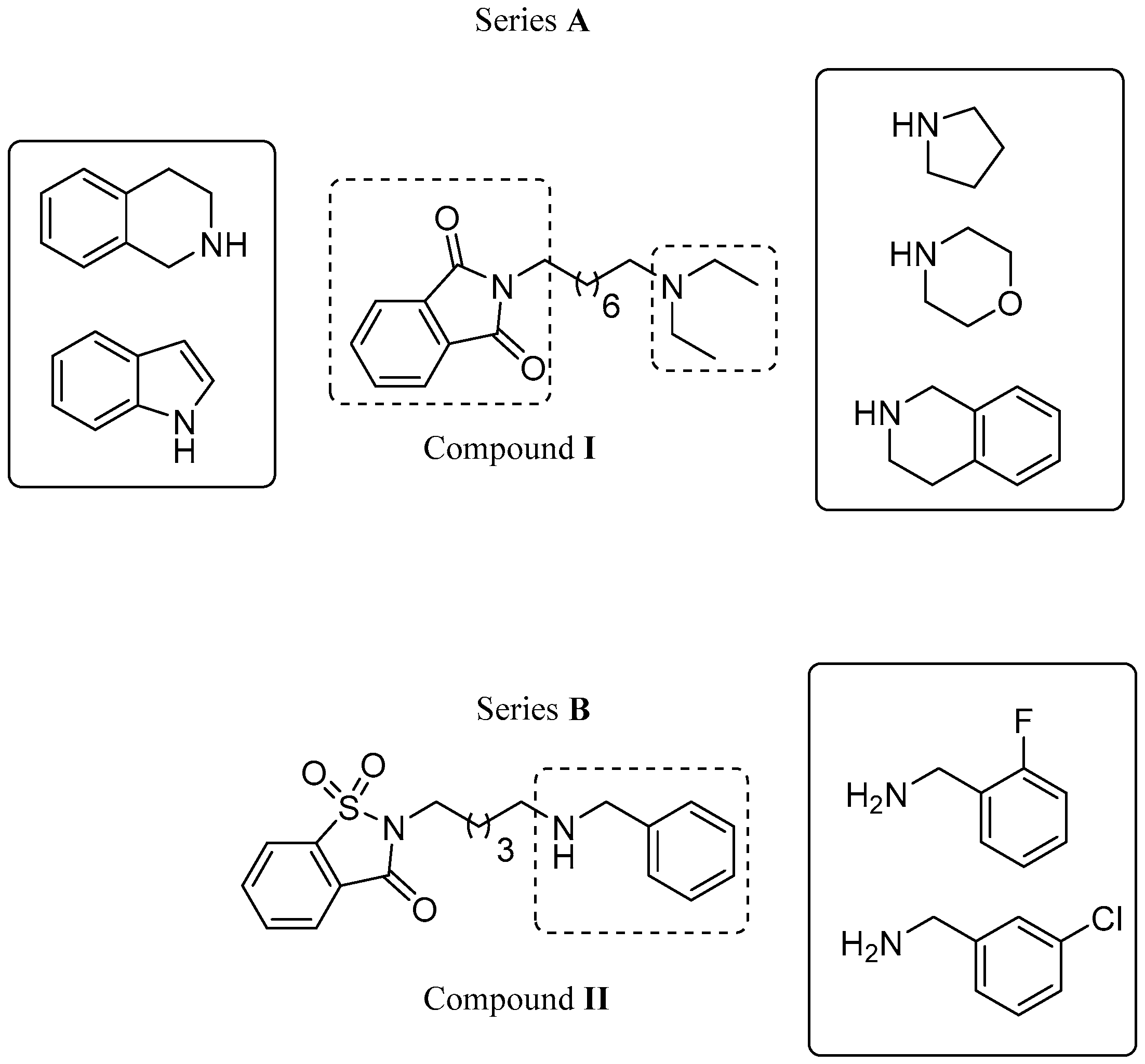
2.1. Design
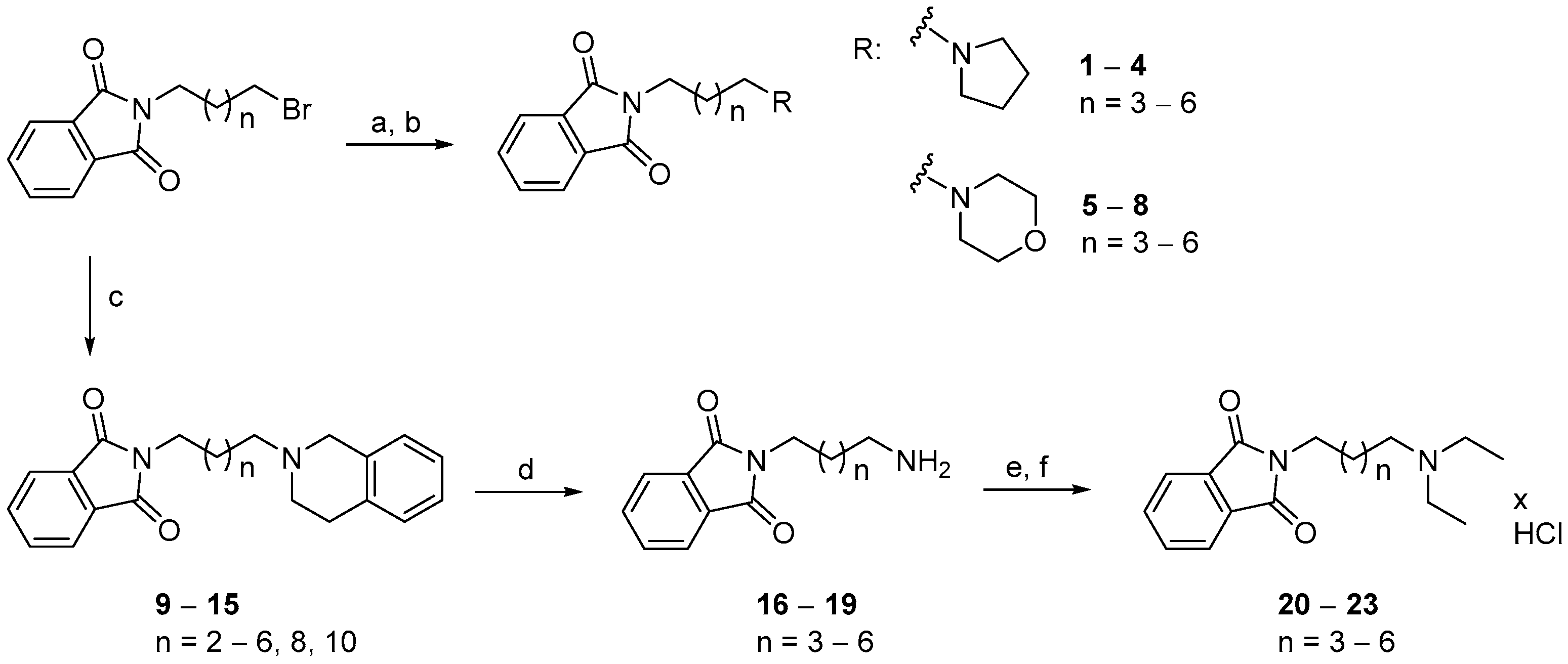
2.2. Chemistry
2.3. Biological Evaluation
2.3.1. Cholinesterase Inhibitory Potency
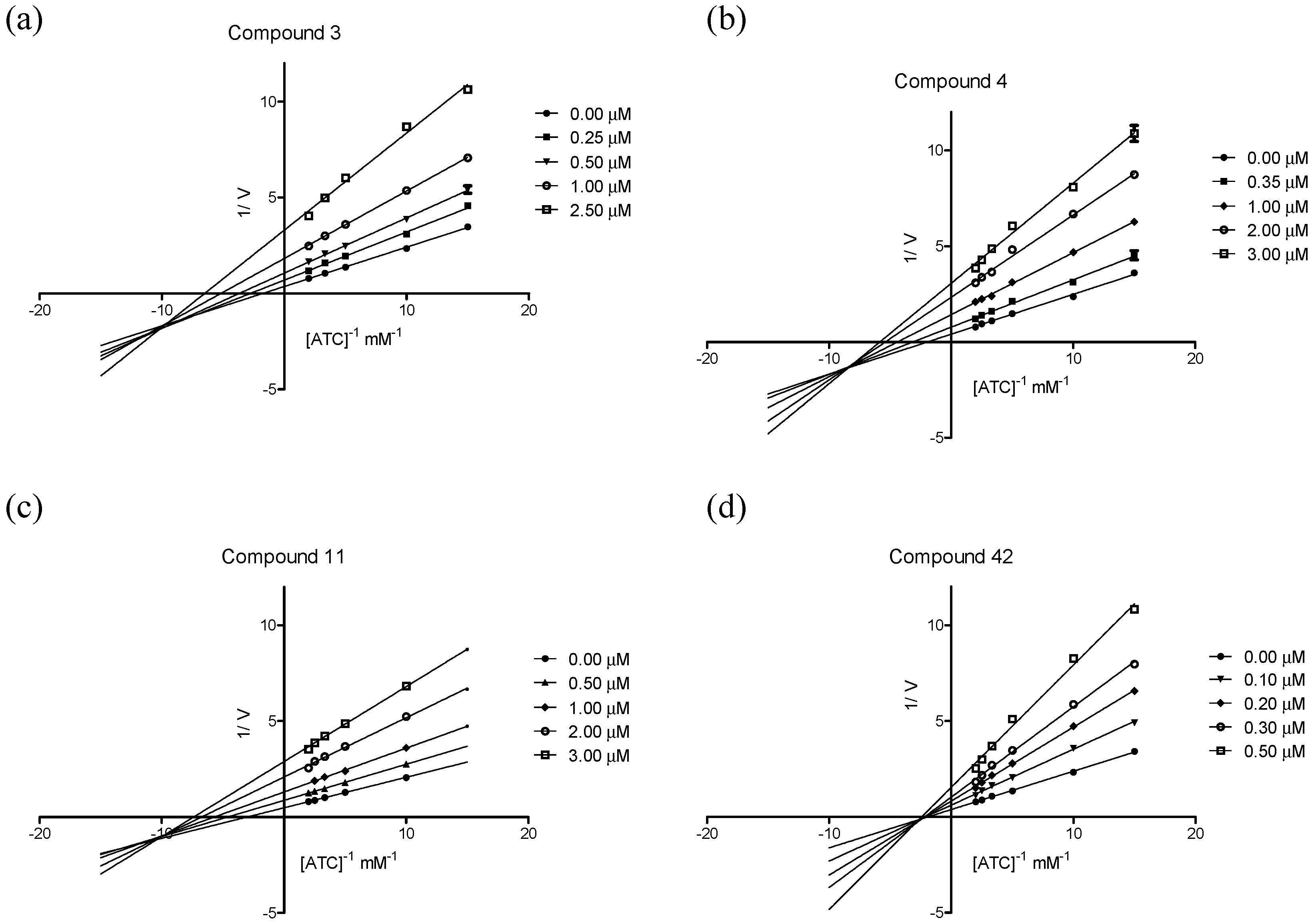
2.3.2. Kinetic Studies of AChE Inhibition
2.3.3. Aβ1–42 Aggregation Inhibitory Potency
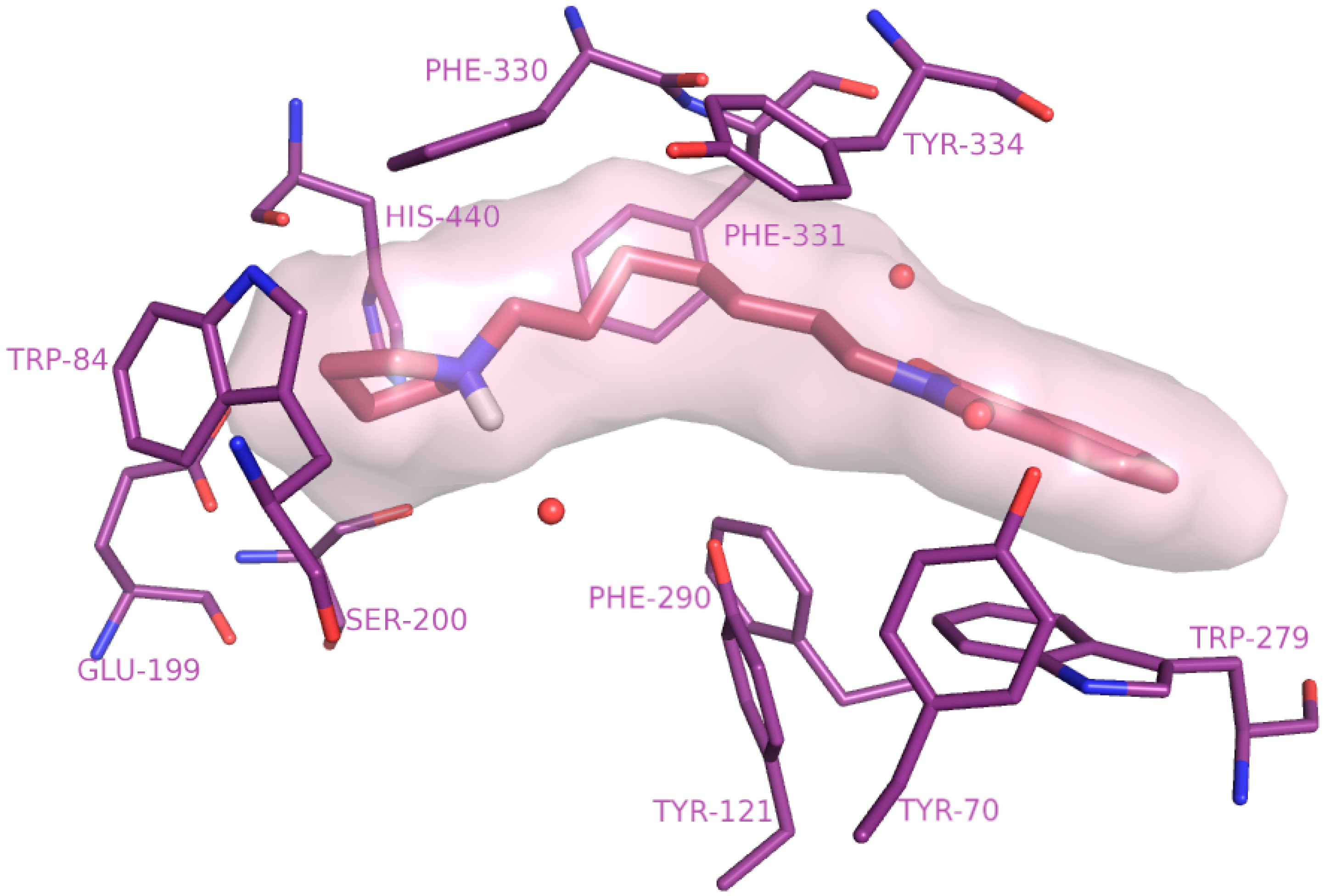
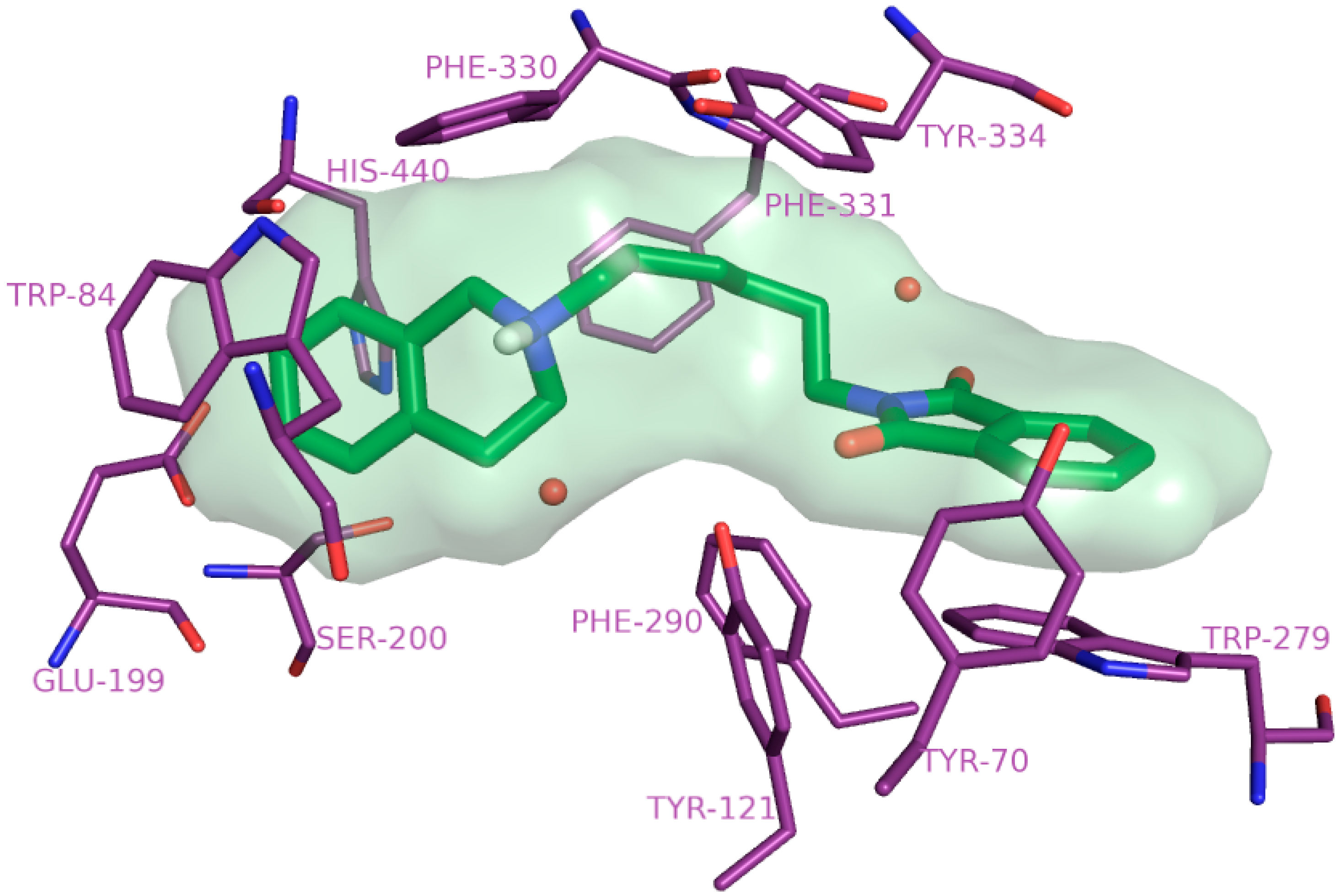
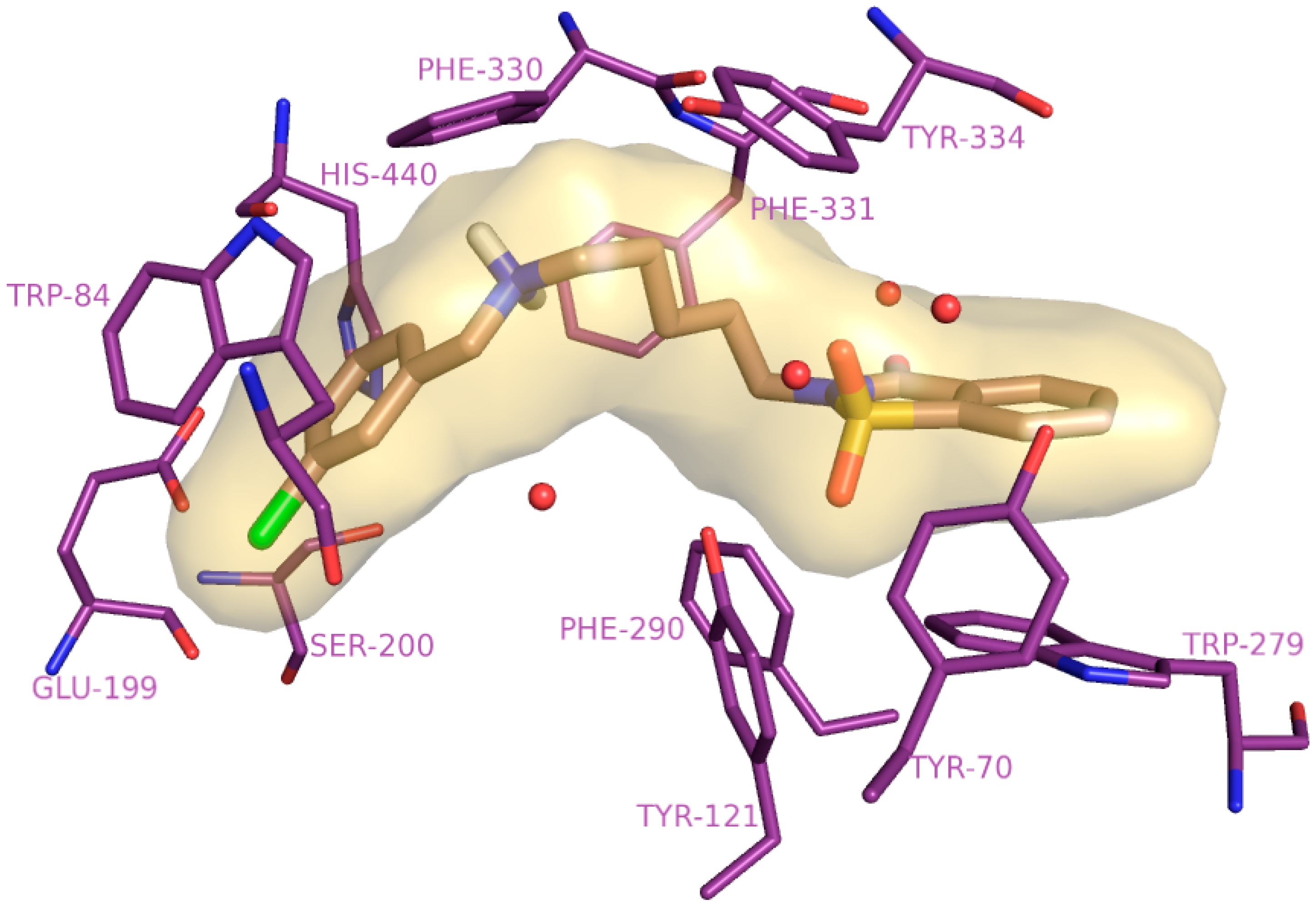
2.4. Molecular Modelling Studies
2.5. Blood–Brain Barrier Permeability Assay
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. General Procedure for the Preparation of Hydrochloride Salts
3.1.3. General Procedure for the Synthesis of Compounds (1–8)
3.1.4. General Procedure for the Synthesis of Compounds (9–15)
3.1.5. General Procedure for the Synthesis of Compounds (20–23)
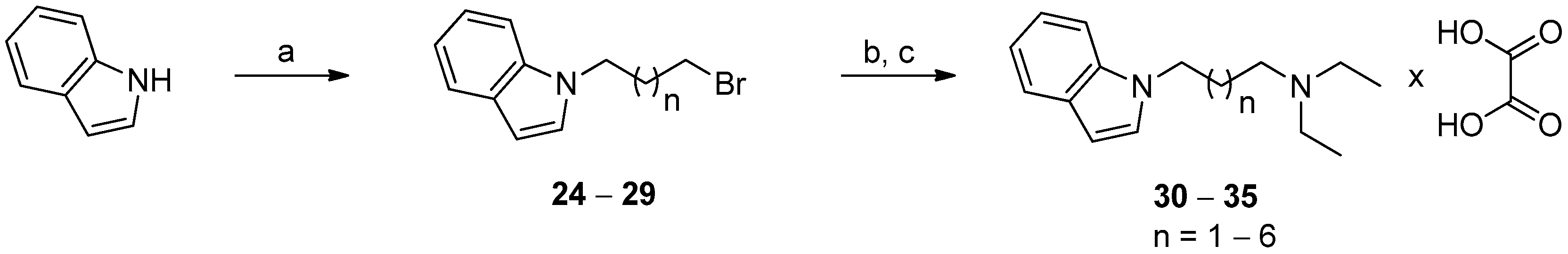
3.1.6. General Procedure for the Synthesis of Diethylamine Derivatives of N-alkyl-1H-indole (30–35)
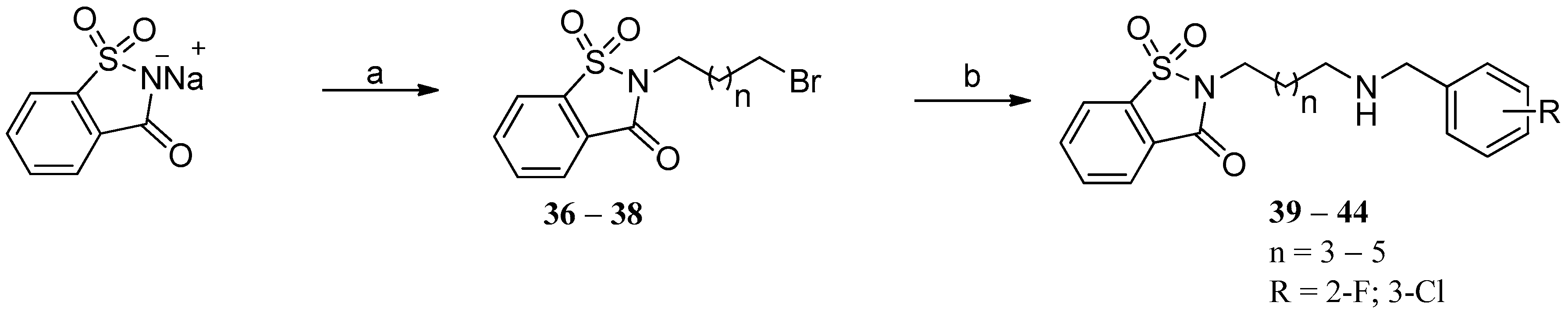
3.1.7. General Procedure for the Synthesis of Compounds (39–44)
3.2. Molecular Modelling
3.3. Biological Evaluation
3.3.1. In vitro Inhibition of AChE and BuChE
3.3.2. Kinetic Characterization of EeAChE Inhibition
3.3.3. In vitro Inhibition of Aβ1–42 Aggregation
3.3.4. PAMPA-BBB Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ach | acetylcholine |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| Aβ | amyloid beta peptide |
| BBB | blood-brain barrier |
| BuChE | butyrylcholinesterase |
| BuChE | butyrylcholinesterase |
| CAS | catalytic anionic site in acetylcholinesterase |
| CNS | central nervous system |
| EeAChE | acetylcholinesterase from electric eel |
| EqBuChE | equine serum butyrylcholinesterase |
| MTDL | multi-target-directed ligands |
| NFTs | neurofibrillary tangles |
| PAMPA | parallel artificial membrane permeation assay |
| PAS | peripheral anionic site in acetylcholinesterase |
| TcAChE | acetylcholinesterase from Torpedo californica |
References
- Prince, M.; Prina, M.; Guerchet, M. World Alzheimer Report 2013 Journey of Caring: An Analysis of Long-Term Care for Dementia; Alzheimer’s Disease International: London, UK, 2013; pp. 1–92. [Google Scholar]
