Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis †



1
Department of Drug Sciences, University of Catania, V.le A. Doria, 95125 Catania, Italy
2
Department of Chemical Sciences, University of Catania, V.le A. Doria, 95125 Catania, Italy
3
Institute of Pharmaceutical Science, King’s College London, Stamford Street, London SE1 9NH, UK
4
King’s Forensics, School of Population Health & Environmental Sciences, King’s College London, Franklin-Wilkins Building, 150 Stamford Street, London SE1 9NH, UK
*
Authors to whom correspondence should be addressed.
†
In memory of Professor Carmela Spatafora, a friend, colleague and distinguished scientist, on the second anniversary of her premature death.
Molecules 2018, 23(9), 2183; https://doi.org/10.3390/molecules23092183
Submission received: 23 August 2018
/
Revised: 26 August 2018
/
Accepted: 28 August 2018
/
Published: 30 August 2018
(This article belongs to the Special Issue QSAR and QSPR: Recent Developments and Applications)
Abstract
:Two 3D quantitative structure–activity relationships (3D-QSAR) models for predicting Cannabinoid receptor 1 and 2 (CB1 and CB2) ligands have been produced by way of creating a practical tool for the drug-design and optimization of CB1 and CB2 ligands. A set of 312 molecules have been used to build the model for the CB1 receptor, and a set of 187 molecules for the CB2 receptor. All of the molecules were recovered from the literature among those possessing measured Ki values, and Forge was used as software. The present model shows high and robust predictive potential, confirmed by the quality of the statistical analysis, and an adequate descriptive capability. A visual understanding of the hydrophobic, electrostatic, and shaping features highlighting the principal interactions for the CB1 and CB2 ligands was achieved with the construction of 3D maps. The predictive capabilities of the model were then used for a scaffold-hopping study of two selected compounds, with the generation of a library of new compounds with high affinity for the two receptors. Herein, we report two new 3D-QSAR models that comprehend a large number of chemically different CB1 and CB2 ligands and well account for the individual ligand affinities. These features will facilitate the recognition of new potent and selective molecules for CB1 and CB2 receptors.
1. Introduction
The receptors of the endocannabinoid system are the cannabinoid receptors. In the human body, this system is involved in different physiological processes, including memory, mood, pain-sensation, and appetite [1]. The cannabinoid receptors are part of the G protein-coupled receptor (GPCR) family, with two known subtypes: CB1 and CB2 receptors, both expressed in the membrane of the cells [2]. Since the discovery of the ∆9-tetrahydrocannabinol (∆9-THC), the main psychoactive component of Cannabis sativa, these receptors have been studied for their implication in different pathological and physiological conditions [3,4,5,6,7]. Interestingly, the type1 cannabinoid receptor is the most expressed GPCR in the human central and peripheral nervous system and is also expressed throughout the body [8]. CB1 selective agonists have shown therapeutic potential in a wide range of disorders, including multiple sclerosis, pain, inflammation, and neurodegenerative disorders [9,10,11]. On the other hand, such ligands also possess the potential of being abused for recreational purposes, and can give rise to mild to severe psychotropic effects. Indeed, in the last decade, a large number of synthetic cannabinoid receptor ligands have appeared on the recreational drug scene [12]. Conversely, the CB2 receptors distribution is more limited through the body, particularly high is their expression in the cells of the immune system, and among them with high levels in B lymphocytes and natural killer cells [13]. The exact physiology of CB2 receptors is still not completely understood but a number of preclinical studies support the advantage of using CB2 ligands for the treatment of different pathophysiological disorders, such as chronic pain, maintenance of bone density, reducing the progression of atherosclerotic lesions, asthma, autoimmune and inflammatory diseases, and multiple sclerosis [14,15]. The distribution of CB2 in the central nervous system is limited, therefore CB2 selective ligands do not possess significant psychoactive properties. Thus, discovering potent and selective ligands for either CB1 or CB2 represents an important—and yet only partially met—research goal.
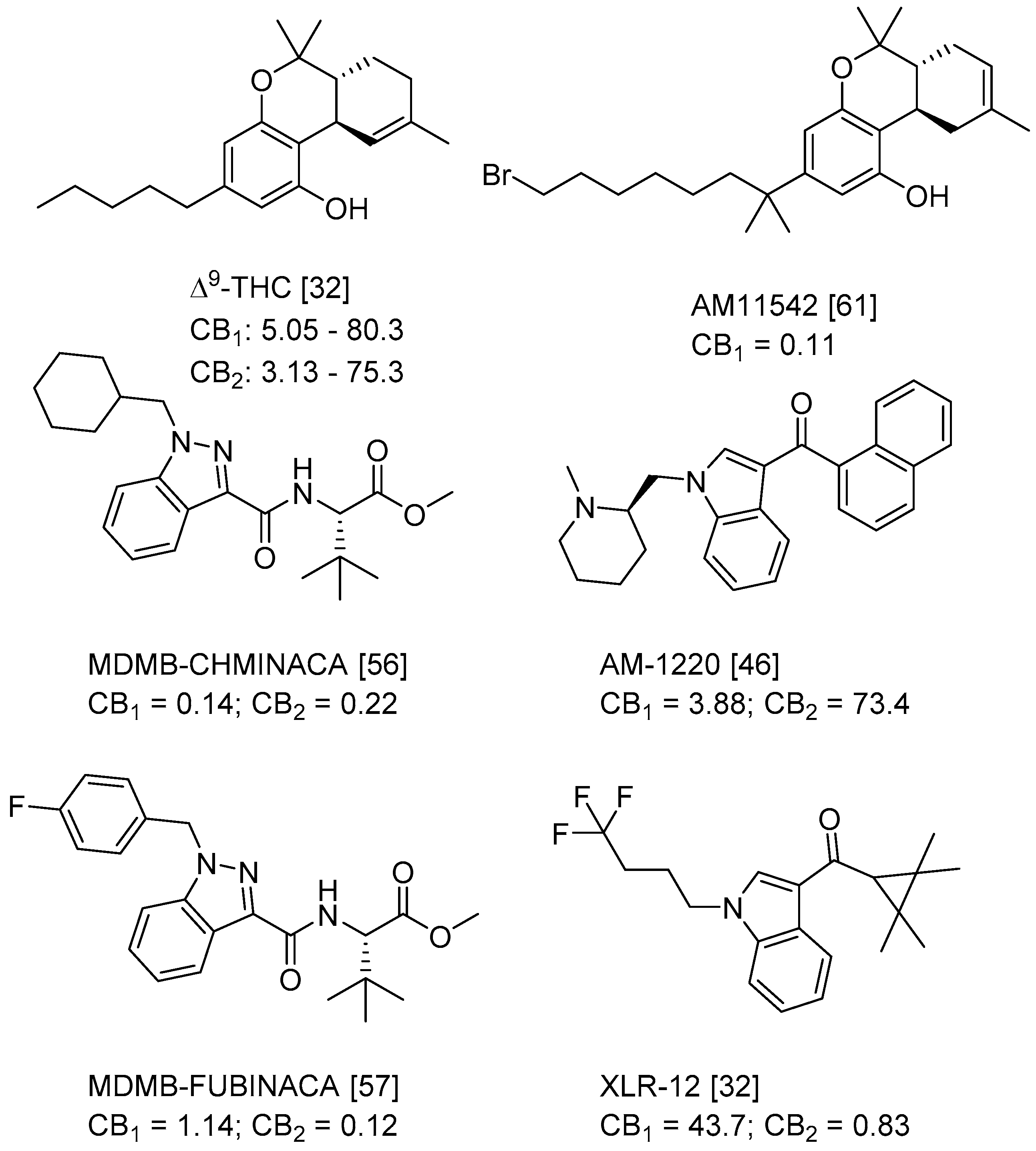
QSAR models are used to help predicting or understanding patterns in the chemical, pharmacological/pharmaceutical and biological sciences [16,17,18,19]. There have been several attempts to build QSAR models for the CB1 and CB2 receptors, but all of them have been produced only using a restricted number of compounds, and only using compounds with similar structure, resulting in good models for restricted classes of cannabinoid receptors binders [20,21,22,23,24,25]. To facilitate the research and investigation among chemical datasets for CB1 and CB2 ligands capabilities, selectivity, and potency, herein we report the development of two different 3D-QSAR models. The models were created using a set of 312 CB1 receptor ligands, and 187 CB2 receptor ligands. Details of all the compounds having experimentally determined Ki values were retrieved from the literature (i.e., anandamide analogues, benzoyl/alkoyl-indoles, cyclohexylphenols, dibenzopyrans, indazole derivatives, indazole-carboxylates, indazole-carboxamides, indole-carboxylates, indole-carboxamides, naphthoyl-benzimidazoles, naphthoyl-indazoles, naphthoyl-indoles, naphthoyl-naphthalenes, naphthoyl-pyrroles and phenylacetyl-indoles) [26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. Figure 1 details the structures of some example of CB1 and CB2 ligands. Among them, the natural derived ∆9-THC; AM11542, which is the unique tetrahydrocannabinol derivative co-crystallized with the CB1 receptor; the two most potent CB1 and CB2 agonists (MDMB-CHMINACA and MDMB-FUBINACA), and the two most selective compounds for the CB1 and CB2 receptor (AM-1220 and XLR-12).
Even if there is an interest of the researchers in using the two cannabinoids receptors as pharmacological targets, the majority of the cannabinoids ligands nowadays are known because they are the most common substances used as new psychoactive drugs of abuse; indeed, an elaborate variety of synthetic cannabinoids ligands are designed in an attempt to avoid the legal restrictions on cannabis and spread into the black market (e.g., MDMB-CHMINACA and MDMB-FUBINACA can be defined as synthetic cannabinoids belonging to the a recently emerged superfamily of “New Psychoactive Substances” (NPS) [12]).
