
Hepatitis E Pathogenesis


 and
and 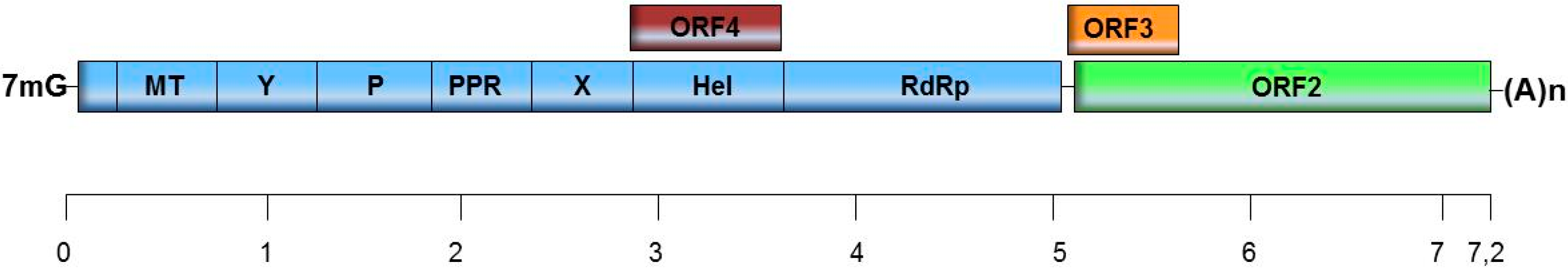{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
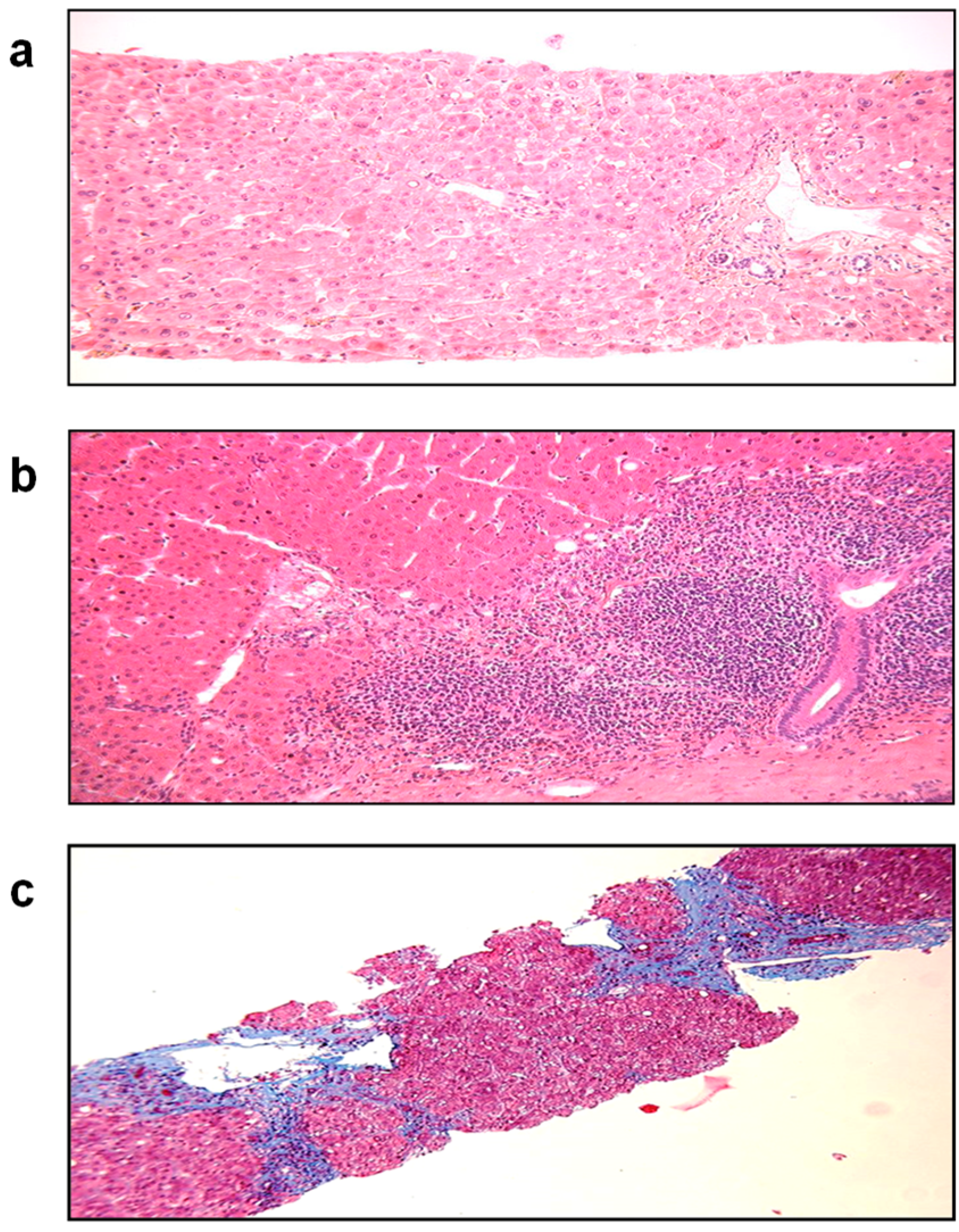
Abstract
:1. Introduction
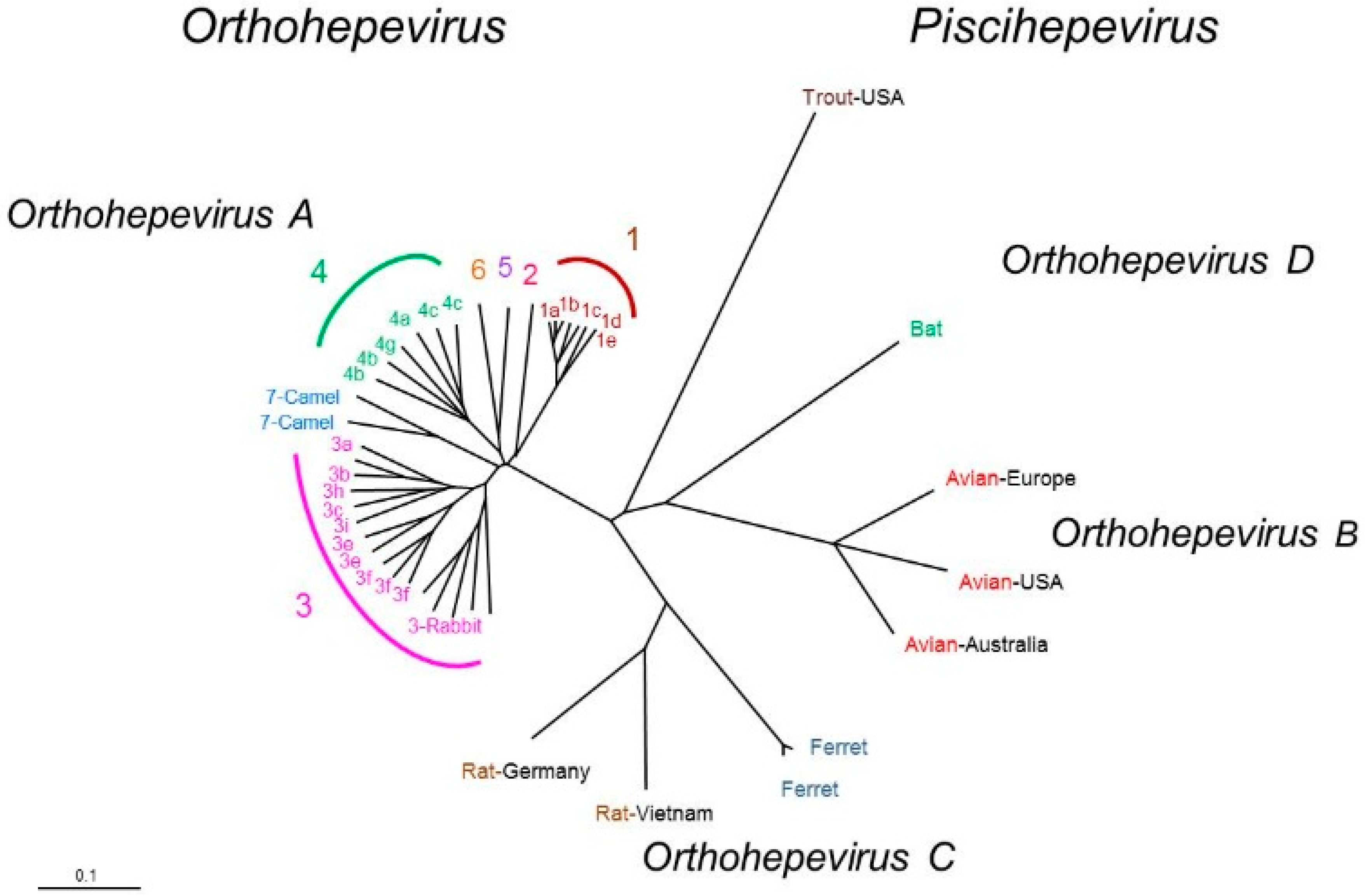
2. HEV and Classification
3. Clinical Course of HEV Infection
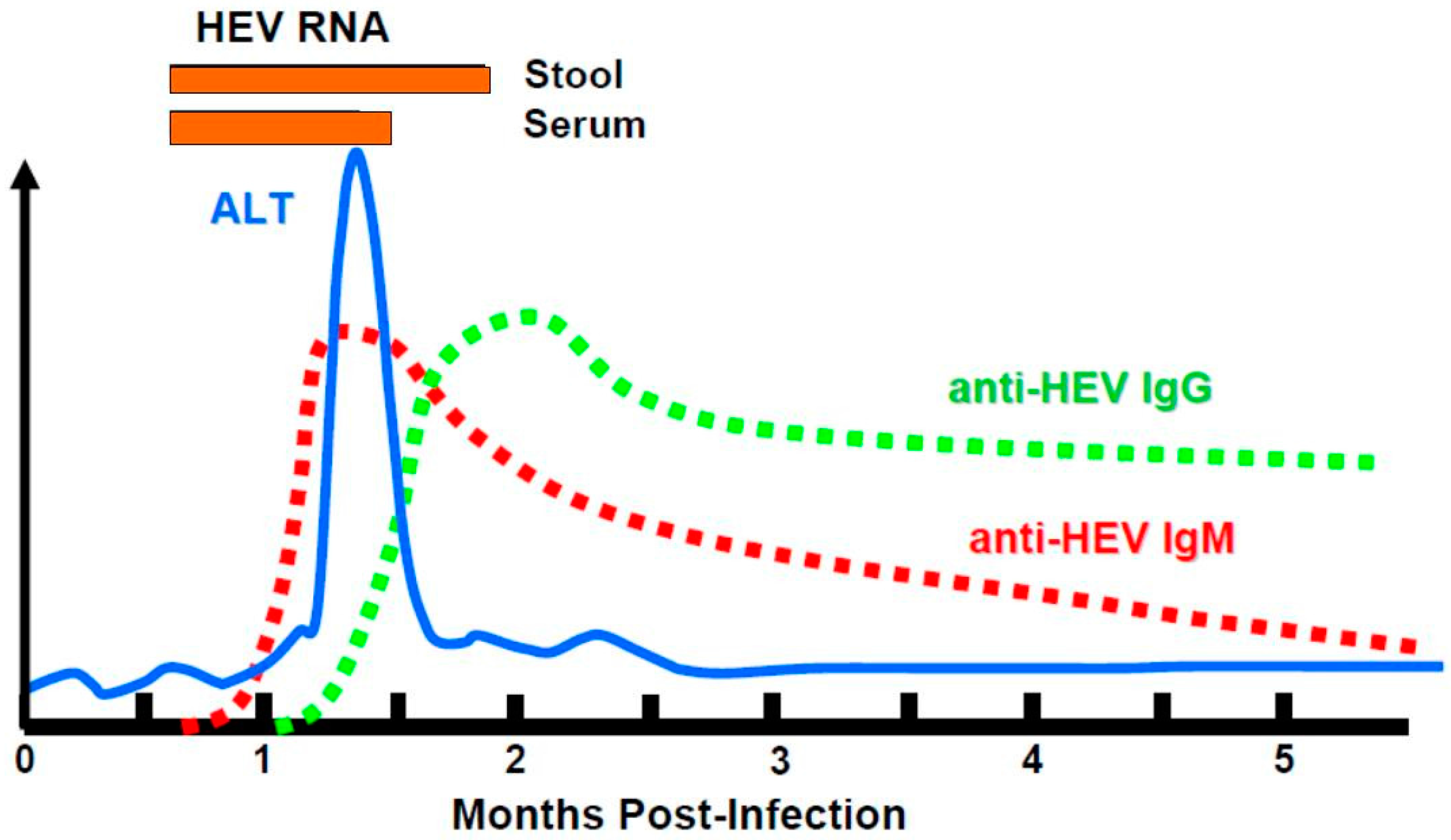
3.1. Natural History
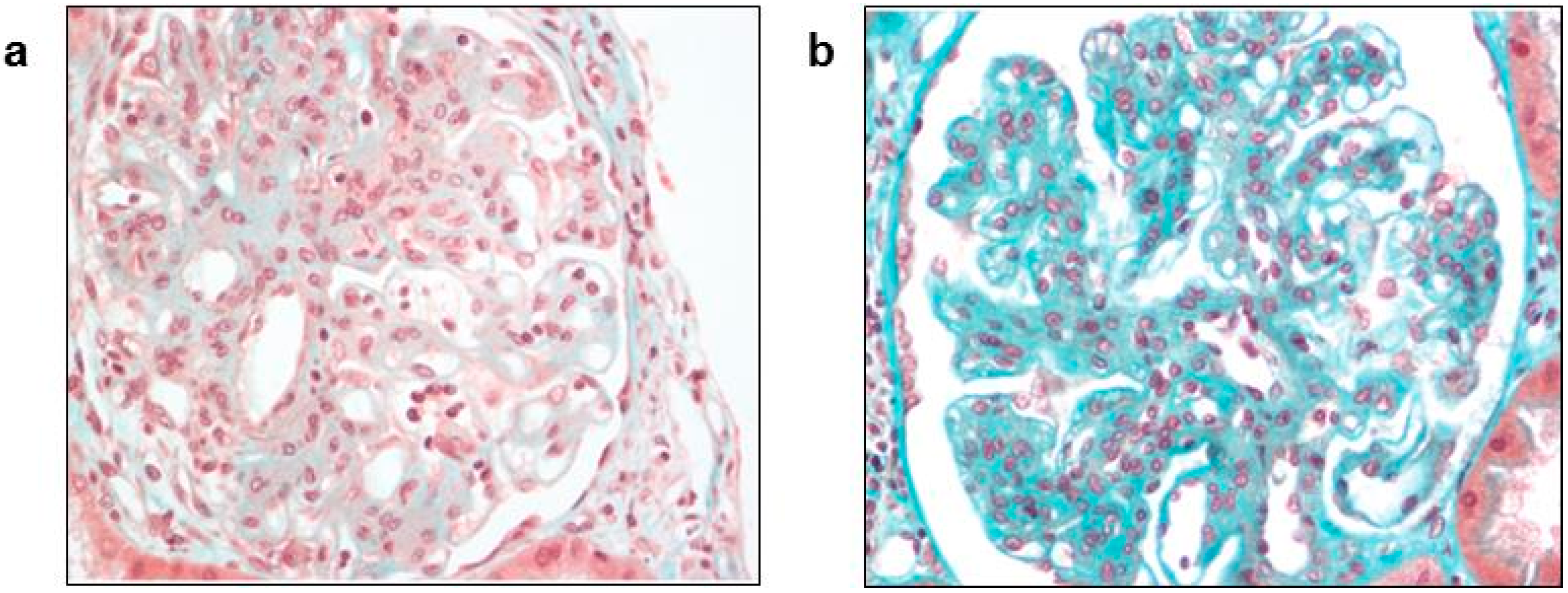
3.2. Extra-Hepatic Manifestations
4. Pathogenesis
4.1. Genetic Susceptibility to HEV
4.2. Innate Immune Response
4.3. Addaptive Response
5. Pathogenesis of Fulminant Hepatitis
5.1. Fulminant Hepatitis E in the General Population
5.2. Immunopathogenesis in Pregnant Women
6. Pathogenesis of Chronic Infection in Immunocompromised Patients
7. Animal and in vitro Models
7.1. Animal Models
7.2. Cell Culture Systems for HEV Replication
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kamar, N.; Dalton, H.R.; Abravanel, F.; Izopet, J. Hepatitis E virus infection. Clin. Microbiol. Rev. 2014, 27, 116–138. [Google Scholar] [CrossRef] [PubMed]
- Holla, R.P.; Ahmad, I.; Ahmad, Z.; Jameel, S. Molecular virology of hepatitis E virus. Semin. Liver Dis. 2013, 33, 3–14. [Google Scholar] [PubMed]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Ct, R.K.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Nguyen, H.; Torian, U.; Purcell, R.H. ORF3 protein of hepatitis E virus is not required for replication, virion assembly, or infection of hepatoma cells in vitro. J. Virol. 2006, 80, 10457–10464. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Nguyen, H.T.; Torian, U.; Burke, D.; Engle, R.; Purcell, R.H. Release of genotype 1 hepatitis E virus from cultured hepatoma and polarized intestinal cells depends on open reading frame 3 protein and requires an intact PXXP motif. J. Virol. 2010, 84, 9059–9069. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Jirintai; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; et al. Hepatitis E Virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: characterization of HEV virions in blood circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked Viruses That Aren’t Always Naked: Quasi-Enveloped Agents of Acute Hepatitis. Annu. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Lemon, S.M. Peek-a-boo: Membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 2014, 22, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; International Committee on Taxonomy of Viruses Hepeviridae Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95 Pt 10, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.J.; et al. Proposed reference sequences for Hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J. Zoonotic and foodborne transmission of hepatitis E virus. Semin. Liver Dis. 2013, 33, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Izopet, J.; Dubois, M.; Bertagnoli, S.; Lhomme, S.; Marchandeau, S.; Boucher, S.; Kamar, N.; Abravanel, F.; Guerin, J.L. Hepatitis E virus strains in rabbits and evidence of a closely related strain in humans, France. Emerg. Infect. Dis. 2012, 18, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Tan, B.H.; Chi-Yuan Teo, E.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R. Hepatitis E: Clinical presentation in disease-endemic areas and diagnosis. Semin. Liver Dis. 2013, 33, 30–40. [Google Scholar] [PubMed]
