
Detection and Quantification of SARS-CoV-2 Receptor Binding Domain Neutralization by a Sensitive Competitive ELISA Assay

 , , ,
, , , 
Abstract
: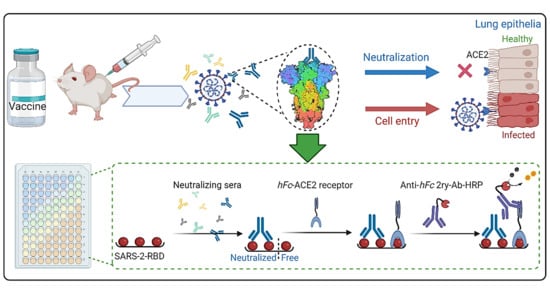
1. Introduction
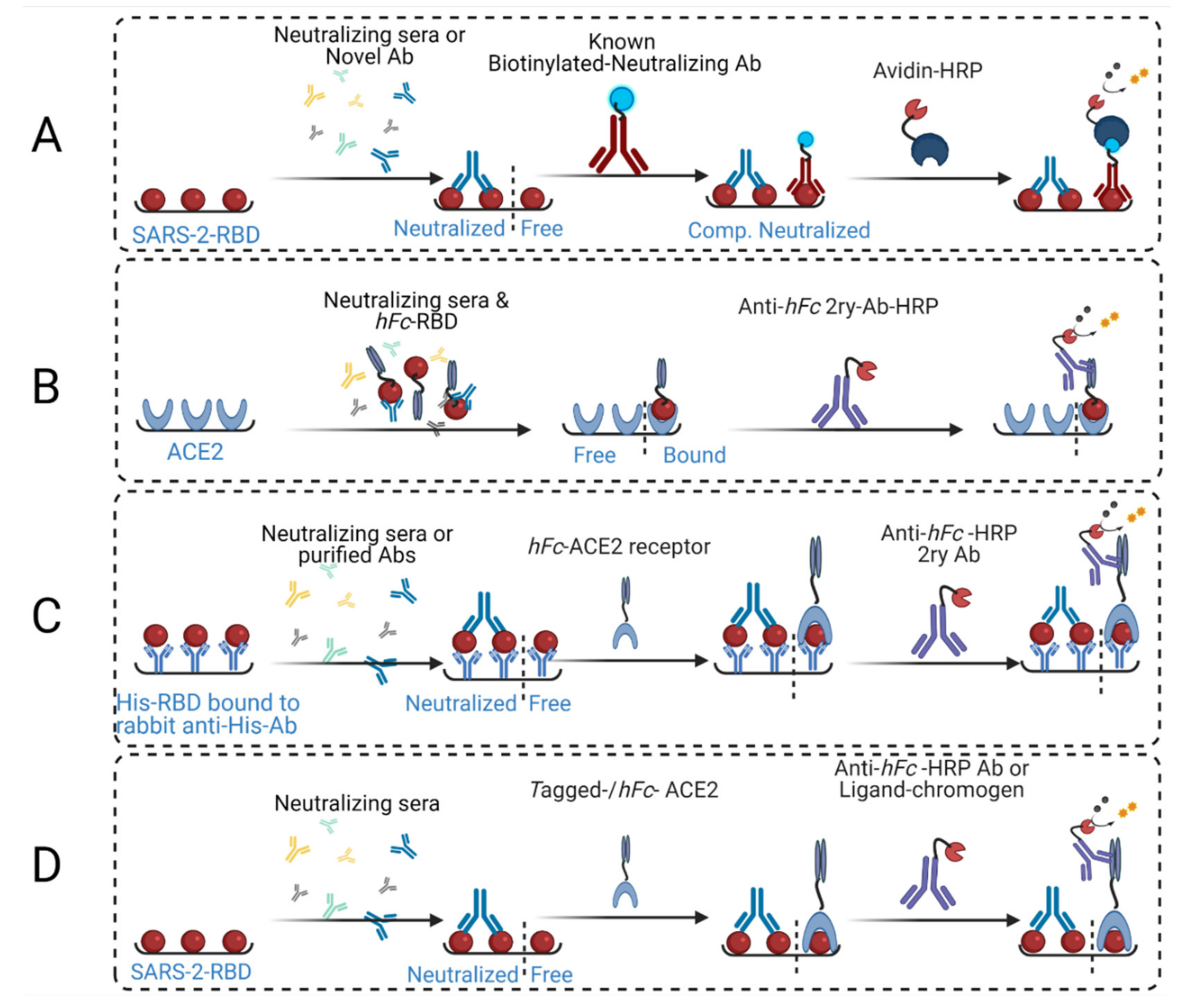
2. Comparison to Other Competitive ELISA Assays
3. Development of cELISA Assay
4. Evaluation of Conventional Validation Parameters
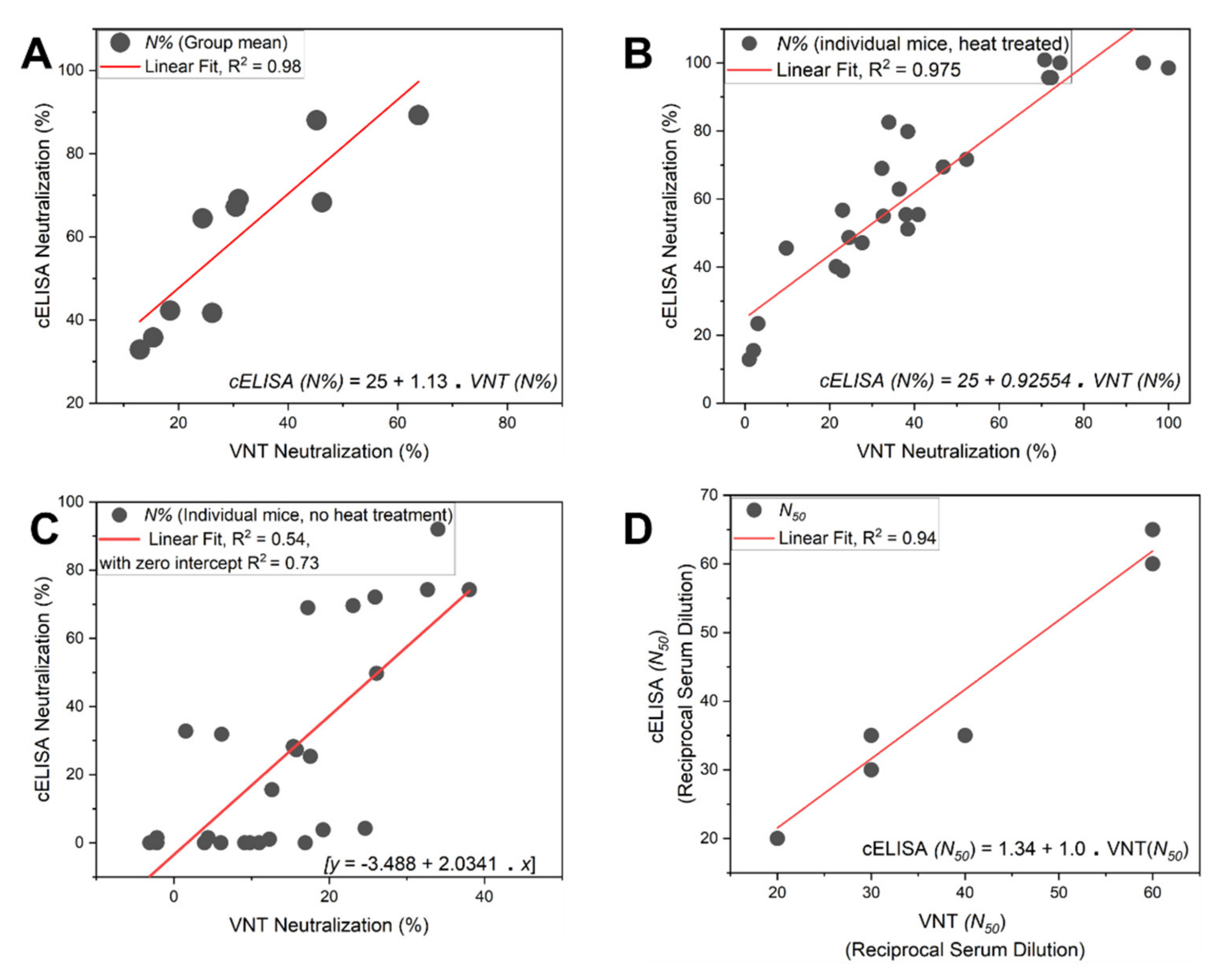
5. Virus Neutralization Assay: Orthogonal Validation
6. Assay Limitations
7. Reagents and Equipment
7.1. Reagents
- Recombinant SARS-CoV-2 spike-RBD-His (RBD, Sino Biological, Beijing, China, Cat. No. 40592-V08H);
- PBS tablets (Gibco, Carlsbad, CA, USA, Cat. No. 18912014);
- Tween 20 (Sigma-Aldrich, Victoria, Australia, Cat. No. P1379);
- Recombinant human ACE2 with human IgG-Fc (hFc-ACE2, Sino Biological, Beijing, China, Cat. No. 10108-H02H);
- Sodium hydrogen carbonate (Chem Supply, South Australia, Australia, Cat. No. SA001-500G);
- Sodium carbonate (Chem Supply, Cat. No. SA099-500G);
- Bovine serum albumin (BSA), heat shock fraction (Sigma-Aldrich, Cat. No. A7906-10G);
- Deionized water (resistivity ~18 MΩ cm);
- Sulfuric acid, 98% (Sigma-Aldrich, Cat. No. 258105)—CAUTION, sulfuric acid is a strong acid and must be handled inside of a fume hood;
- Secondary antibody (2ry Ab): Peroxidase (HRP)-conjugated goat anti-human IgG-Fc Ab (Sigma-Aldrich, Cat. No. A0170);
- SigmaFast o-phenylenediamine dihydrochloride tablets (OPD, Sigma-Aldrich, Cat. No. P9187).