Unlike previously published models, the one reported here includes a wider range of chemically different compound (sub)classes. Moreover, the 3D-QSAR models generated, ushered by the scaffold-hopping analysis, have been employed to design different new series of potentially high-affinity and selective ligands of the CB1 and CB2 receptors. Forge, and Spark software (Cresset) were used for the development of the 3D-QSAR models and for the scaffold-hopping analysis, respectively [47]. Most of the 3D-QSAR methodologies, like CoMFA or CoMSIA, can calculate the molecular properties at the interception points of this 3D grid only after the surrounding the space (with a 3D grid) of the aligned molecules [48,49]. Differently, the software used by us has the ability to calculate the molecular properties only in particular positions that are defined directly from the field points (describing the electrostatic potential and the volume of each molecule, generated by a force field) of the aligned molecules in the training set, yielding exceptional results [50,51,52].
2. Results and Discussion
2.1. Statistical Analysis and Results
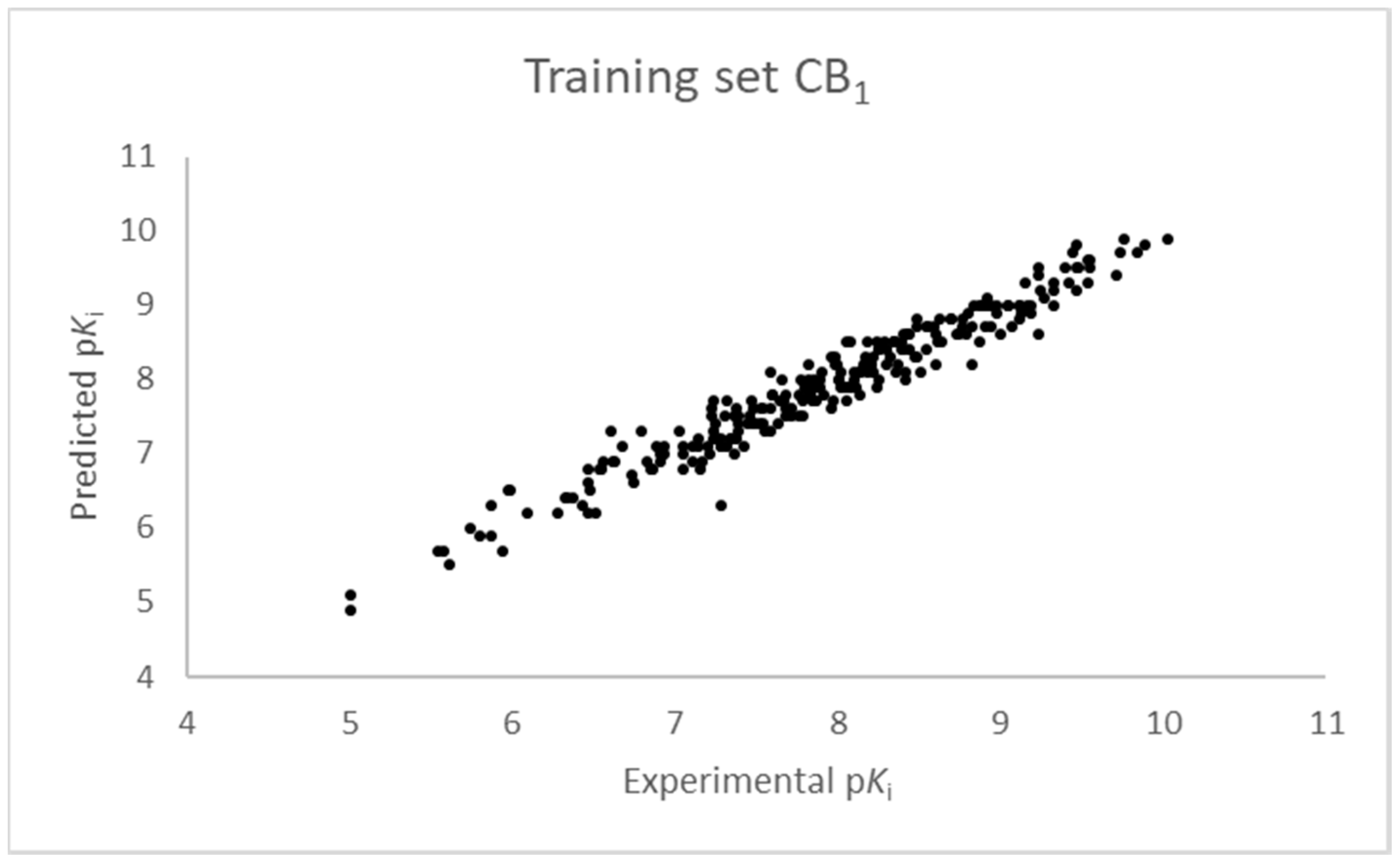
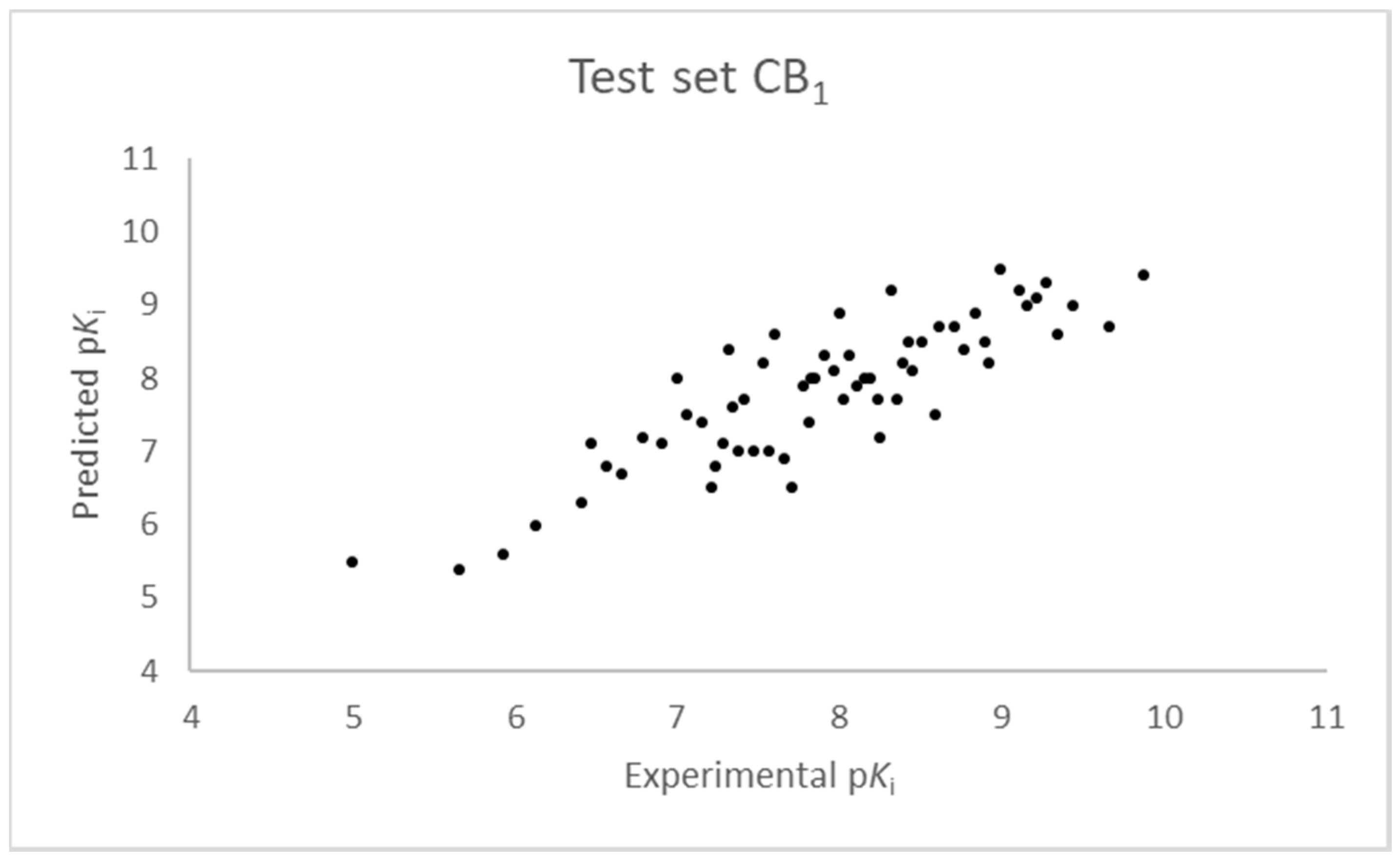