- Wimo, A.; Jönsson, L.; Bond, J.; Prince, M.; Winblad, B. The worldwide economic impact of dementia 2010. Alzheimer’s Dement. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers. Res. Ther. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Godyń, J.; Jończyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2015, 68, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 308. [Google Scholar] [CrossRef]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A Structural Motif of Acetylcholinesterase That Promotes Amyloid β-Peptide Fibril Formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.; Moore, S.W. Identification of a structural site on acetylcholinesterase that promotes neurite outgrowth and binds laminin-1 and collagen IV. Biochem. Biophys. Res. Commun. 2004, 319, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase Accelerates Assembly of Amyloid-β-Peptides into Alzheimer’s Fibrils: Possible Role of the Peripheral Site of the Enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Akasofu, S.; Kimura, M.; Kosasa, T.; Sawada, K.; Ogura, H. Study of neuroprotection of donepezil, a therapy for Alzheimer’s disease. Chem. Biol. Interact. 2008, 175, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Akaike, A.; Takada-Takatori, Y.; Kume, T.; Izumi, Y. Mechanisms of Neuroprotective Effects of Nicotine and Acetylcholinesterase Inhibitors: Role of α4 and α7 Receptors in Neuroprotection. J. Mol. Neurosci. 2009, 40, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Fuller, S.J.; Martins, R.N. The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 2015, 84, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Kan, M.J.; Lee, J.E.; Wilson, J.G.; Everhart, A.L.; Brown, C.M.; Hoofnagle, A.N.; Jansen, M.; Vitek, M.P.; Gunn, M.D.; Colton, C.A. Arginine Deprivation and Immune Suppression in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 5969–5982. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Więckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef] [PubMed]
- Dias, K.S.T.; Viegas, C., Jr. Multi-Target Directed Drugs: A Modern Approach for Design of New Drugs for the treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Agis-Torres, A.; Sölhuber, M.; Fernandez, M.; Sanchez-Montero, J.M. Multi-Target-Directed Ligands and other Therapeutic Strategies in the Search of a Real Solution for Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Calza, L.; Antonio Baldassarro, V.; Giuliani, A.; Lorenzini, L.; Fernandez, M.; Mangano, C.; Sivilia, S.; Alessandri, M.; Gusciglio, M.; Torricella, R.; et