Ascorbic Acid Mitigates D-galactose-Induced Brain Aging by Increasing Hippocampal Neurogenesis and Improving Memory Function

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drug Treatment
2.3. Body Weight
2.4. 5-bromo-2′-Deoxyuridine Administration
2.5. Tissue Processing
2.6. Immunohistochemistry
2.7. Double Immunofluorescence
2.8. Quantification of Histology
2.9. Western Blot Analysis
2.10. Object Location Task
2.11. Statistical Analyses
3. Results
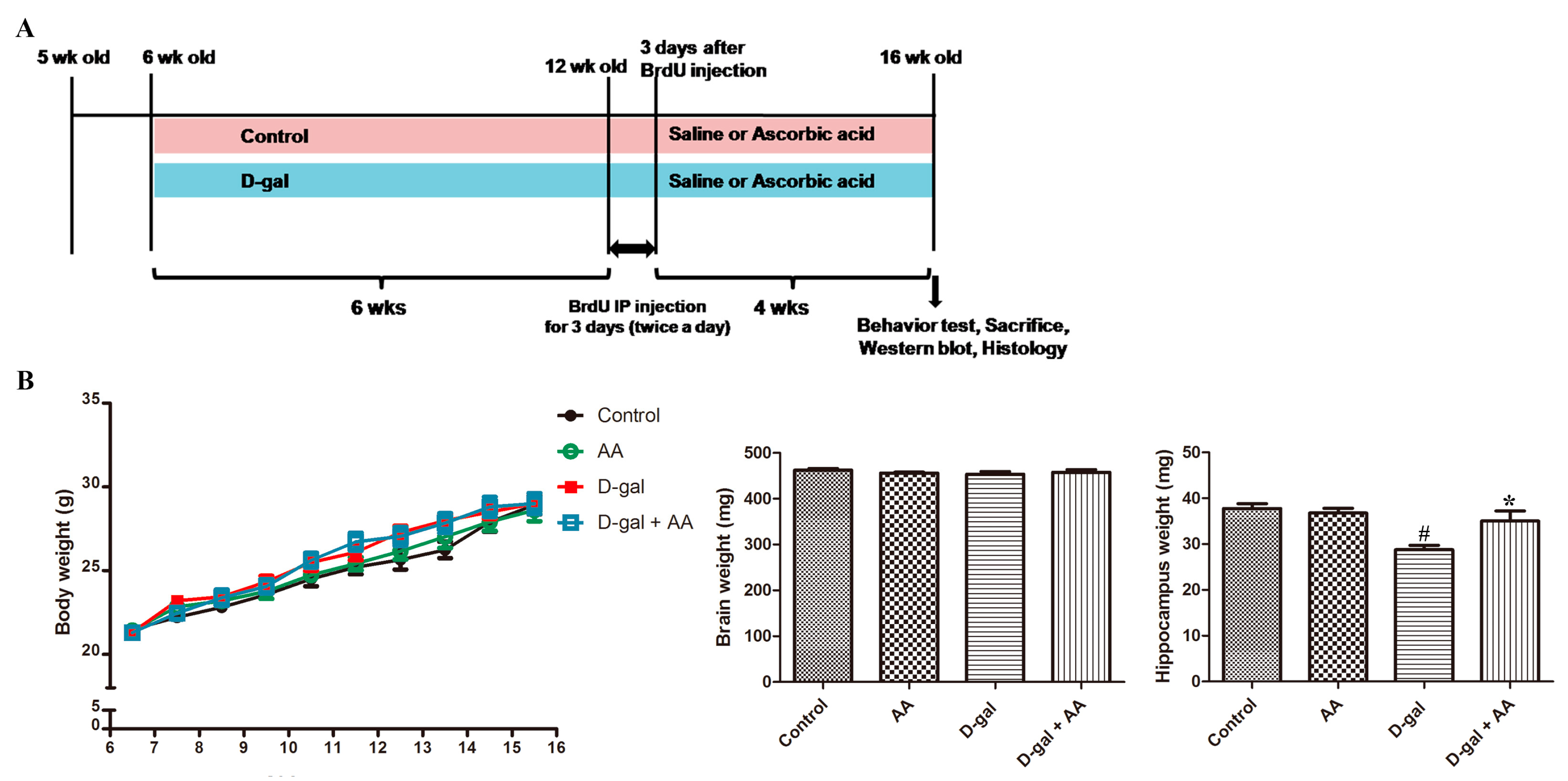
3.1. Effects of Oral Administration of Ascorbic Acid on Body and Brain Weights in D-gal-Treated Mice
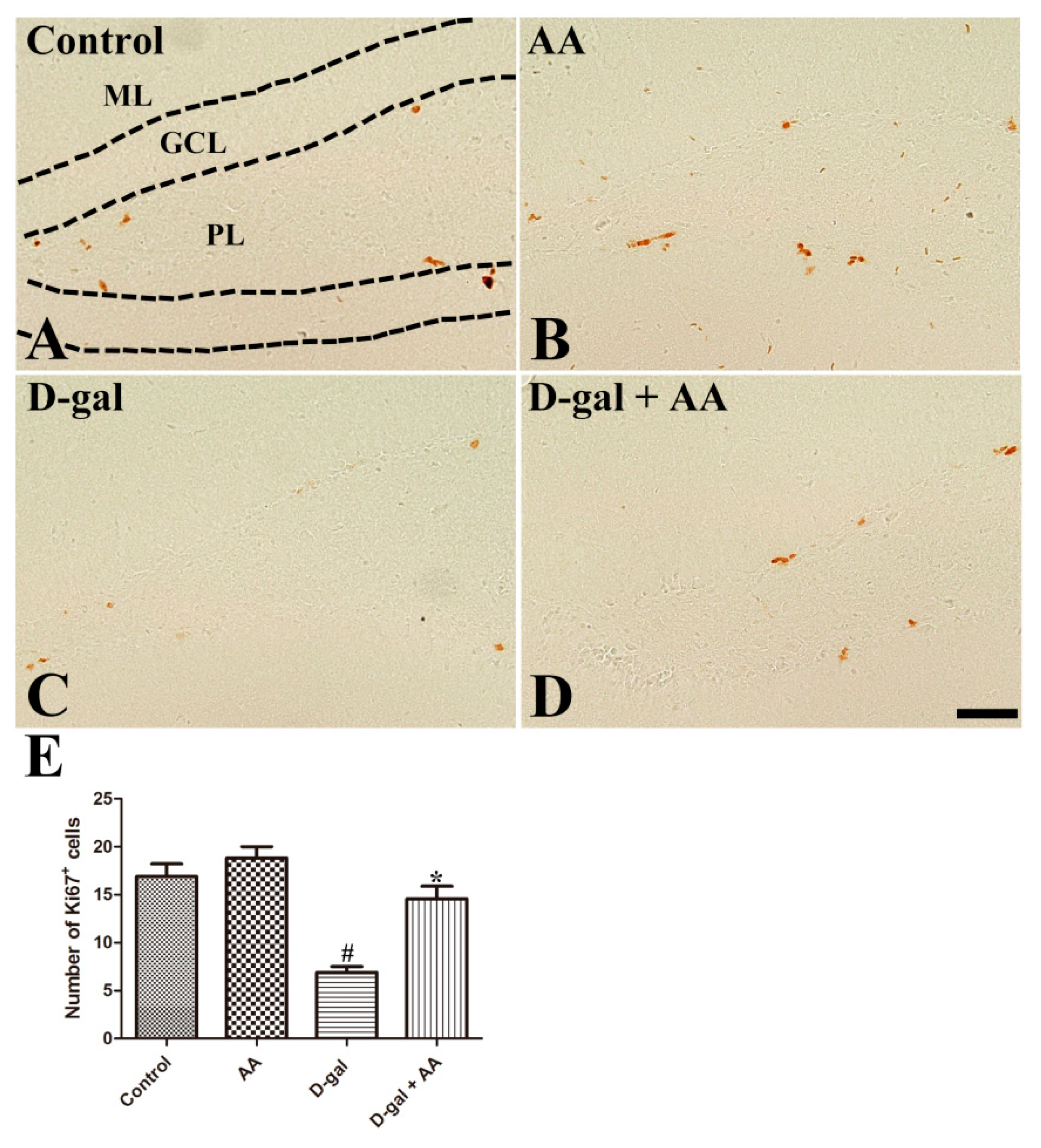
3.2. Effects of Oral Administration of Ascorbic Acid on Cell Proliferation in D-gal-Treated Mice
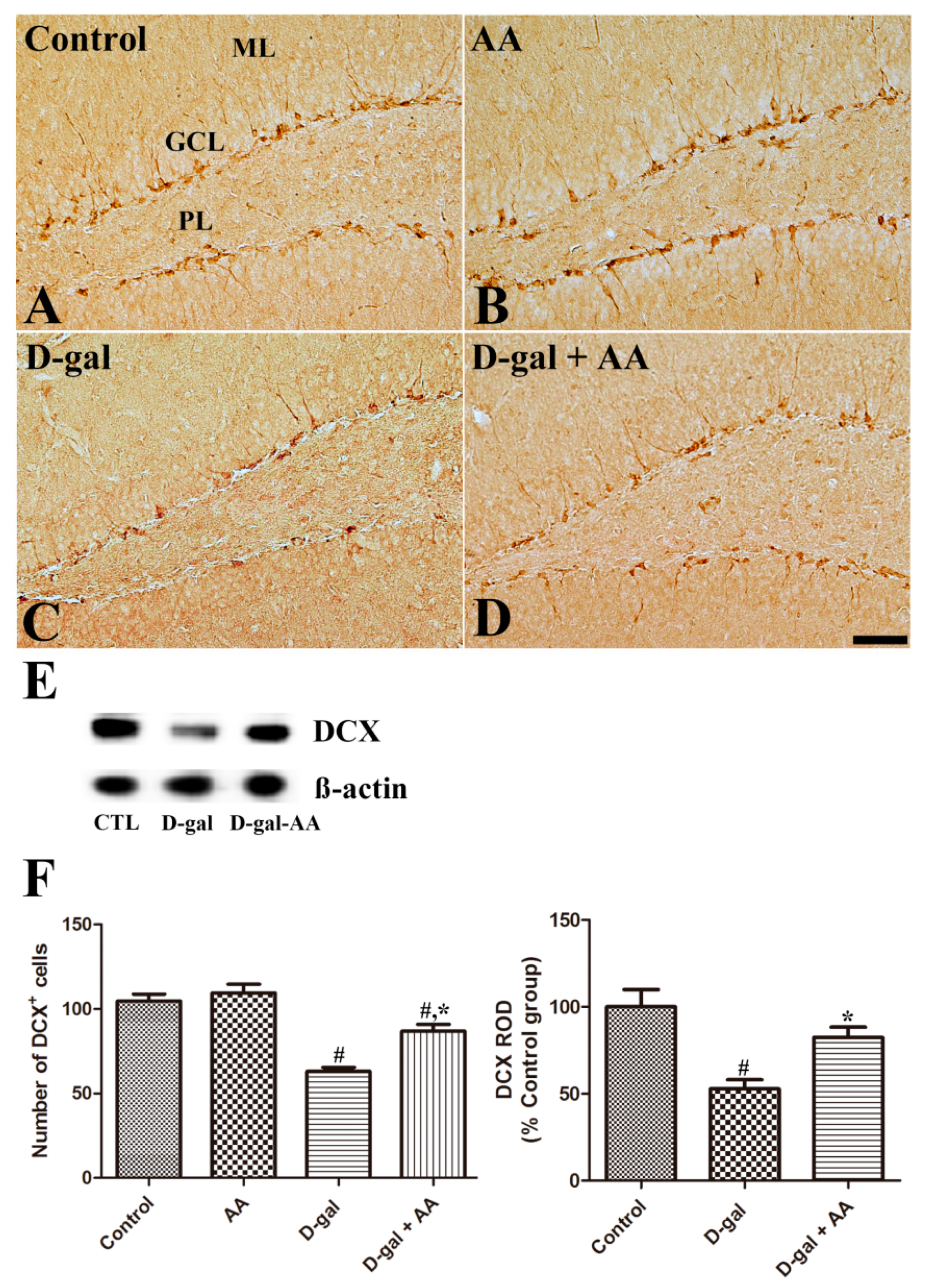
3.3. Effects of Oral Administration of Ascorbic Acid on Neuroblast Differentiation in D-gal-Treated Mice
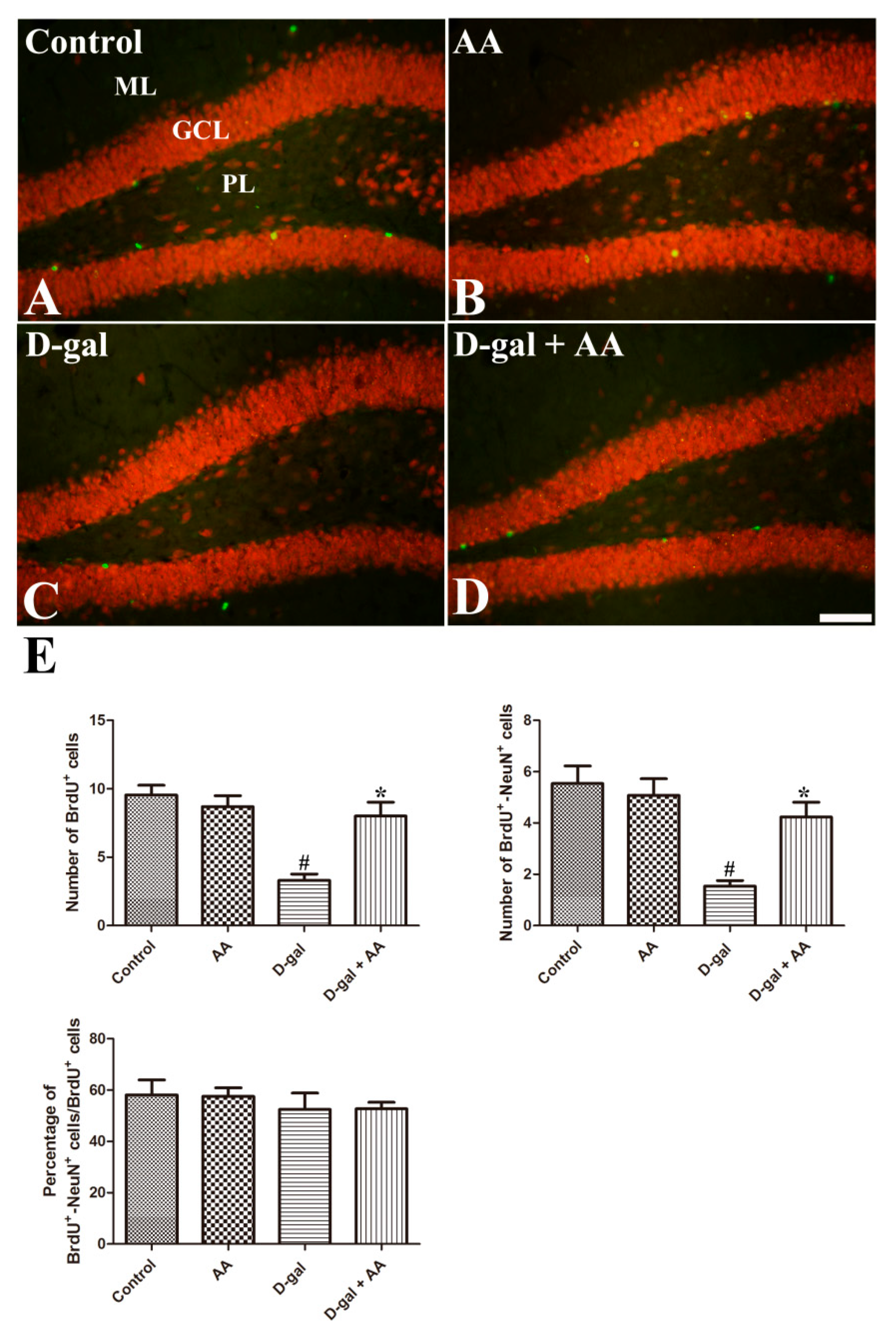
3.4. Effects of Oral Administration of Ascorbic Acid on Neuronal Maturation in D-gal-Treated Mice
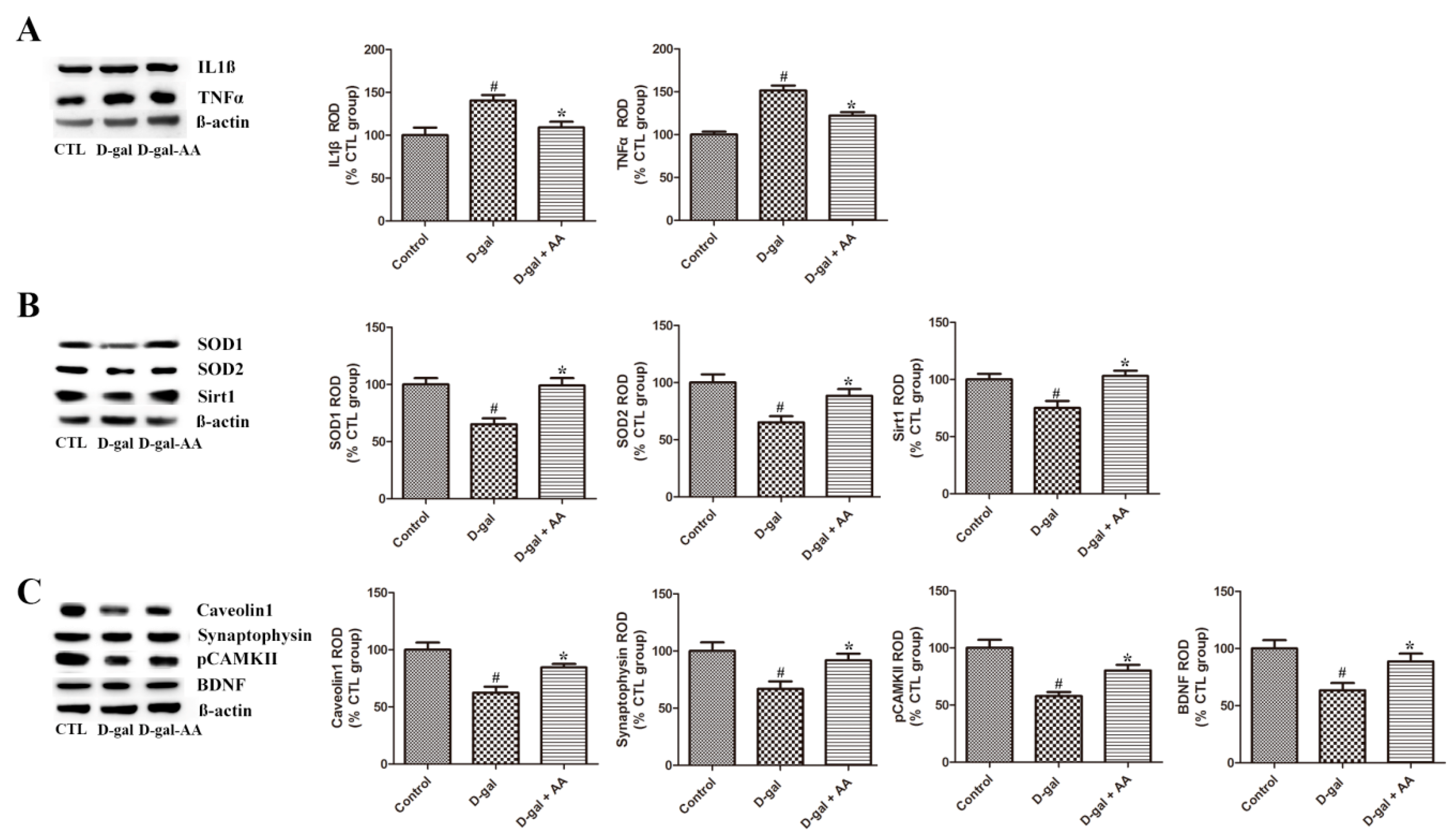
3.5. Effects of Oral Administration of Ascorbic Acid on SOD1 and SOD2 Expression in D-gal-Treated Mice
3.6. Effects of Oral Administration of Ascorbic Acid on IL1β and TNFα Expression in D-gal-Treated Mice
3.7. Effects of Oral Administration of Ascorbic Acid on Sirt1 and Caveolin-1 Expression in D-gal-Treated Mice
3.8. Effects of Oral Administration of Ascorbic Acid on Synaptophysin and pCAMK II Expression in D-gal-Treated Mice
3.9. Effects of Oral Administration of Ascorbic Acid on BDNF Expression in D-gal-Treated Mice
3.10. Effects of Oral Administration of Ascorbic Acid on Phosphorylated cAMP Response Element-Binding Protein (pCREB) Expression in D-gal-Treated Mice
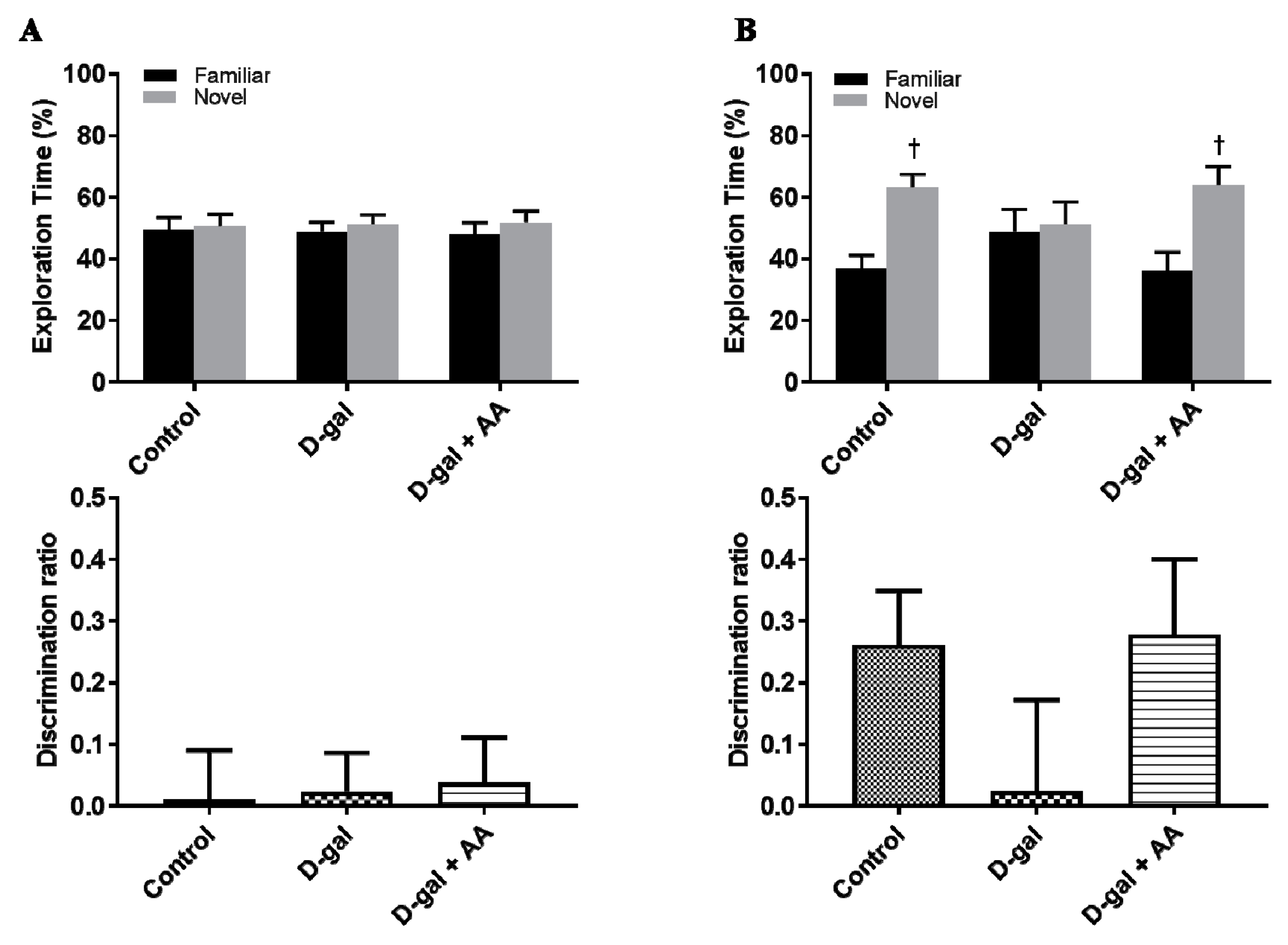