- Mansuy, J.M.; Abravanel, F.; Miedouge, M.; Mengelle, C.; Merviel, C.; Dubois, M.; Kamar, N.; Rostaing, L.; Alric, L.; Moreau, J.; et al. Acute hepatitis E in south-west France over a 5-year period. J. Clin. Virol. 2009, 44, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S.; Kamili, S.; Khuroo, M.S. Clinical course and duration of viremia in vertically transmitted hepatitis E virus (HEV) infection in babies born to HEV-infected mothers. J. Viral Hepat. 2009, 16, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Singhal, A.; Panda, S.K.; Acharya, S.K. A 20-year single-center experience with acute liver failure during pregnancy: Is the prognosis really worse? Hepatology 2008, 48, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Hazeldine, S.; Banks, M.; Ijaz, S.; Bendall, R. Locally acquired hepatitis E in chronic liver disease. Lancet 2007, 369, 1260. [Google Scholar] [CrossRef]
- Peron, J.M.; Bureau, C.; Poirson, H.; Mansuy, J.M.; Alric, L.; Selves, J.; Dupuis, E.; Izopet, J.; Vinel, J.P. Fulminant liver failure from acute autochthonous hepatitis E in France: Description of seven patients with acute hepatitis E and encephalopathy. J. Viral Hepat. 2007, 14, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Peron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, E.B.; van den Berg, A.P.; Porte, R.J.; Benne, C.A.; Vennema, H.; Reimerink, J.H.; Koopmans, M.P. Chronic hepatitis E virus infection in liver transplant recipients. Liver Transpl. 2008, 14, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Gerolami, R.; Moal, V.; Colson, P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N. Engl. J. Med. 2008, 358, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Kaba, M.; Moreau, J.; Brouqui, P. Hepatitis E in an HIV-infected patient. J. Clin. Virol. 2009, 45, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Tedder, R.S.; Ijaz, S. Persistent carriage of hepatitis E virus in patients with HIV infection. N. Engl. J. Med. 2009, 361, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Kenfak-Foguena, A.; Schoni-Affolter, F.; Burgisser, P.; Witteck, A.; Darling, K.E.; Kovari, H.; Kaiser, L.; Evison, J.M.; Elzi, L.; Gurter-De La Fuente, V.; et al. Hepatitis E Virus seroprevalence and chronic infections in patients with HIV, Switzerland. Emerg. Infect. Dis. 2011, 17, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Ollier, L.; Tieulie, N.; Sanderson, F.; Heudier, P.; Giordanengo, V.; Fuzibet, J.G.; Nicand, E. Chronic hepatitis after hepatitis E virus infection in a patient with non-Hodgkin lymphoma taking rituximab. Ann. Intern. Med. 2009, 150, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Shimizu, Y.K.; Tanaka, T.; Kuroda, K.; Arakawa, Y.; Takahashi, K.; Mishiro, S.; Shimizu, K.; Moriyama, M. Persistent infection of hepatitis E virus transmitted by blood transfusion in a patient with T-cell lymphoma. Hepatol. Res. 2007, 37, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Peron, J.M.; Mansuy, J.M.; Recher, C.; Bureau, C.; Poirson, H.; Alric, L.; Izopet, J.; Vinel, J.P. Prolonged hepatitis E in an immunocompromised patient. J. Gastroenterol. Hepatol. 2006, 21, 1223–1224. [Google Scholar] [CrossRef] [PubMed]
- Tavitian, S.; Peron, J.M.; Huynh, A.; Mansuy, J.M.; Ysebaert, L.; Huguet, F.; Vinel, J.P.; Attal, M.; Izopet, J.; Recher, C. Hepatitis E virus excretion can be prolonged in patients with hematological malignancies. J. Clin. Virol. 2010, 49, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Rostaing, L.; Legrand-Abravanel, F.; Izopet, J. How should hepatitis E virus infection be defined in organ-transplant recipients? Am. J. Transplant. 2013, 13, 1935–1936. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Mansuy, J.M.; Cointault, O.; Selves, J.; Abravanel, F.; Danjoux, M.; Otal, P.; Esposito, L.; Durand, D.; Izopet, J.; et al. Hepatitis E virus-related cirrhosis in kidney- and kidney-pancreas-transplant recipients. Am. J. Transplant. 2008, 8, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Kamar, N.; van Eijk, J.J.; McLean, B.N.; Cintas, P.; Bendall, R.P.; Jacobs, B.C. Hepatitis E virus and neurological injury. Nat. Rev. Neurol. 