7.2. Equipment
- Polystyrene microtiter ELISA high affinity plates, 96 flat bottom wells (Sarstedt, South Australia, Australia, Cat. No. 82.1581.200);
- Magnetic bar and stirrer;
- Micropipettes;
- Auto-ELISA (Viaflo ASSIST, Integra Biosciences, Victoria, Australia) equipped with multichannel pipette (Biotools Pty Ltd., Queensland, Australia);
- Sterile pipette tips and Eppendorf tubes;
- Falcon tubes, 50 mL (BD, Cat. No. 352070);
- SpectraMax 250 microplate reader (Molecular Devices, San Jose, CA, USA);
- Originpro 2020 (OriginLab Corporation, Northampton, MA, USA) or Prism v8.3 software (GraphPad Software Inc., San Diego, CA, USA) for function fitting and data analysis;
- Airtight plastic storage box for ELISA microtiter plates.
7.3. Reagent Setup
- hFc-ACE2 (12.5 µg/plate);
- RBD (5 µg/plate);
- Wash buffer, PBST (800 mL/plate);
- Bovine serum albumin (BSA) (0.675 g/plate);
- HRP-conjugated goat anti-human IgG-Fc Ab (3.3 µL/plate);
- OPD tablets (0.5 tablet/plate);
- Sulfuric acid (98%) (0.27 mL/plate).
Example Reagent Set-Up for Five Plates, including the Calibration Curve Plate
- 1.
- Prepare carbonate coating buffer (10 mL/plate):
- 2.
- Prepare PBST wash buffer (800 mL/plate):
- 3.
- Prepare 2% w/v BSA block solution (25 mL/plate):
- 4.
- Prepare 0.5% w/v BSA solution (35 mL/plate):
- 5.
- Prepare hFc-ACE2 solution (10 mL/plate):
- 6.
- Prepare 2ry Ab solution (10 mL/plate):
- 7.
- Prepare OPD substrate (10 mL/plate) (prepare fresh before addition):
- 8.
- Prepare 1N sulfuric acid (10 mL/plate):
8. Assay Protocol
8.1. Plate Coating and Blocking (4.5 h)
- Dissolve RBD in a suitable volume of distilled water to prepare 1 mg/mL stock solution in a 1 mL Eppendorf tube.
- Dilute RBD to a final concentration of 0.5 µg/mL RBD in carbonate buffer in a 50 mL Falcon tube, or other suitable container, by transferring the necessary volume from the stock solution prepared in Step 1.
- Add a volume of 100 µL of RBD (0.5 µg/mL) in carbonate buffer to coat each well of the ELISA sample plates (S-plates), giving a total RBD amount of 50 ng/well.