al. From the Multifactorial Nature of Alzheimer’s Disease to Multitarget Therapy: The Contribution of the Translational Approach. Curr. Top. Med. Chem. 2013, 13, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, M.; Mendes, E.; Perry, M.; Francisco, A.; Marco-Contelles, J. The Multifactorial Nature of Alzheimer’s Disease for Developing Potential Therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- León, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef] [PubMed]
- Sola, I.; Aso, E.; Frattini, D.; López-González, I.; Espargaró, A.; Sabaté, R.; Di Pietro, O.; Luque, F.J.; Clos, M.V.; Ferrer, I.; et al. Novel Levetiracetam Derivatives That Are Effective against the Alzheimer-like Phenotype in Mice: Synthesis, in Vitro, ex Vivo, and in Vivo Efficacy Studies. J. Med. Chem. 2015, 58, 6018–6032. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Brus, B.; Coquelle, N.; Sosič, I.; Šink, R.; Brazzolotto, X.; Mravljak, J.; Colletier, J.P.; Gobec, S. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 2015, 23. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Catto, M.; Tasso, B.; Novelli, F.; Canu, C.; Iusco, G.; Pisani, L.; de Stradis, A.; Denora, N.; Sparatore, A.; et al. Multitarget Therapeutic Leads for Alzheimer’s Disease: Quinolizidinyl Derivatives of Bi- and Tricyclic Systems as Dual Inhibitors of Cholinesterases and β-Amyloid (Aβ) Aggregation. ChemMedChem 2015, 10, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Farina, R.; Pisani, L.; Catto, M.; Nicolotti, O.; Gadaleta, D.; Denora, N.; Soto-Otero, R.; Mendez-Alvarez, E.; Passos, C.S.; Muncipinto, G.; et al. Structure-Based Design and Optimization of Multitarget-Directed 2 H -Chromen-2-one Derivatives as Potent Inhibitors of Monoamine Oxidase B and Cholinesterases. J. Med. Chem. 2015, 58, 5561–5578. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.S.; Wang, X.; Jiang, N.; Yu, W.; Wang, K.D.G.; Lan, J.S.; Li, Z.R.; Kong, L.Y. Multi-target tacrine-coumarin hybrids: Cholinesterase and monoamine oxidase B inhibition properties against Alzheimer’s disease. Eur. J. Med. Chem. 2015, 95, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Bolea, I.; Juárez-Jiménez, J.; de los Rı́os, C.; Chioua, M.; Pouplana, R.; Luque, F.J.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, Biological Evaluation, and Molecular Modeling of Donepezil and N-[(5-(Benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine Hybrids as New Multipotent Cholinesterase/Monoamine Oxidase Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2011, 54, 8251–8270. [Google Scholar] [PubMed]