2016, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, B.; van der Eijk, A.A.; Pas, S.D.; Hunter, J.G.; Madden, R.G.; Tio-Gillen, A.P.; Dalton, H.R.; Jacobs, B.C. Guillain-Barre syndrome associated with preceding hepatitis E virus infection. Neurology 2014, 82, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Van Eijk, J.J.; Madden, R.G.; van der Eijk, A.A.; Hunter, J.G.; Reimerink, J.H.; Bendall, R.P.; Pas, S.D.; Ellis, V.; van Alfen, N.; Beynon, L.; et al. Neuralgic amyotrophy and hepatitis E virus infection. Neurology 2014, 82, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Cintas, P.; Garrouste, C.; Uro-Coste, E.; Cointault, O.; Rostaing, L. Hepatitis E virus-induced neurological symptoms in a kidney-transplant patient with chronic hepatitis. Am. J. Transplant. 2010, 10, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Helsen, N.; Debing, Y.; Paeshuyse, J.; Dallmeier, K.; Boon, R.; Coll, M.; Sancho-Bru, P.; Claes, C.; Neyts, J.; Verfaillie, C.M. Stem cell-derived hepatocytes: A novel model for hepatitis E virus replication. J. Hepatol. 2016, 64, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Weclawiak, H.; Guilbeau-Frugier, C.; Legrand-Abravanel, F.; Cointault, O.; Ribes, D.; Esposito, L.; Cardeau-Desangles, I.; Guitard, J.; Sallusto, F.; et al. Hepatitis E virus and the kidney in solid-organ transplant patients. Transplantation 2012, 93, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Guinault, D.; Ribes, D.; Delas, A.; Milongo, D.; Abravanel, F.; Puissant-Lubrano, B.; Izopet, J.; Kamar, N. Hepatitis E Virus-Induced Cryoglobulinemic Glomerulonephritis in a Nonimmunocompromised Person. Am. J. Kidney Dis. 2016, 67, 660–663. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G. Renal involvement in hepatitis C infection: Cryoglobulinemic glomerulonephritis. Kidney Int. 1998, 54, 650–671. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Zhao, C.; Huang, W.; Harrison, T.J.; Zhang, H.; Geng, K.; Wang, Y. Detection and assessment of infectivity of hepatitis E virus in urine. J. Hepatol. 2016, 64, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.P.; Kasorndorkbua, C.; Halbur, P.G.; Haqshenas, G.; Guenette, D.K.; Toth, T.E.; Meng, X.J. Evidence of extrahepatic sites of replication of the hepatitis E virus in a swine model. J. Clin. Microbiol. 2001, 39, 3040–3046. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Purcell, R.H. Fields Virology 2013, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2242–2258. [Google Scholar]
- Tsarev, S.A.; Tsareva, T.S.; Emerson, S.U.; Govindarajan, S.; Shapiro, M.; Gerin, J.L.; Purcell, R.H. Successful passive and active immunization of cynomolgus monkeys against hepatitis E. Proc. Natl. Acad. Sci. USA 1994, 91, 10198–10202. [Google Scholar] [CrossRef] [PubMed]
- Hishiki, T.; Shimizu, Y.; Tobita, R.; Sugiyama, K.; Ogawa, K.; Funami, K.; Ohsaki, Y.; Fujimoto, T.; Takaku, H.; Wakita, T.; et al. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 2010, 84, 12048–12057. [Google Scholar] [CrossRef] [PubMed]
- Rogee, S.; le Gall, M.; Chafey, P.; Bouquet, J.; Cordonnier, N.; Frederici, C.; Pavio, N. Quantitative proteomics identifies host factors modulated during acute hepatitis E virus infection in the swine model. J. Virol. 2015, 89, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yesupriya, A.; Chang, M.H.; Teshale, E.; Teo, C.G. Apolipoprotein E and protection against hepatitis E viral infection in American non-Hispanic blacks. Hepatology 2015, 62, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Ji, Z.S. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 1999, 40, 1–16. [Google Scholar] [PubMed]
- Liao, Z.; Graham, D.R.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retrovir. 2003, 19, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Pischke, S.; Hartl, J.; Roque-Afonso, A.M.; Mallet, V. Apolipoprotein E epsilon3 and epsilon4 are associated with a lower exposure to hepatitis E virus in American non-Hispanic blacks. Hepatology 2015, 62, 1346–1352. [Google Scholar]