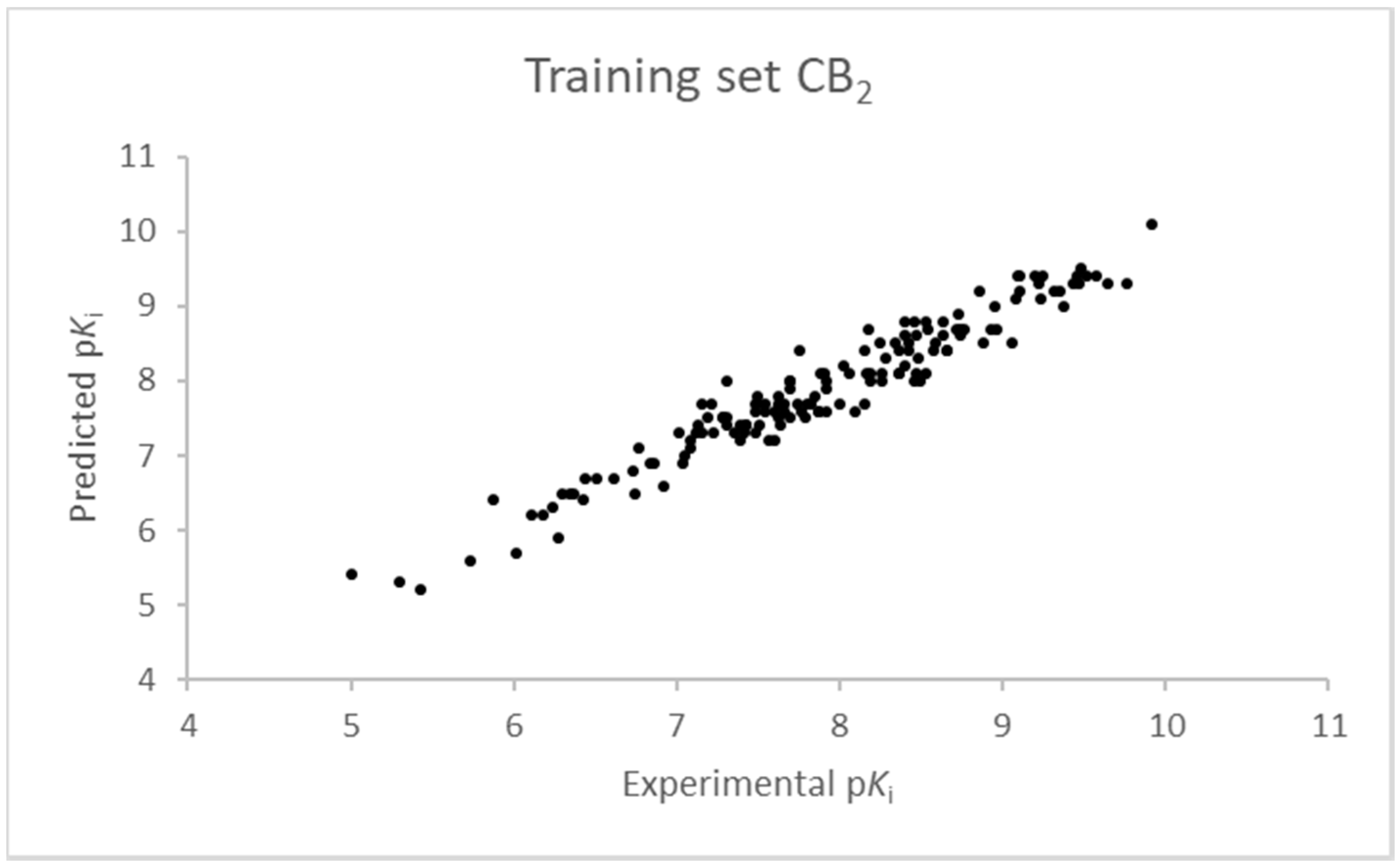
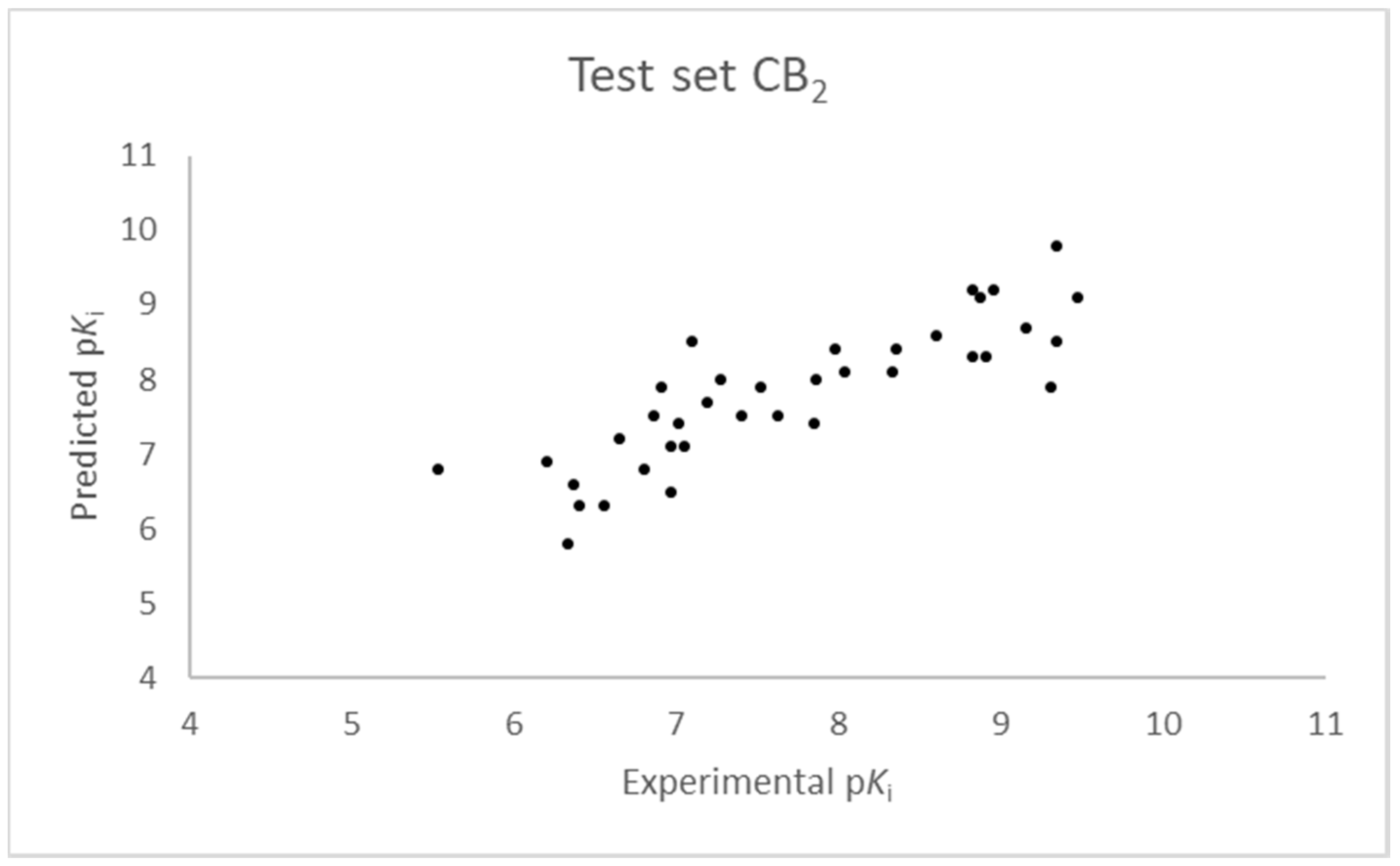
The SIMPLS algorithm (partial least squares regression method) is used by the software for the calculation of the model [53,54]. More information on the methodology used during the build and the validation of the two models are reported in the Supplementary Materials. The results of the 3D-QSAR models are shown in Figure 2, Figure 3, Figure 4 and Figure 5. The 10-component model for the CB1 receptor shows both optimum predictive and descriptive capabilities, demonstrated by the optimum r2 (0.95, training set) and q2 (0.62, cross-validated training set) values (Figure S5, Supplementary Materials) [55]. In parallel, excellent correlations were achieved by the 7-component model for the CB2 receptor, with a r2 of 0.93 and a q2 of 0.72 (Figure S6, Supplementary Materials). An adequate distribution of the experimental vs. predicted affinities values is shown by the graphs in Figure 2 and Figure 4 for the training set and in Figure 3 and Figure 5 for the test set. In this case, only a few outliers were revealed and excellent cross-validated r2 (0.72 and 0.73 for the CB1 and the CB2 receptor, respectively) are calculated.