3.11. Effects of Oral Administration of Ascorbic Acid on Spatial Memory in D-gal-Treated Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yu, Y.H.; Wang, S.T.; Ren, J.; Camer, D.; Hua, Y.Z.; Zhang, Q.; Huang, J.; Xue, D.L.; Zhang, X.F.; et al. Chlorogenic acid protects D-galactose-induced liver and kidney injury via antioxidation and anti-inflammation effects in mice. Pharm. Biol. 2016, 54, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef]
- Wu, W.; Wang, X.; Xiang, Q.; Meng, X.; Peng, Y.; Du, N.; Liu, Z.; Sun, Q.; Wang, C.; Liu, X. Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct. 2014, 5, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Bao, M.; Li, D.; Li, Y.M. Advanced glycation in D-galactose induced mouse aging model. Mech. Ageing Dev. 1999, 108, 239–251. [Google Scholar] [CrossRef]
- Banji, D.; Banji, O.J.; Dasaroju, S.; Kranthi, K.C. Curcumin and piperine abrogate lipid and protein oxidation induced by D-galactose in rat brain. Brain Res. 2013, 1515, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.M.; Chang, H.H.; Kuo, W.W.; Lin, H.J.; Yeh, Y.L.; Padma Viswanadha, V.; Tsai, C.C.; Chen, R.J.; Chang, H.N.; Huang, C.Y. Anti-apoptotic and pro-survival effect of alpinate oxyphyllae fructus (AOF) in a d-galactose-induced aging heart. Int. J. Mol. Sci. 2016, 17, 466. [Google Scholar] [CrossRef]
- Budni, J.; Garcez, M.L.; Mina, F.; Bellettini-Santos, T.; da Silva, S.; Luz, A.P.D.; Schiavo, G.L.; Batista-Silva, H.; Scaini, G.; Streck, E.L.; et al. The oral administration of D-galactose induces abnormalities within the mitochondrial respiratory chain in the brain of rats. Metab. Brain Dis. 2017, 32, 811–817. [Google Scholar] [CrossRef]
- Nam, S.M.; Choi, J.H.; Yoo, D.Y.; Kim, W.; Jung, H.Y.; Kim, J.W.; Kang, S.Y.; Park, J.; Kim, D.W.; Kim, W.J.; et al. Valeriana officinalis extract and its main component, valerenic acid, ameliorate D-galactose-induced reductions in memory, cell proliferation, and neuroblast differentiation by reducing corticosterone levels and lipid peroxidation. Exp. Gerontol. 2013, 48, 1369–1377. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.M.; Hu, B.; Cheng, W.; Zheng, Y.L.; Zhang, Z.F.; Ye, Q.; Fan, S.H.; Shan, Q.; Wang, Y.J. Chronic administration of troxerutin protects mouse brain against D-galactose-induced impairment of cholinergic system. Neurobiol. Learn. Mem. 2010, 93, 157–164. [Google Scholar] [CrossRef]
- Barrita, J.; Sánchez, M. Antioxidant role of ascorbic acid and his protective effects on chronic diseases. In Oxidative Stress and Chronic Degenerative Diseases—A Role Antioxidants; Morales-Gonzalez, J.A., Ed.; IntTech Publisher: London, UK, 2013; pp. 449–484. ISBN 978-953-51-1123-8. [Google Scholar]
- Varvara, M.; Bozzo, G.; Celano, G.; Disanto, C.; Pagliarone, C.N.; Celano, G.V. The use of ascorbic acid as a food additive: Technical-legal issues. Ital. J. Food Saf. 2016, 5, 4313. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed]
- Oke, A.F.; May, L.; Adams, R.N. Ascorbic acid distribution patterns in human brain. A comparison with nonhuman mammalian species. Ann. N. Y. Acad. Sci. 1987, 498, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, B.K.; Kanungo, M.S. Ascorbic acid and aging in the rat. Uptake of ascorbic acid by teeth and concentration of various forms of ascorbic acid in different organs. Biochem. J. 1966, 100, 59–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaw, K.T.; Bingham, S.; Welch, A.; Luben, R.; Wareham, N.; Oakes, S.; Day, N. Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: A prospective population study. Lancet 2001, 357, 657–663. [Google Scholar] [CrossRef]
- Nam, S.M.; Hwang, H.; Seo, M.; Chang, B.J.; Kim, H.J.; Choi, S.H.; Rhim, H.; Kim, H.C.; Cho, I.H.; Nah, S.Y. Gintonin attenuates D-galactose-induced hippocampal senescence by improving long-term hippocampal potentiation, neurogenesis, and cognitive functions. Gerontology 2018, 64, 562–575. [Google Scholar] [CrossRef]