- Wilson, A.G.; Symons, J.A.; McDowell, T.L.; McDevitt, H.O.; Duff, G.W. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc. Natl. Acad. Sci. USA 1997, 94, 3195–3199. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Arankalle, V.A. Association of polymorphisms in the promoter regions of TNF-alpha (−308) with susceptibility to hepatitis E virus and TNF-alpha (−1031) and IFN-gamma (+874) genes with clinical outcome of hepatitis E infection in India. J. Hepatol. 2011, 55, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Boon, D.; McDonald, S.L.; Myers, T.G.; Tomioka, K.; Nguyen, H.; Engle, R.E.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: Similarities and differences. J. Virol. 2010, 84, 11264–11278. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zafrullah, M.; Mixson-Hayden, T.; Dai, X.; Liang, J.; Meng, J.; Kamili, S. Suppression of interferon-alpha signaling by hepatitis E virus. Hepatology 2012, 55, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral Hepat. 2016, 23, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Ma, Z.; Wang, R.; Yu, Y.; Kannan, H.; Fredericksen, B.; Zhang, Y.J. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 2014, 88, 8696–8705. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.J. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.B.; Chatterjee, S.N.; Arankalle, V.A.; Lole, K.S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 2013, 8, e63793. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aggarwal, R.; Jameel, S.; Puri, P.; Gupta, V.K.; Ramesh, V.S.; Bhatia, S.; Naik, S. Cellular immune responses in acute hepatitis E virus infection to the viral open reading frame 2 protein. Viral Immunol. 2007, 20, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aggarwal, R.; Bhagat, M.R.; Chowdhury, A.; Naik, S. Alterations in natural killer cells and natural killer T cells during acute viral hepatitis E. J. Viral Hepat. 2008, 15, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.B.; Gupta, P.; Durgapal, H.; Rath, S.; Gupta, S.D.; Acharya, S.K.; Panda, S.K. Study of cellular immune response against Hepatitis E virus (HEV). J. Viral Hepat. 2011, 18, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Purcell, R.H. Recombinant vaccines for hepatitis E. Trends Mol. Med. 2001, 7, 462–466. [Google Scholar] [CrossRef]
- Tang, X.; Yang, C.; Gu, Y.; Song, C.; Zhang, X.; Wang, Y.; Zhang, J.; Hew, C.L.; Li, S.; Xia, N.; et al. Structural basis for the neutralization and genotype specificity of hepatitis E virus. Proc. Natl. Acad. Sci. USA 2011, 108, 10266–10271. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.C.; Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Wang, H.; Yang, C.L.; Jiang, H.M.; Cai, J.P.; et al. Efficacy and safety of a recombinant hepatitis E vaccine in healthy adults: A large-scale, randomised, double-blind placebo-controlled, phase 3 trial. Lancet 2010, 376, 895–902. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.F.; Huang, S.J.; Wu, T.; Hu, Y.M.; Wang, Z.Z.; Wang, H.; Jiang, H.M.; Wang, Y.J.; Yan, Q.; et al. Long-term efficacy of a hepatitis E vaccine. N. Engl. J. Med. 2015, 372, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.P.; Tsarev, S.A.; Iqbal, M.; Ticehurst, J.; Emerson, S.; Ahmed, A.; Duncan, J.; Rafiqui, A.R.; Malik, I.A.; Purcell, R.H.; et al. Epidemic hepatitis E in Pakistan: Patterns of serologic response and evidence that antibody to hepatitis E virus protects against disease. J. Infect. Dis. 1994, 170, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Arankalle, V.A.; Chadha, M.S.; Chobe, L.P. Long-term serological follow up and cross-challenge studies in rhesus monkeys experimentally infected with hepatitis E virus. J. Hepatol. 