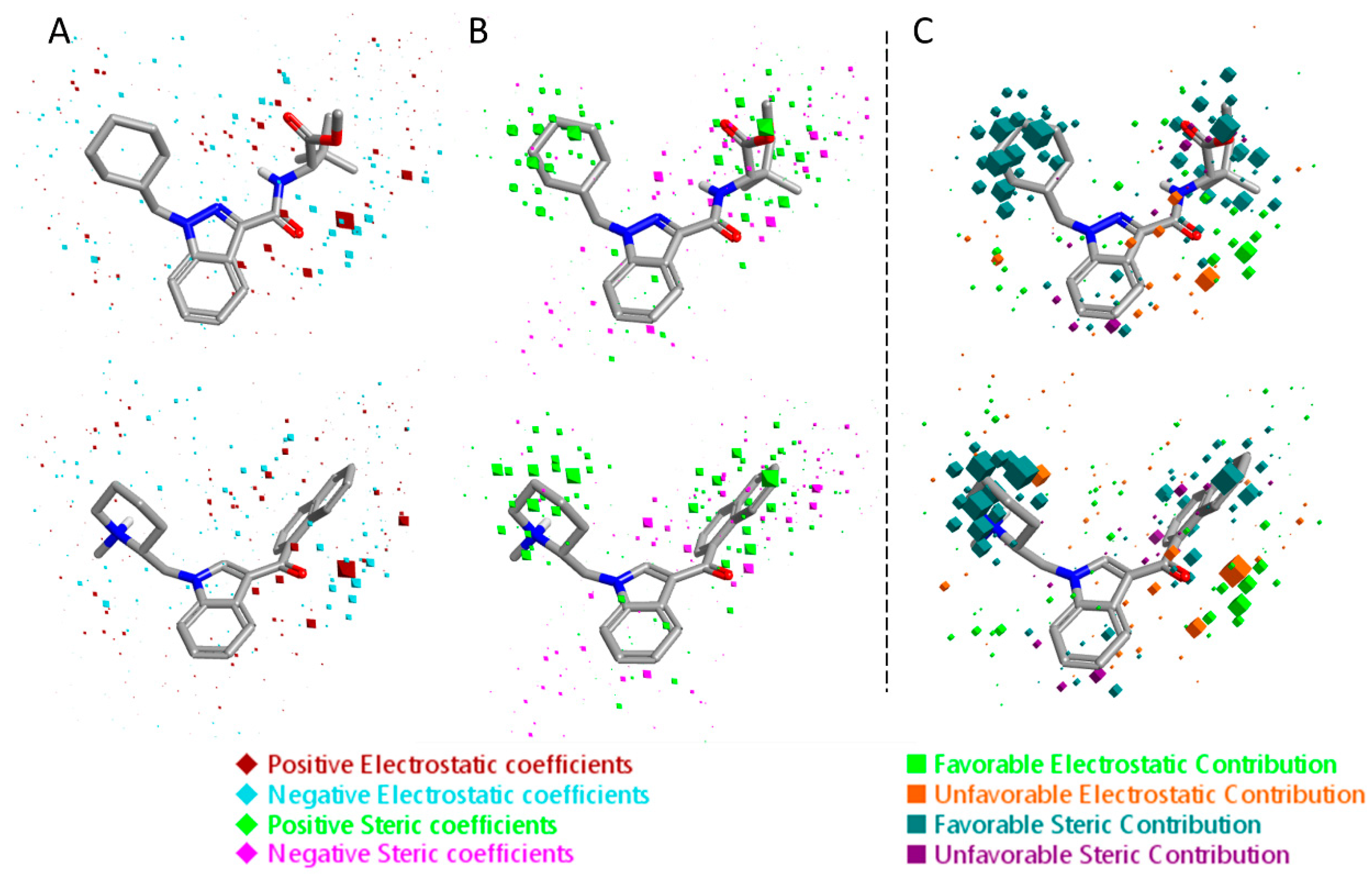
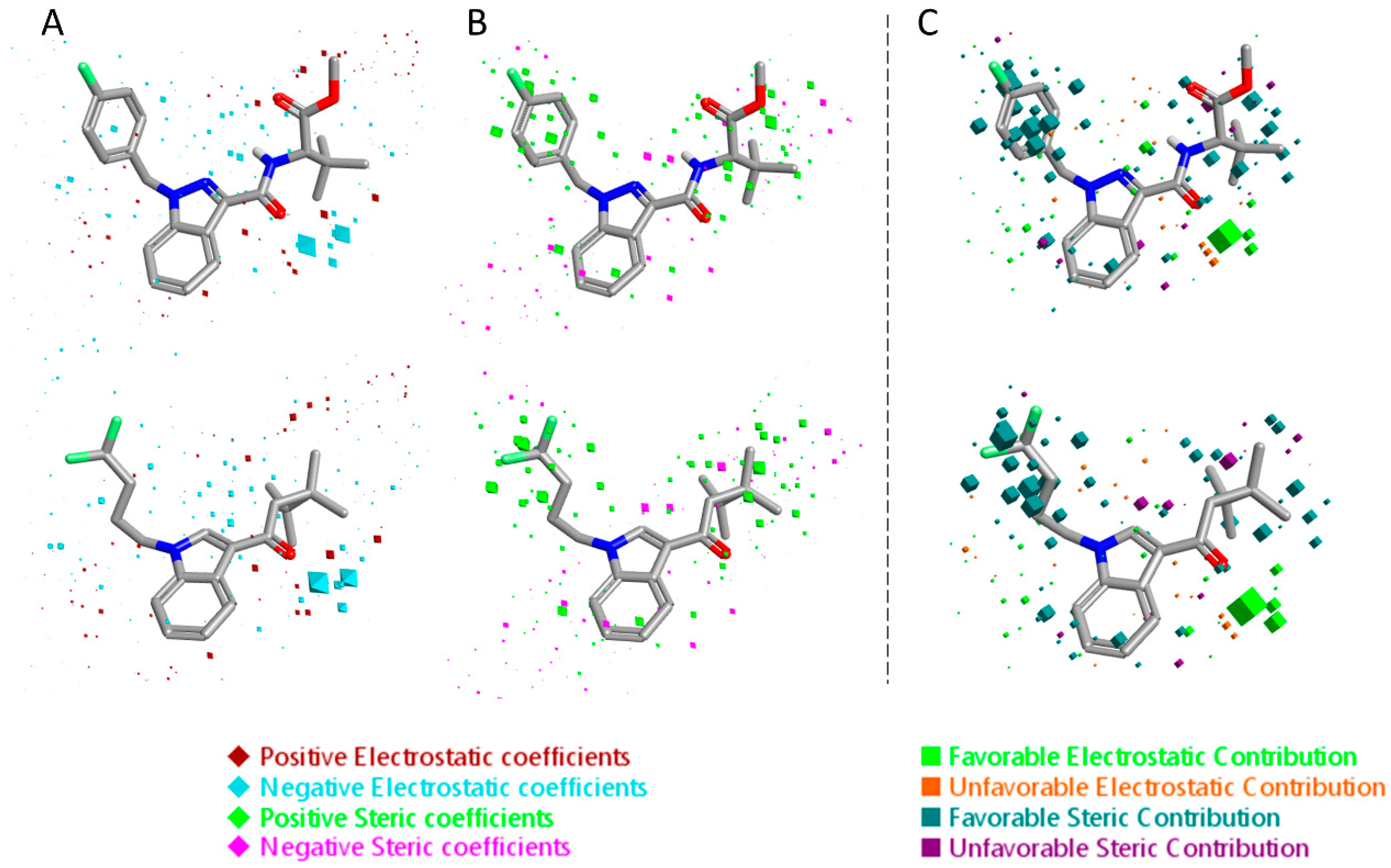
The 3D visualizations of the QSAR models are shown in Figure 6 and Figure 7, where the 3D-QSAR coefficients for the two models are superimposed to the most potent and the most selective compounds for the two cannabinoid receptors. These 3D-QSAR models are characterized by both electrostatic and steric effects, with important differences that encompass the different selectivity of the individual classes of studied compounds toward the CB1 and CB2 receptors. The model overviews show the areas where the QSAR model indicates that the molecular fields have a strong impact on ligand–receptor affinity. The larger the points (depicted as octahedrons), the stronger the correlation between the fields is (electrostatic and steric) in that position. The higher affinity related to the electrostatic potential is depicted in red for the positive values and in blue for the negative ones. For the steric bulk, the green area leads to higher receptor affinity, whereas the violet area leads to lower affinity.
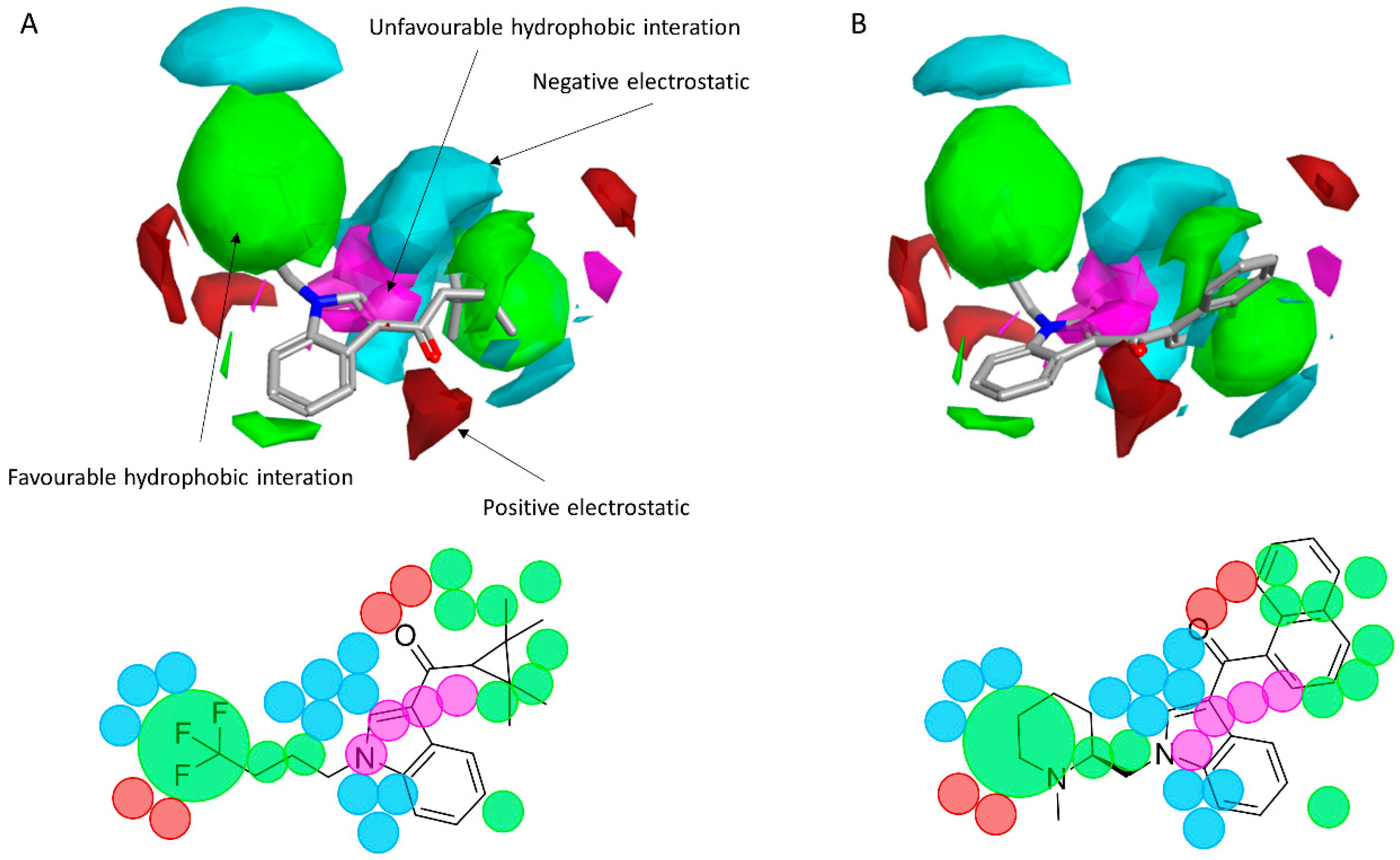
To reveal the key features of the studied set of compounds against the targeted cannabinoid receptors, activity-atlas (AA) was used to perform a structure–activity relationship (SAR) study. AA is a visualization software implemented in Forge that is able to summarize structure–activity data into 3D maps in a qualitative manner through a Bayesian statistics approach. Figure 8 and Figure 9 illustrate the results of the AA calculations for the CB1 and CB2 receptor, respectively. The model map is superimposed to the two most selective CB1 and CB2 receptor ligands, AM-1220 and XLR-12, respectively. The different colors on the maps derived from the AA calculations highlight electrostatic, hydrophobic, and shape features of the different set of compounds. A more positive electrostatic field increases the receptor-affinity in the red region, whereas in the blue area, a more negative electrostatic field increases the affinity. The green and the violet areas account for the steric and bulk/hydrophobic interactions. In the green area, a steric/bulk interaction improves the binding affinity; in the violet area, a steric/bulk interaction decreases the affinity. The two selected molecules (AM-1220 and XLR-12) can be dissected in three different regions: (i) the substituent at the N1 position of the heterocyclic nucleus; (ii) the substituent at the 3-position of the heterocyclic nucleus; (iii) the central core. Such molecular features are typically described nowadays to distinguish most of the novel and emerging synthetic cannabinoids [12]. Each of these moieties possesses a set of information for the CB1 or CB2 affinity/selectivity. Regarding the substitution of the N1 position, both models describe a wide favorable hydrophobic interaction area and in both models a negative electrostatic area is located at the end of the alkyl chain, suggesting that the introduction of electronegative atoms increases the affinity for both CB receptors. The occupancy of this area is fundamental for the affinity in both receptors; indeed, ligands that do not bear any substituent in this area result in low potency (e.g., JWH-042, Ki = 10,000 nM for CB1 and 5050 nM for CB2). On the other hand, this molecular portion appears to be linked with selectivity towards the CB1 receptor. A positive electrostatic region surrounds the initial part of that area, and a favorable interaction seems to be relevant for the selectivity toward the CB1 receptor (e.g., AM-1220). The same positive electrostatic area in the CB2 model doesn’t have any relevant interaction with potent and/or selective compounds for that receptor. For both models, there is not a strong SAR in the central core region, but a small favorable shape/hydrophobic region (green) is located near the positions 5 and 6 of the indole ring for the CB2 receptor, which means that small substituents in this region may increase the selectivity. The 2-position of the heterocyclic nucleus is located near an area in which the hydrophobic interactions are unfavorable. This area is larger for the CB1 model. Thus, in this case, the introduction of a small substituent may be better tolerated by the CB2 receptor. In the 3-position of the heterocyclic nucleus, the two models clearly describe different scenarios. For the CB1 receptor, the substituent in this branch is located within an unfavorable hydrophobic region; for the CB2 receptor, the situation is the opposite. This means that bulkier and hydrophobic substituents will raise the selectivity for the CB2 receptor and small/hydrophilic substituent will raise the selectivity