- Colomina, M.T.; Gómez, M.; Domingo, J.L.; Corbella, J. Lack of maternal and developmental toxicity in mice given high doses of aluminium hydroxide and ascorbic acid during gestation. Pharmacol. Toxicol. 1994, 74, 236–239. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Olajide, O.J.; Yawson, E.O.; Gbadamosi, I.T.; Arogundade, T.T.; Lambe, E.; Obasi, K.; Lawal, I.T.; Ibrahim, A.; Ogunrinola, K.Y. Ascorbic acid ameliorates behavioural deficits and neuropathological alterations in rat model of Alzheimer’s disease. Environ. Toxicol. Pharmacol. 2017, 50, 200–211. [Google Scholar] [CrossRef]
- Nam, S.M.; Kim, Y.N.; Kim, J.W.; Kyeong, D.S.; Lee, S.H.; Son, Y.; Shin, J.H.; Kim, J.; Yi, S.S.; Yoon, Y.S.; et al. Hairy and enhancer of split 6 (Hes6) deficiency in mouse impairs neuroblast differentiation in dentate gyrus without affecting cell proliferation and integration into mature neurons. Cell. Mol. Neurobiol. 2016, 36, 57–67. [Google Scholar] [CrossRef]
- Franklin, K.B.J.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates, 4th ed.; Academic Press: San Diego, CA, USA, 2012; ISBN 9780123910578. [Google Scholar]
- Fukui, K.; Onodera, K.; Shinkai, T.; Suzuki, S.; Urano, S. Impairment of learning and memory in rats caused by oxidative stress and aging, and changes in antioxidative defense systems. Ann. N. Y. Acad. Sci. 2001, 928, 168–175. [Google Scholar] [CrossRef]
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of caveolin-1 accelerates neurodegeneration and aging. PLoS ONE 2010, 5, e15697. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Duan, W.; Mattson, M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002, 82, 1367–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Kuang, S.; Li, H.; Ran, D.; Yang, J. cAMP/PKA-CREB-BDNF signaling pathway in hippocampus mediates cyclooxygenase 2-induced learning/memory deficits of rats subjected to chronic unpredictable mild stress. Oncotarget 2017, 8, 35558–35572. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.P.; Chen, T.; Yin, N.N.; Han, Y.M.; Yuan, F.; Duan, Y.J.; Shen, F.; Zhang, Y.H.; Chen, Z.B. Puerarin ameliorates D-galactose induced enhanced hippocampal neurogenesis and tau hyperphosphorylation in rat brain. J. Alzheimers Dis. 2016, 51, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.M.; Sieber, S.; Wyatt, N.; Lizzi, J.; Flannery, T.; Sibbit, B.; Sanghvi, S. Increasing neurogenesis with fluoxetine, simvastatin and ascorbic acid leads to functional recovery in ischemic stroke. Recent Pat. Drug Deliv. Formul. 2015, 9, 158–166. [Google Scholar] [CrossRef]
- Cao, N.; Liu, Z.; Chen, Z.; Wang, J.; Chen, T.; Zhao, X.; Ma, Y.; Qin, L.; Kang, J.; Wei, B.; et al. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Res. 2012, 22, 219–236. [Google Scholar] [CrossRef]
- Shin, D.M.; Ahn, J.I.; Lee, K.H.; Lee, Y.S.; Lee, Y.S. Ascorbic acid responsive genes during neuronal differentiation of embryonic stem cells. Neuroreport 2004, 15, 1959–1963. [Google Scholar] [CrossRef]
- Ochiai, Y.; Kaburagi, S.; Obayashi, K.; Ujiie, N.; Hashimoto, S.; Okano, Y.; Masaki, H.; Ichihashi, M.; Sakurai, H. A new lipophilic pro-vitamin C, tetra-isopalmitoyl ascorbic acid (VC-IP), prevents UV-induced skin pigmentation through its anti-oxidative properties. J. Dermatol. Sci. 2006, 44, 37–44. [Google Scholar] [CrossRef]
- Chang, B.J.; Jang, B.J.; Son, T.G.; Cho, I.H.; Quan, F.S.; Choe, N.H.; Nahm, S.S.; Lee, J.H. Ascorbic acid ameliorates oxidative damage induced by maternal low-level lead exposure in the hippocampus of rat pups during gestation and lactation. Food Chem. Toxicol. 2012, 50, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Colla, A.; de Oliveira Balen, G.; dos Santos, D.B.; Budni, J.; de Freitas, A.E.; Farina, M.; Severo Rodrigues, A.L. Ascorbic acid treatment, similarly to fluoxetine, reverses depressive-like behavior and brain oxidative damage induced by chronic unpredictable stress. J. Psychiatr. Res. 