1999, 30, 199–204. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Yao, X.; Liang, Z.L.; Wu, T.; Li, J.X.; Yan, Q.; et al. Protection against hepatitis E virus infection by naturally acquired and vaccine-induced immunity. Clin. Microbiol. Infect. 2014, 20, O397–O405. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Lhomme, S.; Chapuy-Regaud, S.; Mansuy, J.M.; Muscari, F.; Sallusto, F.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus reinfections in solid-organ-transplant recipients can evolve into chronic infections. J. Infect. Dis. 2014, 209, 1900–1906. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.M.; Aggarwal, R.; Kumar, D.; Jameel, S.; Naik, S. Effector T cells immune reactivity among patients with acute hepatitis E. J. Viral Hepat. 2011, 18, e603–e608. [Google Scholar] [CrossRef] [PubMed]
- TrehanPati, N.; Sukriti, S.; Geffers, R.; Hissar, S.; Riese, P.; Toepfer, T.; Guzman, C.A.; Sarin, S.K. Gene expression profiles of T cells from hepatitis E virus infected patients in acute and resolving phase. J. Clin. Immunol. 2011, 31, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, A.S.; Das, R.; Rathod, S.B.; Gurav, Y.K.; Arankalle, V.A. Peripheral T regulatory cells and cytokines in hepatitis E infection. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Gisa, A.; Suneetha, P.V.; Behrendt, P.; Pischke, S.; Bremer, B.; Falk, C.S.; Manns, M.P.; Cornberg, M.; Wedemeyer, H.; Kraft, A.R. Cross-genotype-specific T-cell responses in acute hepatitis E virus (HEV) infection. J. Viral Hepat. 2016, 23, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Aggarwal, R.; Naik, S.R.; Saraswat, V.; Ghoshal, U.C.; Naik, S. Hepatitis E virus is responsible for decompensation of chronic liver disease in an endemic region. Indian J. Gastroenterol. 2004, 23, 59–62. [Google Scholar] [PubMed]
- Jeblaoui, A.; Haim-Boukobza, S.; Marchadier, E.; Mokhtari, C.; Roque-Afonso, A.M. Genotype 4 hepatitis e virus in france: An autochthonous infection with a more severe presentation. Clin. Infect. Dis. 2013, 57, e122–e126. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P. Hepatitis E virus and fulminant hepatitis--a virus or host-specific pathology? Liver Int. 2015, 35, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Saravanabalaji, S.; Tripathy, A.S.; Dhoot, R.R.; Chadha, M.S.; Kakrani, A.L.; Arankalle, V.A. Viral load, antibody titers and recombinant open reading frame 2 protein-induced TH1/TH2 cytokines and cellular immune responses in self-limiting and fulminant hepatitis e. Intervirology 2009, 52, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aggarwal, R.; Sachdeva, S.; Alam, M.I.; Jameel, S.; Naik, S. Adaptive immune responses during acute uncomplicated and fulminant hepatitis E. J. Gastroenterol. Hepatol. 2011, 26, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, V.; Goel, A.; Rawat, A.; Naik, S.; Aggarwal, R. Histological and immunohistochemical features in fatal acute fulminant hepatitis E. Indian J. Pathol. Microbiol. 2012, 55, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Anty, R.; Ollier, L.; Peron, J.M.; Nicand, E.; Cannavo, I.; Bongain, A.; Giordanengo, V.; Tran, A. First case report of an acute genotype 3 hepatitis E infected pregnant woman living in South-Eastern France. J. Clin. Virol. 2012, 54, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Tabatabai, J.; Wenzel, J.J.; Soboletzki, M.; Flux, C.; Navid, M.H.; Schnitzler, P. First case report of an acute hepatitis E subgenotype 3c infection during pregnancy in Germany. J. Clin. Virol. 2014, 61, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. The Th1/Th2 paradigm. Immunol. Today 1997, 18, 263–266. [Google Scholar] [CrossRef]