- Pau, A.; Catto, M.; Pinna, G.; Frau, S.; Murineddu, G.; Asproni, B.; Curzu, M.M.; Pisani, L.; Leonetti, F.; Loza, M.I.; et al. Multitarget-directed tricyclic pyridazinones as G protein-coupled receptor ligands and cholinesterase inhibitors. ChemMedChem 2015, 10, 1054–1070. [Google Scholar] [CrossRef] [PubMed]
- Rochais, C.; Lecoutey, C.; Gaven, F.; Giannoni, P.; Hamidouche, K.; Hedou, D.; Dubost, E.; Genest, D.; Yahiaoui, S.; Freret, T.; et al. Novel Multitarget-Directed Ligands (MTDLs) with Acetylcholinesterase (AChE) Inhibitory and Serotonergic Subtype 4 Receptor (5-HT4R) Agonist Activities As Potential Agents against Alzheimer’s Disease: The Design of Donecopride. J. Med. Chem. 2015, 58, 3172–3187. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.S.; Lan, J.S.; Wang, X.B.; Jiang, N.; Dong, G.; Li, Z.R.; Wang, K.D.G.; Guo, P.P.; Kong, L.Y. Multifunctional tacrine-trolox hybrids for the treatment of Alzheimer’s disease with cholinergic, antioxidant, neuroprotective and hepatoprotective properties. Eur. J. Med. Chem. 2015, 93, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Ignasik, M.; Bajda, M.; Guzior, N.; Prinz, M.; Holzgrabe, U.; Malawska, B. Design, synthesis and evaluation of novel 2-(aminoalkyl)-isoindoline-1,3-dione derivatives as dual-binding site acetylcholinesterase inhibitors. Arch. Pharm. 2012, 345, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Bajda, M.; Skrok, M.; Kurpiewska, K.; Lewiński, K.; Brus, B.; Pišlar, A.; Kos, J.; Gobec, S.; Malawska, B. Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and β-amyloid aggregation inhibitors with neuroprotective properties. Eur. J. Med. Chem. 2015, 92, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Bajda, M.; Rakoczy, J.; Brus, B.; Gobec, S.; Malawska, B. Isoindoline-1,3-dione derivatives targeting cholinesterases: Design, synthesis and biological evaluation of potential anti-Alzheimer’s agents. Bioorg. Med. Chem. 2015, 23, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Szałaj, N.; Bajda, M.; Dudek, K.; Brus, B.; Gobec, S.; Malawska, B. Multiple