toward the CB1 receptor. Furthermore, the same region in the CB2 model is contoured by a bigger negative electrostatic area, which means that the adding of an electronegative atom should increase the CB2 affinity. Also, it is interesting to note the different distribution of the 3D maps, among the two models, in the areas around the oxygen of the carbonyl group. In the CB1 model the oxygen is located near an area in which positive electrostatic coefficients will raise the affinity; this means that the oxygen atom in that position bears an unfavorable electrostatic contribution to the resultant ligand-receptor affinity. Conversely, the same oxygen in the CB2 model is located in an area where the negative electrostatic coefficient raises the potency; in fact, a relevant favorable electrostatic contribution is described by the model for the oxygen atom of the carbonyl group. Therefore, despite the carbonyl group present in most compounds designed as a CB1 ligand, the selection of a different connecting group might be exploited to improve the affinity for the CB1 receptor.
2.2. Finding Bioisosteres
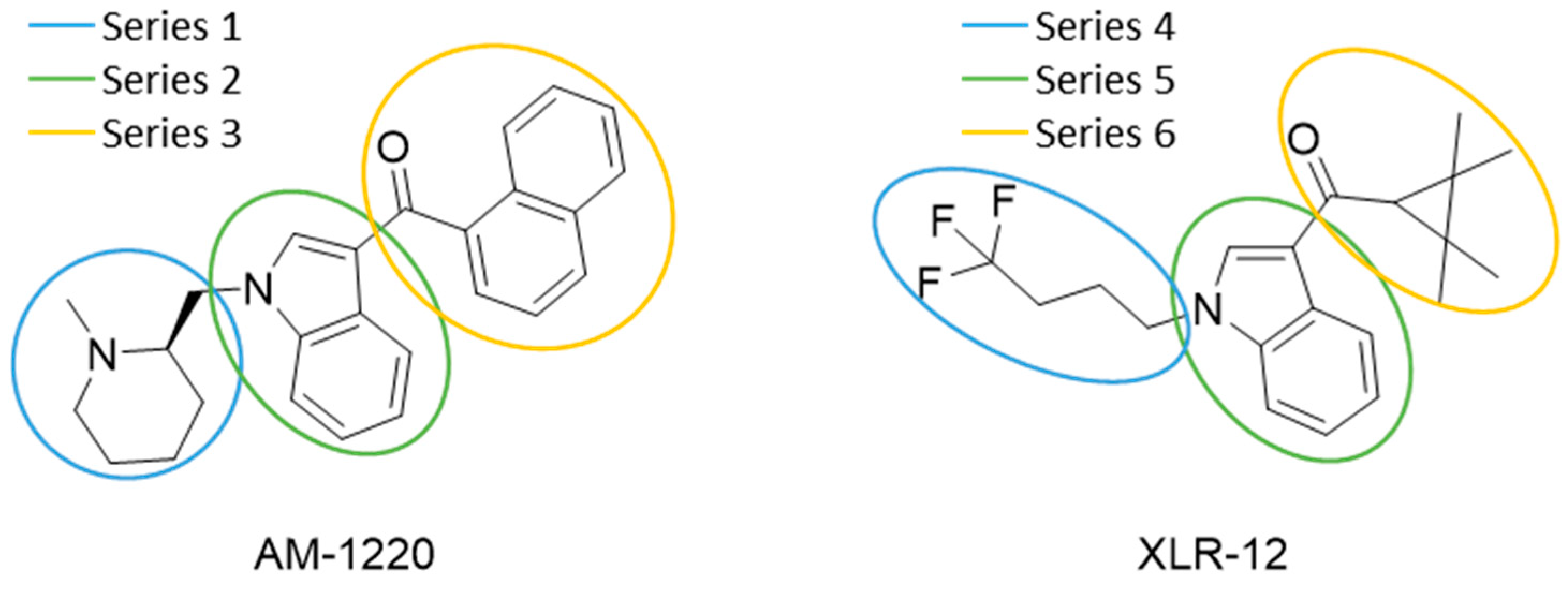
Five hundred compounds were generated for each substitution, as reported in Figure 10, for a total of 3000 analogues, which were then evaluated by the superposition on the 3D-QSAR models. The top-scored compounds, by means of the CB1/CB2 predicted pKi, are reported in Table 1 and Table 2, while the full set of compounds is reported in the Supplementary Materials. Overall, the results indicate that the bioisosteric replacement and the following 3D-QSAR model evaluation generated new structures with the appropriate chemical features for the binding to the CB1 and the CB2 receptors. All the nine groups of the virtually evaluated compounds resulted in a series of molecules more potent than their precursors. The most interesting results for the CB1 receptor were achieved from the molecules belonging to the series 3 (Table 1). The most potent and selective compound for the CB1 receptor is the one in which the substituent at the 3-position of the heterocyclic nucleus was replaced by a 3-sulfonyl-indole. This molecule presents a predicted Ki of 1.3 and 199.5 nM for the CB1 and CB2 receptor, respectively, with a selectivity index (the selectivity index is defined as KiCB2/KiCB1) of 154.5 compared to an index of 18.8 for the original AM-1220 ligand (the higher the selectivity index, the higher the selectivity for CB1 receptor). The increase in selectivity has been achieved with a simple substitution of the group in 3-position to the central core. Further work around this area may provide additional molecular features to further increase selectivity for the CB1 receptor using scaffolds that have not been used yet. Interestingly, none of the compounds derived from the scaffold-hopping of XLR-12 (Table 1, series 4–6) achieve better selectivity upon the CB1 model.
On the other hand, in the case of the molecules virtually evaluated for the CB2 affinity, the most interesting results, in terms of affinity and selectivity, were achieved by four compounds present in series 4 and 6 (Table 2). The two compounds in the series 4 showed a predicted Ki of 12.6 and 15.8 nM for the CB1 receptor, and of 0.063 and 0.079 nM for the CB2 receptor. The calculated selectivity index is 0.005 for both compounds. The two compounds in series 6 both present a predicted Ki of 15.8 nM for the CB1 receptor, and of 0.063 and 0.079 nM for the CB2 receptor, with a calculated selectivity index of 0.004 and 0.005, respectively (the lower the selectivity index the higher the selectivity for CB2 receptor). Considering that the most selective molecule for CB2 receptor in our dataset has a selectivity index of 0.019, we can consider our results promising, and we can conclude that maintaining the central core unchanged, a large increase in the selectivity can be achieved by an appropriate substitution of the substituent at the N1 and at the 3-position of the heterocyclic nucleus.
3. Materials and Methods
3.1. Biological Data
The chemical structures of the 312 CB1 receptor ligands, and of the 187 CB2 receptor ligands were selected from the literature that reports the experimental Ki values, primarily involving assays where isolated CB1 and/or CB2 receptors were incubated with a predetermined fixed concentration of radiolabeled cannabinoid ([3H] CP 55,490) [26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,56,57]. The binding affinity data of the selected dataset were converted into their negative decimal logarithm pKi (pKi = −logKi). Collected pKi values fall into a range 5.00–10.03 and 5.00–9.92 for the CB1 and CB2 receptors, respectively.