- Pal, R.; Aggarwal, R.; Naik, S.R.; Das, V.; Das, S.; Naik, S. Immunological alterations in pregnant women with acute hepatitis E. J. Gastroenterol. Hepatol. 2005, 20, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, R.; Patra, S.; David, P.; Vyas, A.; Khanam, A.; Hissar, S.; Gupta, E.; Kumar, G.; Kottilil, S.; Maiwall, R.; et al. Impaired monocyte-macrophage functions and defective Toll-like receptor signaling in hepatitis E virus-infected pregnant women with acute liver failure. Hepatology 2015, 62, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Devi, S.G.; Kar, P.; Agarwal, S.; Husain, S.A.; Gupta, R.K.; Sharma, S. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 2014, 65, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.D.; Das, B.C.; Kumar, A.; Gondal, R.; Kumar, D.; Kar, P. High viral load and deregulation of the progesterone receptor signaling pathway: Association with hepatitis E-related poor pregnancy outcome. J. Hepatol. 2011, 54, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Check, J.H.; Arwitz, M.; Gross, J.; Peymer, M.; Szekeres-Bartho, J. Lymphocyte immunotherapy (LI) increases serum levels of progesterone induced blocking factor (PIBF). Am. J. Reprod. Immunol. 1997, 37, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Druckmann, R.; Druckmann, M.A. Progesterone and the immunology of pregnancy. J. Steroid Biochem. Mol. Biol. 2005, 97, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Kmush, B.L.; Labrique, A.; Li, W.; Klein, S.L.; Schulze, K.; Shaikh, S.; Ali, H.; Engle, R.E.; Wu, L.; Purcell, R.H.; et al. The Association of Cytokines and Micronutrients with Hepatitis E Virus Infection During Pregnancy and the Postpartum Period in Rural Bangladesh. Am. J. Trop. Med. Hyg. 2016, 94, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Jilani, N.; Das, B.C.; Husain, S.A.; Baweja, U.K.; Chattopadhya, D.; Gupta, R.K.; Sardana, S.; Kar, P. Hepatitis E virus infection and fulminant hepatic failure during pregnancy. J. Gastroenterol. Hepatol. 2007, 22, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Yang, C.; Yu, W.; Zhao, X.; Zhao, C.; He, Z.; Jing, S.; Wang, H.; Huang, F. Pregnancy serum facilitates hepatitis E virus replication in vitro. J. Gen. Virol. 2015, 96 Pt 5, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Jilani, N.; Husain, S.A.; Pasha, S.T.; Anand, R.; Rai, A.; Das, B.C. Does hepatitis E viral load and genotypes influence the final outcome of acute liver failure during pregnancy? Am. J. Gastroenterol. 2008, 103, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Garrouste, C.; Haagsma, E.B.; Garrigue, V.; Pischke, S.; Chauvet, C.; Dumortier, J.; Cannesson, A.; Cassuto-Viguier, E.; Thervet, E.; et al. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology 2011, 140, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- McAlister, V.C.; Haddad, E.; Renouf, E.; Malthaner, R.A.; Kjaer, M.S.; Gluud, L.L. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: A meta-analysis. Am. J. Transplant. 2006, 6, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, X.; Debing, Y.; Chen, K.; van der Laan, L.J.; Neyts, J.; Janssen, H.L.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus. Gastroenterology 2014, 146, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, Y.; Metselaar, H.J.; Janssen, H.L.; Peppelenbosch, M.P.; Pan, Q. Rapamycin and everolimus facilitate hepatitis E virus replication: Revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. J. Hepatol. 2014, 61, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Marchese, P.; Castro, M.G.; Lowenstein, P.R.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat. Med. 2005, 11, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Moal, V.; Textoris, J.; Ben Amara, A.; Mehraj, V.; Berland, Y.; Colson, P.; Mege, J.L. Chronic hepatitis E virus infection is specifically associated with an interferon-related transcriptional program. J. Infect. Dis. 2013, 207, 125–132. [Google Scholar] [PubMed]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus quasispecies and the outcome of acute hepatitis E in solid-organ transplant patients. J. Virol. 2012, 86, 10006–10014. [Google Scholar] [CrossRef] [PubMed]