Ligands Targeting Cholinesterases and b-Amyloid: Synthesis, Biological Evaluation of Heterodimeric Compounds with Benzylamine Pharmacophore. Arch. Pharm. Chem. Life Sci. 2015, 348, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Więckowska, A.; Więckowski, K.; Bajda, M.; Brus, B.; Sałat, K.; Czerwińska, P.; Gobec, S.; Filipek, B.; Malawska, B. Synthesis of new N-benzylpiperidine derivatives as cholinesterase inhibitors with β-amyloid anti-aggregation properties and beneficial effects on memory in vivo. Bioorg. Med. Chem. 2015, 23, 2445–2457. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-β peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, X.; Cheng, B.; Huang, K. How our bodies fight amyloidosis: Effects of physiological factors on pathogenic aggregation of amyloidogenic proteins. Arch. Biochem. Biophys. 2015, 568, 46–55. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H. Biotin-avidin interaction-based screening assay for Alzheimer’s beta-peptide oligomer inhibitors. Anal. Biochem. 2006, 356, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Bajda, M.; Więckowska, A.; Hebda, M.; Guzior, N.; Sotriffer, C.A.; Malawska, B. Structure-based search for new inhibitors of cholinesterases. Int. J. Mol. Sci. 2013, 14, 5608–5632. [Google Scholar] [CrossRef] [PubMed]
- Sopkova-de Oliveira Santos, J.; Lesnard, A.; Agondanou, J.-H.; Dupont, N.; Godard, A.-M.; Stiebing, S.; Rochais, C.; Fabis, F.; Dallemagne, P.; Bureau, R.; et al. Virtual Screening Discovery of New Acetylcholinesterase Inhibitors Issued from CERMN Chemical Library. J. Chem. Inf. Model. 2010, 50, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood–brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Apelt, J.; Ligneau, X.; Pertz, H.H.; Arrang, J.M.; Ganellin, C.R.; Schwartz, J.C.; Schunack, W.; Stark, H. Development of a new class of nonimidazole histamine H3 receptor ligands with combined inhibitory histamine N-methyltransferase activity. J. Med. Chem. 2002, 45, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.J.; Gunn, B.; Harms, C.S.; Kline, A.D.; Mullaney, J.T.; Nunes, A.; Scarborough, R.M.; Arfsten, A.E.; Skelton, M.A.; Um, S.L.; et al. Non-peptide RGD surrogates which mimic a Gly-Asp β-turn: Potent antagonists of platelet glycoprotein IIb-IIIa. J. Med. Chem. 1997, 40, 2085–2101. [Google Scholar] [CrossRef] [PubMed]