3.2. Molecular Modeling
The structures of the studied molecules were built using Marvin Sketch (ChemAxon, Budapest, Hungary) [58]. The 2D structures were subjected to molecular mechanics energy minimization by Merck molecular force field (MMFF94) using Marvin Sketch [58]. A pH of 7.0 was assumed for the calculation of the protonation states of the molecules. The 3D structures derived from the force field minimization were further optimized at semi-empirical level using the parameterized model number 3 (PM3) Hamiltonian using MOPAC package (Stewart Computational Chemistry, Colorado Springs, CO, USA) (vMOPAC2016) as software [59,60,61].
3.3. Compound Alignment
All the 3D molecules, with their experimentally measured pKi values, were imported into the chemistry software Forge (v10.4.2, Cresset, New Cambridge House, UK). Out of the 312 ligands for the CB1 receptor, 250 molecules (80%) were randomly selected as a training set. The remaining 62 compounds (20%) were used as test set for the model quality evaluation. For the CB2 receptor, we used the same procedure: selecting 150 ligands (80%) as training set and 37 compounds (20%) as test set [62]. For both training and test sets, the selected molecules covered a wide range of biological activities. All the molecules were aligned using AM11542 in its bioactive co-crystallized conformation as a template [63]. The field points electrostatic, shape, and hydrophobic field points of each molecule were generated using the extended electron distribution force field, proprietary of the Cresset group. A maximum common substructure algorithm was used to align the full set of molecules, a customized set-up was used (Figure S2, Supplementary Materials) [64]. Five hundred different conformers were generated for each molecule. The similarity between two different conformers was calculated by means of root-mean-square deviation of atomic positions (RMSD), two conformers with values less than 0.5 Å RMSD are considered identical. The gradient cutoff for conformers minimization was set to 0.1 kcal/mol, and the energy window was set to 2.5 kcal/mol. Conformers with calculated energy (kcal/mol) outside the energy window were rejected. All the alignments’ generated poses were manually checked. All the field points of the training set were analyzed by the software to reduce the number of descriptors to be considered. A distance of 1 Å between the sample points was used to calculate the sample values of each compound, in order to efficaciously described all the areas around the analyzed compound. Other information about the conformation calculation, the alignment, and the build of the model are reported in the Supplementary Materials (Figures S1–S6).
3.4. Bioisosteric Replacement
The bioisosteric replacement study was performed with Spark [65]. We studied the bioisosteric replacement in different portions of the two most selective compounds in our dataset for CB1 and CB2 receptors (Figure 1, AM-1220 and XLR-12). In those two molecules, the substitutions at the N1 and at the 3-position of the heterocyclic nucleus, and the substitution of the central core (Figure 10) were analyzed. Once the new virtual compounds were assembled, the new compounds were scored by the superposition on the different 3D-QSAR models, considering that if the fields of the compounds derived from the bioisosteric replacement are very similar to that of the original compounds, the resulting compounds will have similar biological properties [66,67]. The bioisosteric replacement was performed, in all the cases, using 240,068 fragments selected from ChEMBL, Zinc, and VEHICLe databases (See Supplementary Materials, Figure S7) [68,69,70].
4. Conclusions
Two 3D-QSAR models for CB1 and CB2 binders were developed in this study. The models were built and then used as a tool for the evaluation of different sets of compounds derived from a thorough scaffold-hopping analysis, for the design of novel high-affinity and selective molecules with novel scaffold and improved affinity/selectivity within such class of proteins. The QSAR models were built using Forge as a software and a set of 312 molecules for the CB1 receptor and 187 molecules for the CB2 receptor covering the whole range of different chemical classes of the ligands for these proteins. These two models are the first ones that include—within their descriptive capabilities—a wide range of chemically different compounds identified as cannabinoid receptors binders. The visualization of the models allowed to process the statistical data in a pharmacophoric description for both receptor, describing steric and electrostatic effects and rationalizing both potency and selectivity. The information reported in the 3D-QSAR models can ensure fruitful applications to speed up the design and the identification process of new potent CB1 and CB2 receptor ligands. However, since synthetic cannabinoids currently represent the most common substances used as NPS (from 2008 to 2017, a total of 179 new synthetic cannabinoids were reported to the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), and were the largest group of NPS [71]), and an elaborate variety of new synthetic cannabinoids are constantly designed in an attempt to avoid the legal restrictions on cannabis [12,72,73], the findings of this paper should also warn relevant forensic, public health, and legal authorities that new potent and unidentified scaffolds might be on their way to recreational use.
Supplementary Materials
The following are available online. Statistical analysis information for the model, Figures S1–S7, Tables S1–S16.
Author Contributions
G.F. and V.A. conceived the research and wrote the paper. G.F. designed the QSAR model and the scaffold hopping analysis. O.A. created the database for the QSAR model. A.R. and V.A. revised the paper. All authors reviewed the manuscript.
Funding
This research received no external funding.
Acknowledgments
All authors gratefully acknowledge Prof. Robert Hider and Dave Barlow at King’s College London for kindly reviewing this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aizpurua-Olaizola, O.; Elezgarai, I.; Rico-Barrio, I.; Zarandona, I.; Etxebarria, N.; Usobiaga, A. Targeting the endocannabinoid system: Future therapeutic strategies. Drug Discov. Today 2017, 22, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Pharmacology of cannabinoid cb1 and cb2 receptors. Pharmacol. Ther. 1997, 74, 129–180. [Google Scholar] [CrossRef]
- Valdeolivas, S.; Satta, V.; Pertwee, R.G.; Fernandez-Ruiz, J.; Sagredo, O. Sativex-like combination of phytocannabinoids is neuroprotective in malonate-lesioned rats, an inflammatory model of huntington’s disease: Role of cb1 and cb2 receptors. ACS Chem. Neurosci. 2012, 3, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cabrerizo, R.; Garcia-Fuster, M.J. Opposite regulation of scannabinoid cb1 and cb2 receptors in the prefrontal cortex of rats treated with cocaine during adolescence. Neurosci. Lett. 2016, 615, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Clayton, N.; Marshall, F.H.; Bountra, C.; O’Shaughnessy, C.T. Cb1 and cb2 cannabinoid receptors are implicated in inflammatory pain. Pain 2002, 96, 253–260. [Google Scholar] [CrossRef]
- Elmes, S.J.; Winyard, L.A.; Medhurst, S.J.; Clayton, N.M.; Wilson, A.W.; Kendall, D.A.; Chapman, V. Activation of cb1 and cb2 receptors attenuates the induction and maintenance of inflammatory pain in the rat. Pain 2005, 118, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Mule, F.; Amato, A.; Baldassano, S.; Serio, R. Involvement of cb1 and cb2 receptors in the modulation of cholinergic neurotransmission in mouse gastric preparations. Pharmacol. Res. 2007, 56, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Lichtman, A.H. The endogenous cannabinoid system and its role in nociceptive behavior. J. Neurobiol. 2004, 61, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Ruiz, J.; Romero, J.; Ramos, J.A. Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, huntington’s chorea, alzheimer’s disease, and others. Handb. Exp. Pharmacol. 2015, 231, 233–259. [Google Scholar] [PubMed]
- Pryce, G.; Baker, D. Endocannabinoids in multiple sclerosis and amyotrophic lateral sclerosis. Handb. Exp. Pharmacol. 2015, 231, 213–231. [Google Scholar] [PubMed]
- Abbate, V.; Schwenk, M.; Presley Brandon, C.; Uchiyama, N. The ongoing challenge of novel psychoactive drugs of abuse. Part i. Synthetic cannabinoids (iupac technical report). Pure Appl. Chem. 2018, 90, 1255–1282. [Google Scholar] [CrossRef]
- Galiegue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carriere, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulos, S.; Thakur, G.A.; Nikas, S.P.; Makriyannis, A. Cannabinoid receptors as therapeutic targets. Curr. Pharm. Des. 2006, 12, 1751–1769. [Google Scholar] [PubMed]
- Mackie, K. Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Sigma-2 receptor ligands qsar model dataset. Data Brief 2017, 13, 514–535. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistara, V.; Pittala, V.; Prezzavento, O.; Amata, E. Hyphenated 3d-qsar statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017, 139, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Salerno, L.; Romeo, G.; Prezzavento, O.; Pittala, V.; Rescifina, A. Identification of potentially potent heme oxygenase 1 inhibitors through 3d-qsar coupled to scaffold-hopping analysis. ChemMedChem 2018, 13, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Development of a sigma-2 receptor affinity filter through a monte carlo based qsar analysis. Eur. J. Pharm. Sci. 2017, 106, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Mella-Raipan, J.; Hernandez-Pino, S.; Morales-Verdejo, C.; Pessoa-Mahana, D. 3d-qsar/comfa-based structure-affinity/selectivity relationships of aminoalkylindoles in the cannabinoid cb1 and cb2 receptors. Molecules 2014, 19, 2842–2861. [Google Scholar] [CrossRef] [PubMed]