- Suneetha, P.V.; Pischke, S.; Schlaphoff, V.; Grabowski, J.; Fytili, P.; Gronert, A.; Bremer, B.; Markova, A.; Jaroszewicz, J.; Bara, C.; et al. Hepatitis E virus (HEV)-specific T-cell responses are associated with control of HEV infection. Hepatology 2012, 55, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Legrand-Abravanel, F.; Dalton, H.R.; Izopet, J. Hepatitis E virus-specific T-cell response after transplantation. Hepatology 2012, 55, 1643; author reply 1644. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Barrague, H.; Dorr, G.; Saune, K.; Peron, J.M.; Alric, L.; Kamar, N.; Izopet, J.; Champagne, E. Conventional and innate lymphocytes response at the acute phase of HEV infection in transplanted patients. J. Infect. 2016, 72, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Garrouste, C.; Kamar, N.; Saune, K.; Abravanel, F.; Mansuy, J.M.; Dubois, M.; Rostaing, L.; Izopet, J. Influence of polyproline region and macro domain genetic heterogeneity on HEV persistence in immunocompromised patients. J. Infect. Dis. 2014, 209, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the polyproline region of the hepatitis E virus in immunocompromised patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93 Pt 3, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J.; Purcell, R.H.; Halbur, P.G.; Lehman, J.R.; Webb, D.M.; Tsareva, T.S.; Haynes, J.S.; Thacker, B.J.; Emerson, S.U. A novel virus in swine is closely related to the human hepatitis E virus. Proc. Natl. Acad. Sci. USA 1997, 94, 9860–9865. [Google Scholar] [CrossRef] [PubMed]
- Halbur, P.G.; Kasorndorkbua, C.; Gilbert, C.; Guenette, D.; Potters, M.B.; Purcell, R.H.; Emerson, S.U.; Toth, T.E.; Meng, X.J. Comparative pathogenesis of infection of pigs with hepatitis E viruses recovered from a pig and a human. J. Clin. Microbiol. 2001, 39, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Salines, M.; Barnaud, E.; Andraud, M.; Eono, F.; Renson, P.; Bourry, O.; Pavio, N.; Rose, N. Hepatitis E virus chronic infection of swine co-infected with Porcine Reproductive and Respiratory Syndrome Virus. Vet. Res. 2015, 46, 55. [Google Scholar] [CrossRef] [PubMed]
- Cossaboom, C.M.; Cordoba, L.; Sanford, B.J.; Pineyro, P.; Kenney, S.P.; Dryman, B.A.; Wang, Y.; Meng, X.J. Cross-species infection of pigs with a novel rabbit, but not rat, strain of hepatitis E virus isolated in the United States. J. Gen. Virol. 2012, 93 Pt 8, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Bu, Q.N.; Wang, L.; Han, J.; Du, R.J.; Lei, Y.X.; Ouyang, Y.Q.; Li, J.; Zhu, Y.H.; Lu, F.M.; et al. Transmission of hepatitis E virus from rabbits to cynomolgus macaques. Emerg. Infect. Dis. 2013, 19, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Liu, L.; Wang, L.; Zhang, Y.; Zeng, H.; Liu, P.; Zou, Q.; Wang, L.; Zhuang, H. Experimental infection of pregnant rabbits with hepatitis E virus demonstrating high mortality and vertical transmission. J. Viral Hepat. 2015, 22, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xia, J.; Wang, L.; Wang, Y. Experimental infection of rabbits with genotype 3 hepatitis E virus produced both chronicity and kidney injury. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Yang, T.; Yoshizaki, S.; Ami, Y.; Suzaki, Y.; Ishii, K.; Kishida, N.; Shirakura, M.; Asanuma, H.; Takeda, N.; et al. Ferret hepatitis E virus infection induces acute hepatitis and persistent infection in ferrets. Vet. Microbiol. 