2012, 46, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Tamari, Y.; Nawata, H.; Inoue, E.; Yoshimura, A.; Yoshii, H.; Kashino, G.; Seki, M.; Enomoto, T.; Watanabe, M.; Tano, K. Protective roles of ascorbic acid in oxidative stress induced by depletion of superoxide dismutase in vertebrate cells. Free Radic. Res. 2013, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Shah, S.A.; Ali, T.; Chung, J.I.; Kim, M.O. Anthocyanins reversed D-galactose-induced oxidative stress and neuroinflammation mediated cognitive impairment in adult rats. Mol. Neurobiol. 2017, 54, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, Z.; Liu, J.; Zou, W.; Li, X.; Wang, Y.; An, L. Downregulation of caveolin-1 contributes to the synaptic plasticity deficit in the hippocampus of aged rats. Neural Regen. Res. 2013, 8, 2725–2733. [Google Scholar] [CrossRef]
- Ng, F.; Wijaya, L.; Tang, B.L. SIRT1 in the brain-connections with aging-associated disorders and lifespan. Front. Cell. Neurosci. 2015, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.M.; Jung, S.H.; An, H.T.; Lee, S.; Hong, J.; Park, J.S.; Lee, H.; Lee, H.; Bahn, M.S.; Lee, H.C.; et al. Caveolin 1 deficiency induces premature senescence with mitochondrial dysfunction. Aging Cell 2017, 16, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Pallauf, K.; Rimbach, G.; Rupp, P.M.; Chin, D.; Wolf, I.M. Resveratrol and lifespan in model organisms. Curr. Med. Chem. 2016, 23, 4639–4680. [Google Scholar] [CrossRef] [PubMed]
- Raynes, R.; Brunquell, J.; Westerheide, S.D. Stress inducibility of SIRT1 and its role in cytoprotection and cancer. Genes Cancer 2013, 4, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Li, L.; Zhang, Y.; Yang, J.; Zhang, Y.; Xing, Y. Vitamin C protected human retinal pigmented epithelium from oxidant injury depending on regulating SIRT1. Sci. World J. 2014, 2014, 750634. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; An, R.; Yang, Y.; Yang, X.; Liu, H.; Yue, L.; Li, X.; Lin, Y.; Reiter, R.J.; Qu, Y. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: The role of SIRT1 signaling. J. Pineal Res. 2015, 59, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Liu, X.; Chen, X.; Li, J.; Yang, X.; Fan, J.; Yang, Y.; Chen, N. Ampelopsin attenuates brain aging of D-gal-induced rats through miR-34a-mediated SIRT1/mTOR signal pathway. Oncotarget 2016, 7, 74484–74495. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.Y.; Peng, C.X.; Zhang, L.H. Berberine rescues D-galactose-induced synaptic/memory impairment by regulating the levels of Arc. Pharmacol. Biochem. Behav. 2014, 117, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Hu, Y.; Finley, J.C.; Saldana, M.D.; Bonds, J.A.; Miyanohara, A.; Niesman, I.R.; Ali, S.S.; Murray, F.; Insel, P.A.; et al. Neuron-targeted caveolin-1 protein enhances signaling and promotes arborization of primary neurons. J. Biol. Chem. 2011, 286, 33310–33321. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Tsutsumi, Y.M.; Hu, Y.; Mejia, T.; Mora, R.C.; Insel, P.A.; Roth, D.M.; Drummond, J.C.; Patel, P.M. Caveolin-1 expression is essential for N-methyl-D-aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008, 22, 828–840. [Google Scholar] [CrossRef]
- Karamian, R.; Komaki, A.; Salehi, I.; Tahmasebi, L.; Komaki, H.; Shahidi, S.; Sarihi, A. Vitamin C reverses lead-induced deficits in hippocampal synaptic plasticity in rats. Brain Res. Bull. 2015, 116, 7–15. [Google Scholar] [CrossRef]
- Vogel-Ciernia, A.; Wood, M.A. Examining object location and object recognition memory in mice. Curr. Protoc. Neurosci. 2014, 69, 8–31. [Google Scholar]
- Wimmer, M.E.; Hernandez, P.J.; Blackwell, J.; Abel, T. Aging impairs hippocampus-dependent long-term memory for object location in mice. Neurobiol. Aging 2012, 33, 2220–2224. [Google Scholar] [CrossRef] [Green Version]