- Dehaen, W.; Hassner, A. Cycloadditions. 45. Annulation of heterocycles via intramolecular nitrile oxide-heterocycle cycloaddition reaction. J. Org. Chem. 1991, 56, 896–900. [Google Scholar] [CrossRef]
- Plocki, S.; Aoun, D.; Ahamada-Himidi, A.; Tavarès-Camarinha, F.; Dong, C.-Z.; Massicot, F.; Huet, J.; Adolphe-Pierre, S.; Chau, F.; Godfroid, J.-J.; et al. Molecular Modeling, Design, and Synthesis of Less Lipophilic Derivatives of 3-(4-Tetradecyloxybenzyl)-4H-1,2,4-oxadiazol-5-one (PMS1062) Specific for Group II Enzyme. Eur. J. Org. Chem. 2005, 2005, 2747–2757. [Google Scholar] [CrossRef]
- Choi, J.Y.; Seo, H.N.; Lee, M.J.; Park, S.J.; Park, S.J.; Jeon, J.Y.; Kang, J.H.; Pae, A.N.; Rhim, H.; Lee, J.Y. Synthesis and biological evaluation of novel T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2007, 17, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Dickson, J.K. Jr.; Hodge, C.N.; Mendoza, J.S.; Chen, K. 2-Amido-Thiazole-based Compounds Exhibiting ATP-Utilizing Enzyme Inhibitory Activity, and Compositions, and Uses Thereof. U.S. Patent No 7,410,988, 12 August 2008. [Google Scholar]
- Alisi, M.A.; Brufani, M.; Cesta, M.C.; Filocamo, L.; Gostoli, G.; Lappa, S.; Pagella, P.G.; Ferrari, E.; Maiorana, S.; Marchesini, D. Mediolanium Farmaceutucu S.P.A. Milan, Italy, Aminoalkylcarbamic Esters of Eseroline Suitable for Use As Chlorinesterase Activity Inhibitors. U.S. Patent No 5,302,593, 12 April 1994. [Google Scholar]
- Mokrosz, J.L.; Dereń-Wesołek, A.; Tatarczyńska, E.; Duszyńska, B.; Bojarski, A.J.; Mokrosz, M.J.; Chojnacka-Wójcik, E. 8-[4-[2-(1,2,3,4-Tetrahydroisoquinolinyl)]butyl]-8-azaspiro[4.5]decane-7,9-dione: A New 5-HT1A Receptor Ligand with the Same Activity Profile as Buspirone. J. Med. Chem. 1996, 39, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.; Mitka, K.; Jaśkowska, J.; Duszyńska, B.; Bojarski, A.J. New Arylpiperazines with Flexible versus Partly Constrained Linker as Serotonin 5-HT1A/5-HT7 Receptor Ligands. Arch. Pharm. (Weinheim). 2013, 346, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Tsotinis, A.; Afroudakis, P.A.; Davidson, K.; Prashar, A.; Sugden, D. Design, Synthesis, and Melatoninergic Activity of New Azido- and Isothiocyanato-Substituted Indoles. J. Med. Chem. 2007, 50, 6436–6440. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.M.; Rival, Y.M.; Chayer, S.; Bourguignon, J.J.; Wermuth, C.G. Aminopyridazines as acetylcholinesterase inhibitors. J. Med. Chem. 1999, 42, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Commerford, J.D.; Donahue, H.B. Notes -N-(-Bromoalkyl)saccharins and N,N’-Undecamethylenedisaccharin. J. Org. Chem. 1956, 21, 583–584. [Google Scholar] [CrossRef]