2016, 183, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, R.; She, R.; Soomro, M.H.; Mao, J.; Du, F.; Zhao, Y.; Liu, C. Effect of swine hepatitis E virus on the livers of experimentally infected Mongolian gerbils by swine hepatitis E virus. Virus Res. 2015, 208, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Yoshizaki, S.; Ami, Y.; Suzaki, Y.; Yasuda, S.P.; Yoshimatsu, K.; Arikawa, J.; Takeda, N.; Wakita, T. Susceptibility of laboratory rats against genotypes 1, 3, 4, and rat hepatitis E viruses. Vet. Microbiol. 2013, 163, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.H.; Engle, R.E.; Rood, M.P.; Kabrane-Lazizi, Y.; Nguyen, H.T.; Govindarajan, S.; St Claire, M.; Emerson, S.U. Hepatitis E virus in rats, Los Angeles, California, USA. Emerg. Infect. Dis. 2011, 17, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Gass, S.; Giersch, K.; Groth, A.; Kah, J.; Volz, T.; Rapp, G.; Schobel, A.; Lohse, A.W.; Polywka, S.; et al. Human liver chimeric mice as a new model of chronic hepatitis E virus infection and preclinical drug evaluation. J. Hepatol. 2016, 64, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Van de Garde, M.D.; Pas, S.D.; van der Net, G.; de Man, R.A.; Osterhaus, A.D.; Haagmans, B.L.; Boonstra, A.; Vanwolleghem, T. Hepatitis E Virus (HEV) Genotype 3 Infection of Human Liver Chimeric Mice as a Model for Chronic HEV Infection. J. Virol. 2016, 90, 4394–4401. [Google Scholar] [CrossRef] [PubMed]
- Billam, P.; Pierson, F.W.; Li, W.; LeRoith, T.; Duncan, R.B.; Meng, X.J. Development and validation of a negative-strand-specific reverse transcription-PCR assay for detection of a chicken strain of hepatitis E virus: Identification of nonliver replication sites. J. Clin. Microbiol. 2008, 46, 2630–2634. [Google Scholar] [CrossRef] [PubMed]
- Pudupakam, R.S.; Huang, Y.W.; Opriessnig, T.; Halbur, P.G.; Pierson, F.W.; Meng, X.J. Deletions of the hypervariable region (HVR) in open reading frame 1 of hepatitis E virus do not abolish virus infectivity: Evidence for attenuation of HVR deletion mutants in vivo. J. Virol. 2009, 83, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Pudupakam, R.S.; Kenney, S.P.; Cordoba, L.; Huang, Y.W.; Dryman, B.A.; Leroith, T.; Pierson, F.W.; Meng, X.J. Mutational analysis of the hypervariable region of hepatitis e virus reveals its involvement in the efficiency of viral RNA replication. J. Virol. 2011, 85, 10031–10040. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and evaluation of an efficient cell-culture system for Hepatitis E virus. J. Gen. Virol. 2007, 88 Pt 3, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Okamoto, H. Development and characterization of a genotype 4 hepatitis E virus cell culture system using a HE-JF5/15F strain recovered from a fulminant hepatitis patient. J. Clin. Microbiol. 2009, 47, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Rogee, S.; Talbot, N.; Caperna, T.; Bouquet, J.; Barnaud, E.; Pavio, N. New models of hepatitis E virus replication in human and porcine hepatocyte cell lines. J. Gen. Virol. 2013, 94 Pt 3, 549–558. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lhomme, S.; Marion, O.; Abravanel, F.; Chapuy-Regaud, S.; Kamar, N.; Izopet, J. Hepatitis E Pathogenesis. Viruses 2016, 8, 212. https://doi.org/10.3390/v8080212
Lhomme S, Marion O, Abravanel F, Chapuy-Regaud S, Kamar N, Izopet J. Hepatitis E Pathogenesis. Viruses. 2016; 8(8):212. https://doi.org/10.3390/v8080212
Chicago/Turabian StyleLhomme, Sébastien, Olivier Marion, Florence Abravanel, Sabine Chapuy-Regaud, Nassim Kamar, and Jacques Izopet. 2016. "Hepatitis E Pathogenesis" Viruses 8, no. 8: 212. https://doi.org/10.3390/v8080212