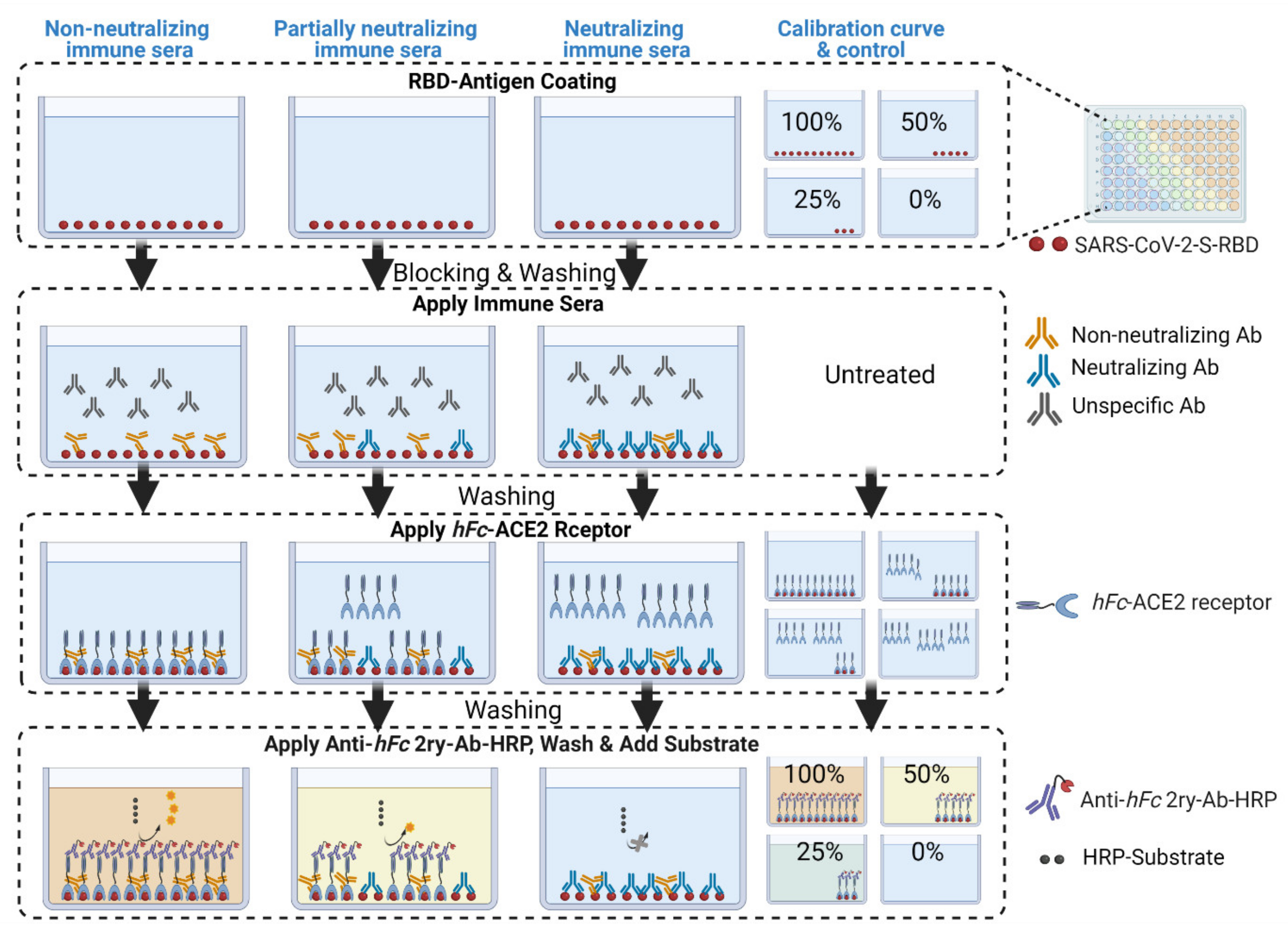
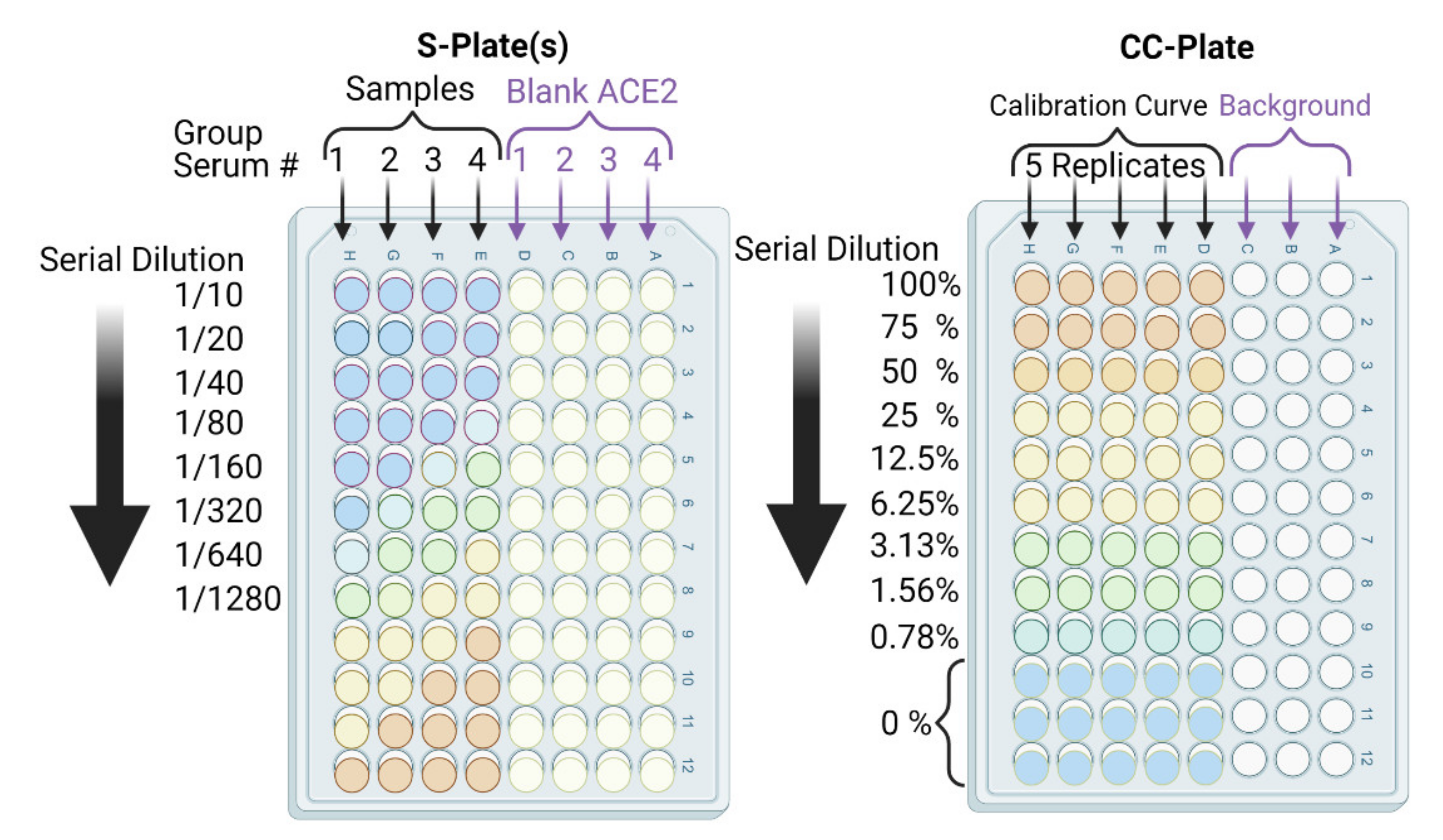
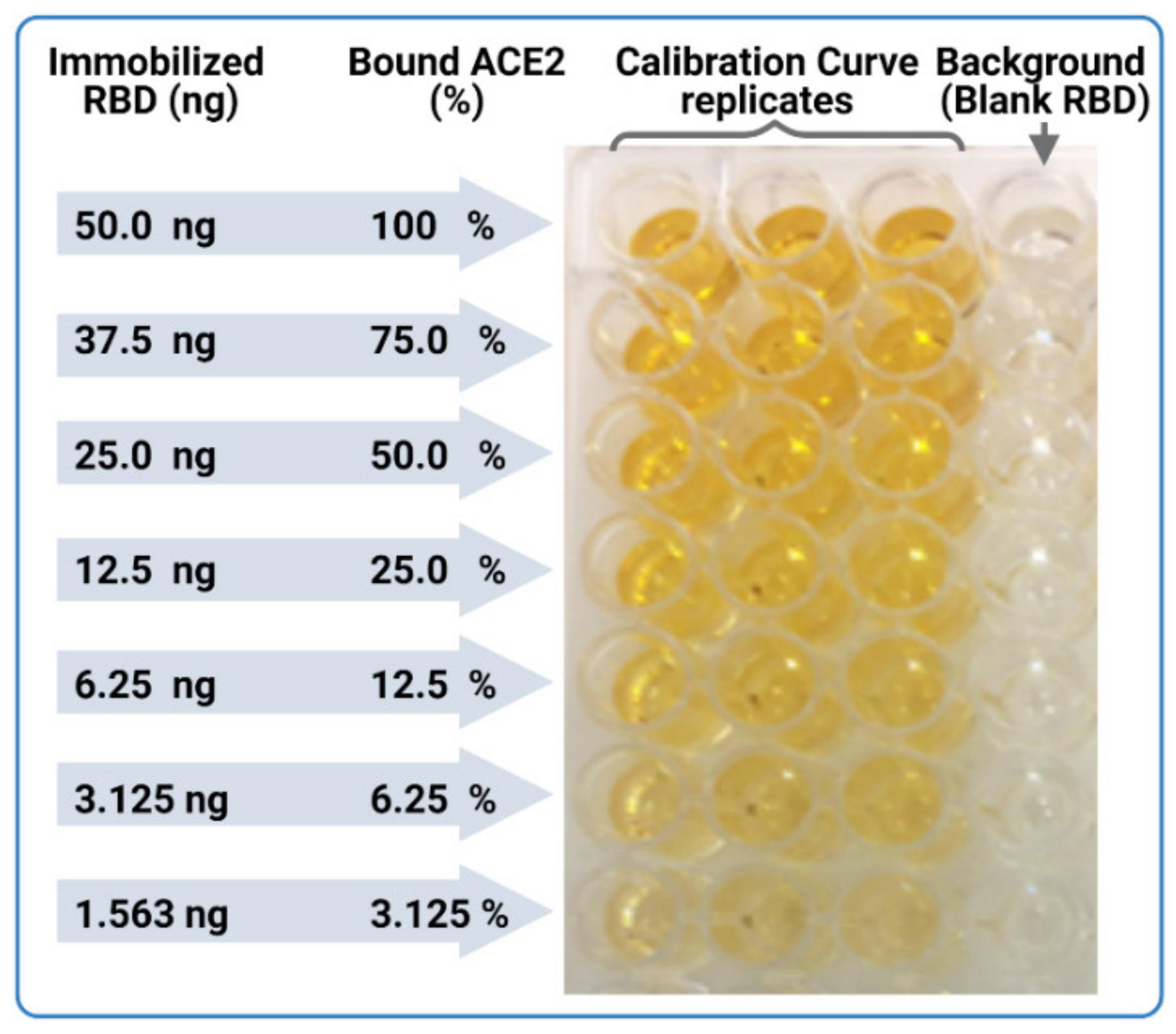
- In parallel, prepare a calibration curve plate (CC-plate) by adding 100 µL of, protein-free, carbonate buffer to five rows (D–H), starting from column 3, down to column 9. Add 25 µL of carbonate buffer to column 2 for five rows (D–H) (Figure 5).
- Coat the wells of the CC-plate by adding 200 µL of the RBD solution prepared in Step 2 to column 1 for five rows (D–H), then conduct two-fold serial dilutions using a multichannel pipette by taking 100 µL from column 1 and transferring it to column 3. Mix the solution by pipetting 50 µL up and down five