- Meng, F.C.; Mao, F.; Shan, W.J.; Qin, F.; Huang, L.; Li, X.S. Design, synthesis, and evaluation of indanone derivatives as acetylcholinesterase inhibitors and metal-chelating agents. Bioorg. Med. Chem. Lett. 2012, 22, 4462–4466. [Google Scholar] [CrossRef] [PubMed]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A.; et al. Quinolizidinyl derivatives of bi- and tricyclic systems as potent inhibitors of acetyl- and butyrylcholinesterase with potential in Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef] [PubMed]
- Shaik, J.B.; Palaka, B.K.; Penumala, M.; Kotapati, K.V.; Devineni, S.R.; Eadlapalli, S.; Darla, M.M.; Ampasala, D.R.; Vadde, R.; Amooru, G.D. Synthesis, pharmacological assessment, molecular modeling and in silico studies of fused tricyclic coumarin derivatives as a new family of multifunctional anti-Alzheimer agents. Eur. J. Med. Chem. 2016, 107, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the selected compounds (39–44) are available from the authors.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compd | R | n | EeAChE IC50 [µM] a | EqBuChE IC50 [µM] a | Aβ1–42 Aggreg. b |
|---|---|---|---|---|---|
| 1 |  | 3 | 0.781 ± 0.009 | >50 c | - |
| 2 | 4 | 0.643 ± 0.006 | >50 c | - | |
| 3 | 5 | 0.276 ± 0.003 | >50 c | - | |
| 4 | 6 | 0.282 ± 0.011 | 3.241 ± 0.141 | 15.17 ± 3.06 | |
| 5 |  | 3 | 18.117 ± 0.402 | >50 c | - |
| 6 | 4 | 17.249 ± 0.474 | >50 c | - | |
| 7 | 5 | 4.114 ± 0.066 | >50 c | 17.06 ± 6.13 | |
| 8 | 6 | 4.503 ± 0.064 | >50 c | - | |
| 9 |  | 2 | 5.596 ± 0.087 | 12.818 ± 0.107 | - |
| 10 | 3 | 1.266 ± 0.011 | 5.314 ± 0.160 | - | |
| 11 | 4 | 0.387 ± 0.003 | 5.124 ± 0.090 | - | |
| 12 | 5 | 0.688 ± 0.017 | 1.169 ± 0.027 | - | |
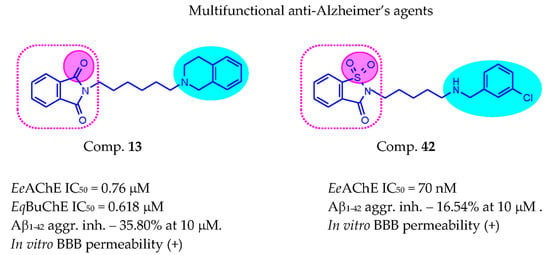
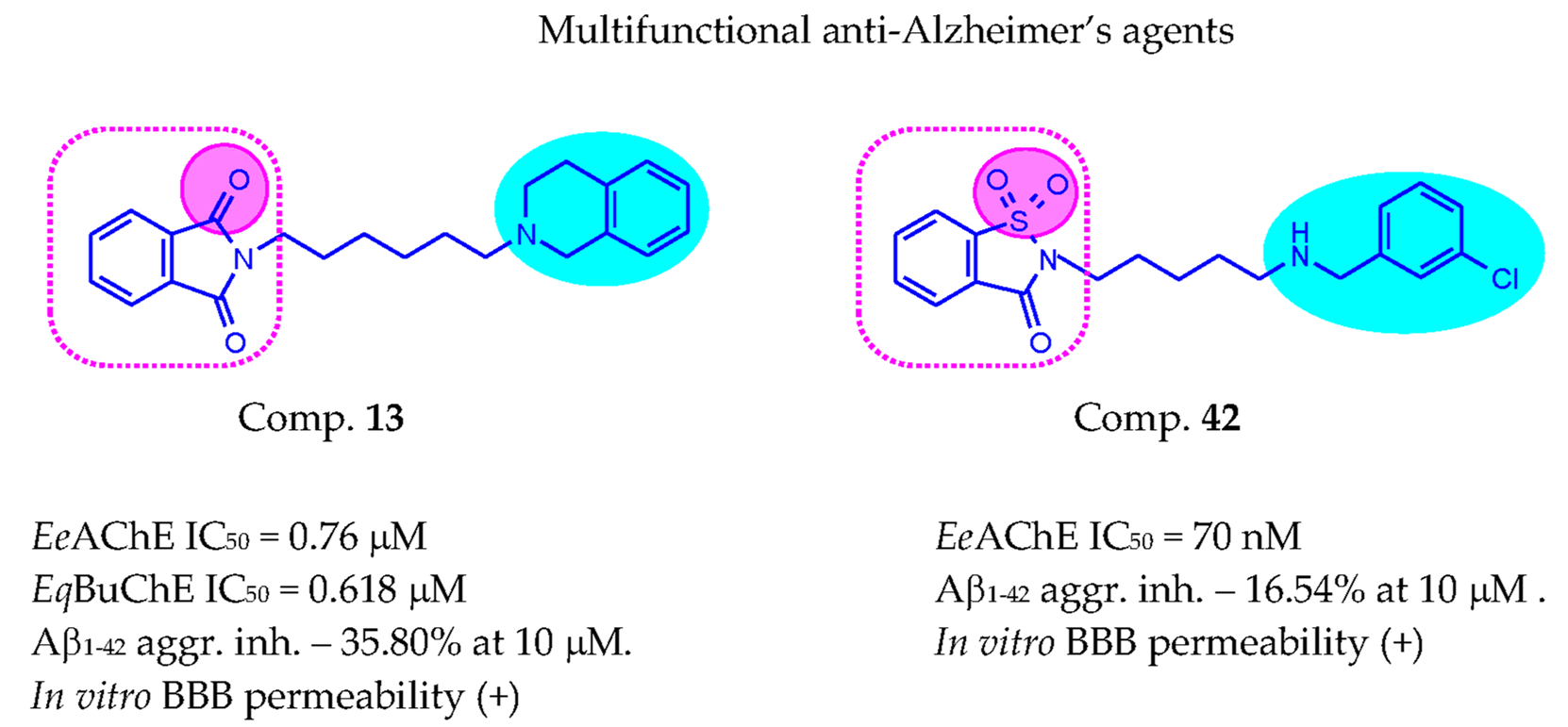
| 13 | 6 | 0.760 ± 0.017 | 0.618 ± 0.009 | 35.80 ± 5.39 | |
| 14 | 8 | >50 c | 1.727 ± 0.038 | - | |
| 15 | 10 | >50 c | 3.662 ± 0.085 | - | |
| 20 |  | 3 | >50 c | >50 c | - |
| 21 | 4 | >50 c | >50 c | - | |