- Noguchi-Shinohara, M.; Abe, C.; Yuki-Nozaki, S.; Dohmoto, C.; Mori, A.; Hayashi, K.; Shibata, S.; Ikeda, Y.; Sakai, K.; Iwasa, K.; et al. Higher blood vitamin C levels are associated with reduction of apolipoprotein E E4-related risks of cognitive decline in women: The Nakajima study. J. Alzheimers Dis. 2018, 63, 1289–1297. [Google Scholar] [CrossRef]
- Harrison, F.E. A critical review of vitamin C for the prevention of age related cognitive decline and Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, J.; Zeevalk, G.D. Cooperative interaction between ascorbate and glutathione during mitochondrial impairment in mesencephalic cultures. J. Neurochem. 2003, 86, 1487–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham, W.B.; Zuckerkandl, E.; Reynolds, R.; Willoughby, R.; Marcuson, R.; Barth, R.; Pauling, L. Effects of intake of L-ascorbic acid on the incidence of dermal neoplasms induced in mice by ultraviolet light. Proc. Natl. Acad. Sci. USA 1982, 79, 7532–7536. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, S.M.; Seo, M.; Seo, J.-S.; Rhim, H.; Nahm, S.-S.; Cho, I.-H.; Chang, B.-J.; Kim, H.-J.; Choi, S.-H.; Nah, S.-Y. Ascorbic Acid Mitigates D-galactose-Induced Brain Aging by Increasing Hippocampal Neurogenesis and Improving Memory Function. Nutrients 2019, 11, 176. https://doi.org/10.3390/nu11010176
Nam SM, Seo M, Seo J-S, Rhim H, Nahm S-S, Cho I-H, Chang B-J, Kim H-J, Choi S-H, Nah S-Y. Ascorbic Acid Mitigates D-galactose-Induced Brain Aging by Increasing Hippocampal Neurogenesis and Improving Memory Function. Nutrients. 2019; 11(1):176. https://doi.org/10.3390/nu11010176
Chicago/Turabian StyleNam, Sung Min, Misun Seo, Jin-Seok Seo, Hyewhon Rhim, Sang-Soep Nahm, Ik-Hyun Cho, Byung-Joon Chang, Hyeon-Joong Kim, Sun-Hye Choi, and Seung-Yeol Nah. 2019. "Ascorbic Acid Mitigates D-galactose-Induced Brain Aging by Increasing Hippocampal Neurogenesis and Improving Memory Function" Nutrients 11, no. 1: 176. https://doi.org/10.3390/nu11010176