- Mella-Raipan, J.A.; Lagos, C.F.; Recabarren-Gajardo, G.; Espinosa-Bustos, C.; Romero-Parra, J.; Pessoa-Mahana, H.; Iturriaga-Vasquez, P.; Pessoa-Mahana, C.D. Design, synthesis, binding and docking-based 3d-qsar studies of 2-pyridylbenzimidazoles--a new family of high affinity cb1 cannabinoid ligands. Molecules 2013, 18, 3972–4001. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Kapou, A.; Kourouli, T.; Andreou, T.; Nikas, S.P.; Nahmias, V.R.; Papahatjis, D.P.; Papadopoulos, M.G.; Mavromoustakos, T. The application of 3d-qsar studies for novel cannabinoid ligands substituted at the c1’ position of the alkyl side chain on the structural requirements for binding to cannabinoid receptors cb1 and cb2. J. Med. Chem. 2007, 50, 2875–2885. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Z.; Han, X.W.; Liu, Q.; Makriyannis, A.; Wang, J.; Xie, X.Q. 3d-qsar studies of arylpyrazole antagonists of cannabinoid receptor subtypes cb1 and cb2. A combined nmr and comfa approach. J. Med. Chem. 2006, 49, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Salo, O.M.; Savinainen, J.R.; Parkkari, T.; Nevalainen, T.; Lahtela-Kakkonen, M.; Gynther, J.; Laitinen, J.T.; Jarvinen, T.; Poso, A. 3d-qsar studies on cannabinoid cb1 receptor agonists: G-protein activation as biological data. J. Med. Chem. 2006, 49, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Romero-Parra, J.; Chung, H.; Tapia, R.A.; Faundez, M.; Morales-Verdejo, C.; Lorca, M.; Lagos, C.F.; Di Marzo, V.; David Pessoa-Mahana, C.; Mella, J. Combined comfa and comsia 3d-qsar study of benzimidazole and benzothiophene derivatives with selective affinity for the cb2 cannabinoid receptor. Eur. J. Pharm. Sci. 2017, 101, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Aung, M.M.; Griffin, G.; Huffman, J.W.; Wu, M.; Keel, C.; Yang, B.; Showalter, V.M.; Abood, M.E.; Martin, B.R. Influence of the n-1 alkyl chain length of cannabimimetic indoles upon cb(1) and cb(2) receptor binding. Drug Alcohol Depend. 2000, 60, 133–140. [Google Scholar] [CrossRef]
- Wiley, J.L.; Compton, D.R.; Dai, D.; Lainton, J.A.; Phillips, M.; Huffman, J.W.; Martin, B.R. Structure-activity relationships of indole- and pyrrole-derived cannabinoids. J. Pharmacol. Exp. Ther. 1998, 285, 995–1004. [Google Scholar] [PubMed]
- Huffman, J.W.; Mabon, R.; Wu, M.J.; Lu, J.; Hart, R.; Hurst, D.P.; Reggio, P.H.; Wiley, J.L.; Martin, B.R. 3-indolyl-1-naphthylmethanes: New cannabimimetic indoles provide evidence for aromatic stacking interactions with the cb(1) cannabinoid receptor. Bioorg. Med. Chem. 2003, 11, 539–549. [Google Scholar] [CrossRef]
- Huffman, J.W.; Padgett, L.W. Recent developments in the medicinal chemistry of cannabimimetic indoles, pyrroles and indenes. Curr. Med. Chem. 2005, 12, 1395–1411. [Google Scholar] [CrossRef] [PubMed]
- Huffman, J.W.; Zengin, G.; Wu, M.J.; Lu, J.; Hynd, G.; Bushell, K.; Thompson, A.L.; Bushell, S.; Tartal, C.; Hurst, D.P.; et al. Structure-activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid cb(1) and cb(2) receptors: Steric and electronic effects of naphthoyl substituents. New highly selective cb(2) receptor agonists. Bioorg. Med. Chem. 2005, 13, 89–112. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.L.; Smith, V.J.; Chen, J.; Martin, B.R.; Huffman, J.W. Synthesis and pharmacology of 1-alkyl-3-(1-naphthoyl)indoles: Steric and electronic effects of 4- and 8-halogenated naphthoyl substituents. Bioorg. Med. Chem. 2012, 20, 2067–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, C.; Schoeder, C.T.; Pillaiyar, T.; Madea, B.; Muller, C.E. Pharmacological evaluation of synthetic cannabinoids identified as constituents of spice. Forensic Toxicol. 2016, 34, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-inverse agonist characterization at cb1 and cb2 cannabinoid receptors of l759633, l759656, and am630. Br. J. Pharmacol. 1999, 126, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Huffman, J.W.; Szklennik, P.V.; Almond, A.; Bushell, K.; Selley, D.E.; He, H.; Cassidy, M.P.; Wiley, J.L.; Martin, B.R. 1-pentyl-3-phenylacetylindoles, a new class of cannabimimetic indoles. Bioorg. Med. Chem. Lett. 2005, 15, 4110–4113. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, N.; Kikura-Hanajiri, R.; Goda, Y. Identification of a novel cannabimimetic phenylacetylindole, cannabipiperidiethanone, as a designer drug in a herbal product and its affinity for cannabinoid cb(1) and cb(2) receptors. Chem. Pharm. Bull. 2011, 59, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.L.; Marusich, J.A.; Lefever, T.W.; Antonazzo, K.R.; Wallgren, M.T.; Cortes, R.A.; Patel, P.R.; Grabenauer, M.; Moore, K.N.; Thomas, B.F. Ab-chminaca, ab-pinaca, and fubimina: Affinity and potency of novel synthetic cannabinoids in producing delta9-tetrahydrocannabinol-like effects in mice. J. Pharmacol. Exp. Ther. 2015, 354, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Showalter, V.M.; Compton, D.R.; Martin, B.R.; Abood, M.E. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (cb2): Identification of cannabinoid receptor subtype selective ligands. J. Pharmacol. Exp. Ther. 1996, 278, 989–999. [Google Scholar] [PubMed]
- Huffman, J.W.; Padgett, L.W.; Isherwood, M.L.; Wiley, J.L.; Martin, B.R. 1-alkyl-2-aryl-4-(1-naphthoyl)pyrroles: New high affinity ligands for the cannabinoid cb1 and cb2 receptors. Bioorg. Med. Chem. Lett. 2006, 16, 5432–5435. [Google Scholar] [CrossRef] [PubMed]
- Franz, F.; Angerer, V.; Moosmann, B.; Auwarter, V. Phase i metabolism of the highly potent synthetic cannabinoid mdmb-chmica and detection in human urine samples. Drug Test. Anal. 2017, 9, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Pryce, G.; Baker, D. Antidote to cannabinoid intoxication: The cb1 receptor inverse agonist, am251, reverses hypothermic effects of the cb1 receptor agonist, cb-13, in mice. Br. J. Pharmacol. 2017, 174, 3790–3794. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Heaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Neliat, G.; Caput, D.; et al. Sr141716a, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef]
- Gatch, M.B.; Forster, M.J. Delta(9)-tetrahydrocannabinol-like effects of novel synthetic cannabinoids in mice and rats. Psychopharmacology 2016, 233, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.; Deng, H.; Zvonok, A.; Cockayne, D.A.; Kwan, J.; Mata, H.P.; Vanderah, T.W.; Lai, J.; Porreca, F.; Makriyannis, A.; et al. Activation of cb2 cannabinoid receptors by am1241 inhibits experimental neuropathic pain: Pain inhibition by receptors not present in the cns. Proc. Natl. Acad. Sci. USA 2003, 100, 10529–10533. [Google Scholar] [CrossRef] [PubMed]
- Buchler, I.P.; Hayes, M.J.; Hegde, S.G.; Hockerman, S.L.; Jones, D.E.; Kortum, S.W.; Rico, J.G.; Tenbrink, R.E.; Wu, K.K. Indazole Derivatives. Patent No. WO2009106980, 3 September 2009. [Google Scholar]
- Makriyannis, A.; Deng, H. Cannabimimetic Indole Derivatives. Patent No. WO200128557, 26 April 2001. [Google Scholar]
- Makriyannis, A.; Deng, H. Cannabimimetic Indole Derivatives. US Patent No. 2008/0090871, 17 April 2008. [Google Scholar]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, M.; Huang, B.; Ji, H.; Yuan, M. Comparative molecular field analysis (comfa) and comparative molecular similarity indices analysis (comsia) studies on alpha(1a)-adrenergic receptor antagonists based on pharmacophore molecular alignment. Int. J. Mol. Sci. 2011, 12, 7022–7037. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.Q.; Jin, H.X.; Yan, X.J.; Zhu, P.; Hu, G.X. 3d-qsar and 3d-qssr studies of thieno[2,3-d]pyrimidin-4-yl hydrazone analogues as cdk4 inhibitors by comfa analysis. Acta Pharmacol. Sin. 2014, 35, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Khan, F. 3d-qsar studies on maslinic acid analogs for anticancer activity against breast cancer cell line mcf-7. Sci. Rep. 2017, 7, 6019. [Google Scholar] [CrossRef] [PubMed]
- Fallarini, S.; Massarotti, A.; Gesu, A.; Giovarruscio, S.; Zabetta, G.C.; Bergo, R.; Giannelli, B.; Brunco, A.; Lombardi, G.; Sorba, G.; et al. In silico-driven multicomponent synthesis of 4,5-and 1,5-disubstituted imidazoles as indoleamine 2,3-dioxygenase inhibitors. MedChemComm 2016, 7, 409–419. [Google Scholar] [CrossRef]
- Lee, J.W.; Hirota, T.; Kumar, A.; Kim, N.J.; Irle, S.; Kay, S.A. Development of small-molecule cryptochrome stabilizer derivatives as modulators of the circadian clock. ChemMedChem 2015, 10, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S. Simpls: An alternative approach to partial least squares regression. Chemom. Intell. Lab. Syst. 1993, 18, 251–263. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. Pls-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Schoeder, C.T.; Hess, C.; Madea, B.; Meiler, J.; Muller, C.E. Pharmacological evaluation of new constituents of “spice”: Synthetic cannabinoids based on indole, indazole, benzimidazole and carbazole scaffolds. Forensic Toxicol. 2018, 36, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Gamage, T.F.; Farquhar, C.E.; Lefever, T.W.; Marusich, J.A.; Kevin, R.C.; McGregor, I.S.; Wiley, J.L.; Thomas, B.F. Molecular and behavioral pharmacological characterization of abused synthetic cannabinoids mmb- and mdmb-fubinaca, mn-18, nnei, cumyl-pica, and 5-fluoro-cumyl-pica. J. Pharmacol. Exp. Ther. 2018, 365, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Lehmann, F.; Hammer, K.; Haile, S.; Axen, E.; Medina, C.; Uppenberg, J.; Svensson, S.; Rondahl, L.; Lundback, T. N-benzyl-indolo carboxylic acids: Design and synthesis of potent and selective adipocyte fatty-acid binding protein (a-fabp) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. Optimization of parameters for semiempirical methods iv: Extension of mndo, am1, and pm3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Alemán, C.; Luque, F.J.; Orozco, M. Suitability of the pm3-derived molecular electrostatic potentials. J. Comput. Chem. 1993, 14, 799–808. [Google Scholar] [CrossRef]
- Qiao, F.; Luo, L.; Peng, H.; Luo, S.; Huang, W.; Cui, J.; Li, X.; Kong, L.; Jiang, D.; Chitwood, D.J.; et al. Characterization of three novel fatty acid- and retinoid-binding protein genes (ha-far-1, ha-far-2 and hf-far-1) from the cereal cyst nematodes heterodera avenae and h. Filipjevi. PLoS ONE 2016, 11, e0160003. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.P.; Leonard, J.T.; Roy, K. Exploring the impact of size of training sets for the development of predictive qsar models. Chemom. Intell. Lab. Syst. 2008, 90, 31–42. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor cb1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Chaudhaery, S.S.; Roy, K.K.; Saxena, A.K. Consensus superiority of the pharmacophore-based alignment, over maximum common substructure (mcs): 3d-qsar studies on carbamates as acetylcholinesterase inhibitors. J. Chem. Inf. Model. 2009, 49, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Olesen, P.H. The use of bioisosteric groups in lead optimization. Curr. Opin. Drug Discov. Dev. 2001, 4, 471–478. [Google Scholar]
- Burger, A. Isosterism and bioisosterism in drug design. Prog. Drug. Res. 1991, 37, 287–371. [Google Scholar] [PubMed]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Zinc—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Kruger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The chembl bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Pitt, W.R.; Parry, D.M.; Perry, B.G.; Groom, C.R. Heteroaromatic rings of the future. J. Med. Chem. 2009, 52, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Emcdda. European Drug Report 2018. Available online: Http://www.Emcdda.Europa.Eu/edr2018_en (accessed on 17 July 2018).