| 22 | 5 | 17.303 ± 0.401 | 37.129 ± 0.878 | - | |
| 23 | 6 | 4.722 ± 0.149 | 14.002 ± 0.343 | <10 d | |
| 30 | | 1 | >50 c | >50 c | - |
| 31 | 2 | >50 c | 25.995 ± 0.813 | - | |
| 32 | 3 | >50 c | 22.571 ± 0.602 | - | |
| 33 | 4 | >50 c | 28.733 ± 0.723 | - | |
| 34 | 5 | 22.185 ± 0.424 | 1.472 ± 0.593 | - | |
| 35 | 6 | 9.557 ± 0.289 | 3.471 ± 0.100 | 16.64 ± 2.74 | |
| 39 |  | 3 | 0.150 ± 0.003 | >50 c | 12.27 ± 2.63 |
| 40 | 4 | 1.145 ± 0.015 | >50 c | - | |
| 41 | 5 | 0.735 ± 0.009 | 4.971 ± 0.127 | - | |
| 42 |  | 3 | 0.070 ± 0.001 | >50 c | 16.54 ± 3.01 |
| 43 | 4 | 0.596 ± 0.006 | 6.535 ± 0.163 | - | |
| 44 | 5 | 0.923 ± 0.012 | 2.459 ± 0.032 | - | |
| Compd. I e | 0.900 ± 0.004 | >50 c | 30.10 ± 5.78 f | ||
| Compd. II e | 0.036 ± 0.001 | >50 c | 22.19 ± 16.6 | ||
| Donepezil | 0.010 ± 0.001 | 1.830 ± 0.176 | 11.48 ± 4.51 | ||
| Tacrine | 0.024 ± 0.001 | 0.002 ± 0.001 | - |
| Compound | Log Pe a | Prediction |
|---|---|---|
| Verapamil | –3.9 | CNS+ |
| Lidocaine | –4.3 | CNS+ |
| Quinidine | –4.0 | CNS+ |
| Progesterone | –3.8 | CNS+ |
| Propranolol | –3.7 | CNS+ |
| Corticosterone | –4.5 | CNS± |
| Theophylline | –6.3 | CNS– |
| 4 | –4.0 | CNS+ |
| 7 | –4.2 | CNS+ |
| 13 | –4.2 | CNS+ |
| 39 | –4.0 | CNS+ |
| 42 | –4.3 | CNS+ |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hebda, M.; Bajda, M.; Więckowska, A.; Szałaj, N.; Pasieka, A.; Panek, D.; Godyń, J.; Wichur, T.; Knez, D.; Gobec, S.; et al. Synthesis, Molecular Modelling and Biological Evaluation of Novel Heterodimeric, Multiple Ligands Targeting Cholinesterases and Amyloid Beta. Molecules 2016, 21, 410. https://doi.org/10.3390/molecules21040410
Hebda M, Bajda M, Więckowska A, Szałaj N, Pasieka A, Panek D, Godyń J, Wichur T, Knez D, Gobec S, et al. Synthesis, Molecular Modelling and Biological Evaluation of Novel Heterodimeric, Multiple Ligands Targeting Cholinesterases and Amyloid Beta. Molecules. 2016; 21(4):410. https://doi.org/10.3390/molecules21040410
Chicago/Turabian StyleHebda, Michalina, Marek Bajda, Anna Więckowska, Natalia Szałaj, Anna Pasieka, Dawid Panek, Justyna Godyń, Tomasz Wichur, Damijan Knez, Stanislav Gobec, and et al. 2016. "Synthesis, Molecular Modelling and Biological Evaluation of Novel Heterodimeric, Multiple Ligands Targeting Cholinesterases and Amyloid Beta" Molecules 21, no. 4: 410. https://doi.org/10.3390/molecules21040410