- Nayak, B.P.; Khajuria, H. Synthetic marijuana is no more marijuana. Asian J. Psychiatr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Palamar, J.J.; Barratt, M.J. Synthetic cannabinoids: Undesirable alternatives to natural marijuana. Am. J. Drug Alcohol Abuse 2016, 42, 371–373. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds not available from the authors. |
Figure 1.
Structure of ∆9-THC, AM11542, MDMB-CHMINACA, AM-1220, MDMB-FUBINACA, and XLR-12 along with their Ki (nM).
Figure 1.
Structure of ∆9-THC, AM11542, MDMB-CHMINACA, AM-1220, MDMB-FUBINACA, and XLR-12 along with their Ki (nM).

Figure 2.
Ten component 3D-QSAR model experimental vs. predicted pKi of the compounds in the training set for the CB1 receptor.
Figure 2.
Ten component 3D-QSAR model experimental vs. predicted pKi of the compounds in the training set for the CB1 receptor.

Figure 3.
Ten component 3D-QSAR model experimental vs. predicted pKi of the compounds in the test set for the CB1 receptor.
Figure 3.
Ten component 3D-QSAR model experimental vs. predicted pKi of the compounds in the test set for the CB1 receptor.

Figure 4.
Seven component 3D-QSAR model experimental vs. predicted pKi of the compounds in the training set for the CB2 receptor.
Figure 4.
Seven component 3D-QSAR model experimental vs. predicted pKi of the compounds in the training set for the CB2 receptor.

Figure 5.
Seven component 3D-QSAR model experimental vs. predicted pKi of the compounds in the test set for the CB2 receptor.
Figure 5.
Seven component 3D-QSAR model experimental vs. predicted pKi of the compounds in the test set for the CB2 receptor.

Figure 6.
Electrostatic and steric coefficients for the CB1 model. (A) Electrostatic coefficients superimposed to MDMB-CHMINACA (up) and AM-1220 (down). (B) Steric coefficients superimposed to MDMB-CHMINACA (up) and AM-1220 (down). (C) Contributions to predicted affinity for MDMB-CHMINACA (up) and AM-1220 (down).
Figure 6.
Electrostatic and steric coefficients for the CB1 model. (A) Electrostatic coefficients superimposed to MDMB-CHMINACA (up) and AM-1220 (down). (B) Steric coefficients superimposed to MDMB-CHMINACA (up) and AM-1220 (down). (C) Contributions to predicted affinity for MDMB-CHMINACA (up) and AM-1220 (down).

Figure 7.
Electrostatic and steric coefficients for the CB2 model. (A) Electrostatic coefficients superimposed to MDMB-FUBINACA (up) and XLR-12 (down). (B) Steric coefficients superimposed to MDMB-FUBINACA (up) and XLR-12 (down). (C) Contributions to predicted affinity for MDMB-FUBINACA (up) and XLR-12 (down).
Figure 7.
Electrostatic and steric coefficients for the CB2 model. (A) Electrostatic coefficients superimposed to MDMB-FUBINACA (up) and XLR-12 (down). (B) Steric coefficients superimposed to MDMB-FUBINACA (up) and XLR-12 (down). (C) Contributions to predicted affinity for MDMB-FUBINACA (up) and XLR-12 (down).

Figure 8.
(A) The CB1 model map superimposed to AM-1220. (B) The CB1 model map is superimposed to XLR-12. Molecular insight of SAR mechanism models, revealing the different lead optimization sites of active compounds. Red color shows positive field region controlling the activity, and blue color the negative ones. Green color shows favorable shape/hydrophobic regions, and purple color the unfavorable ones.
Figure 8.
(A) The CB1 model map superimposed to AM-1220. (B) The CB1 model map is superimposed to XLR-12. Molecular insight of SAR mechanism models, revealing the different lead optimization sites of active compounds. Red color shows positive field region controlling the activity, and blue color the negative ones. Green color shows favorable shape/hydrophobic regions, and purple color the unfavorable ones.

Figure 9.
(A) The CB2 model map is superimposed to XLR-12. (B) The CB2 model map is superimposed to AM-1220. Molecular insight of SAR mechanism models, revealing the different lead optimization sites of active compounds. Red color shows positive field region controlling the activity, and blue color the negative ones. Green color shows favorable shape/hydrophobic regions, and purple color the unfavorable ones.
Figure 9.
(A) The CB2 model map is superimposed to XLR-12. (B) The CB2 model map is superimposed to AM-1220. Molecular insight of SAR mechanism models, revealing the different lead optimization sites of active compounds. Red color shows positive field region controlling the activity, and blue color the negative ones. Green color shows favorable shape/hydrophobic regions, and purple color the unfavorable ones.

Figure 10.
Bioisosteric replacement of selected compounds. The different studied parts of the molecules are highlighted in blue, green, and yellow.
Figure 10.
Bioisosteric replacement of selected compounds. The different studied parts of the molecules are highlighted in blue, green, and yellow.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Chemical structures of the most potent compounds derived from the bioisosteric replacement for the CB1 receptor, the predicted Ki value are presented as pKi.
Table 1.
Chemical structures of the most potent compounds derived from the bioisosteric replacement for the CB1 receptor, the predicted Ki value are presented as pKi.
| Series 1 | Series 2 | Series 3 |
 |  |  |
| CB1 = 9.3 CB2 = 7.9 | CB1 = 8.8 CB2 = 7.9 | CB1 = 8.9 CB2 = 6.7 |
 |  |  |
| CB1 = 9.2 CB2 = 8.3 | CB1 = 8.7 CB2 = 8.0 | CB1 = 8.8 CB2 = 8.0 |
| Series 4 | Series 5 | Series 6 |
 |  |  |
| CB1 = 8.5 CB2 = 9.9 | CB1 = 8.0 CB2 = 9.3 | CB1 = 8.3 CB2 = 8.9 |
 |  |  |
| CB1 = 8.5 CB2 = 9.2 | CB1 = 8.0 CB2 = 9.6 | CB1 = 8.2 CB2 = 8.9 |
Table 2.
Chemical structures of the most potent compounds derived from the bioisosteric replacement for the CB2 receptor, the predicted Ki value are presented as pKi.
Table 2.
Chemical structures of the most potent compounds derived from the bioisosteric replacement for the CB2 receptor, the predicted Ki value are presented as pKi.
| Series 1 | Series 2 | Series 3 |
 |  |  |
| CB2 = 9.4 CB1 = 8.6 | CB2 = 8.7 CB1 = 8.1 | CB2 = 8.3 CB1 = 8.1 |
 |  |  |
| CB2 = 9.4 CB1 = 7.7 | CB2 = 8.6 CB1 = 8.6 | CB2 = 8.2 CB1 = 7.8 |
| Series 4 | Series 5 | Series 6 |
 |  |  |
| CB2 = 10.2 CB1 = 7.9 | CB2 = 9.9 CB1 = 7.8 | CB2 = 10.2 CB1 = 7.8 |
 |  |  |
| CB2 = 10.1 CB1 = 7.8 | CB2 = 9.8 CB1 = 7.3 | CB2 = 10.1 CB1 = 7.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules 2018, 23, 2183. https://doi.org/10.3390/molecules23092183
AMA Style
Floresta G, Apirakkan O, Rescifina A, Abbate V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules. 2018; 23(9):2183. https://doi.org/10.3390/molecules23092183
Chicago/Turabian StyleFloresta, Giuseppe, Orapan Apirakkan, Antonio Rescifina, and Vincenzo Abbate. 2018. "Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis" Molecules 23, no. 9: 2183. https://doi.org/10.3390/molecules23092183