times, then transfer 100 µL to column 4, and repeat until you reach column 9. Discard the last 100 µL you withdraw from column 9. This should leave three blank rows (A–C) across all columns, as well as rows (D–H) in column 2 (Figure 5).
- 6.
- Add 75 µL of the stock RBD solution (from Step 2) to the CC-plate column 2, rows D–H, using a multichannel pipette, then mix the solutions by pipetting 50 µL up and down five times.
- 7.
- Place the plates in an airtight storage container and incubate the S-plates and CC-plate for 90 min at 37 °C.
- 8.
- After incubation, dispose of the solution from the wells of the S-plates and CC-plate and wash the plates three times with deionized water, followed by three more washes with PBST wash buffer. Dry the plates by tapping upside down on paper towels.
- 9.
- Block the CC-plate and S-plates by adding 250 µL of 2% bovine serum albumin (BSA) solution in PBST wash buffer to all wells, including the three empty rows (A–C) of the CC-plate, which provide background OD readings for substrate control (Figure 4).
- 10.
- Place the plates in an airtight storage container and incubate all plates overnight at 4 °C or for 90 min at 37 °C.
- 11.
- Repeat washing Step 8 (Pause Point).
8.2. Addition of Immune Murine Sera (3.5 h)
- 12.
- Add 180 µL of 0.5% BSA solution in PBST to the wells in column 1 of each S-plate, and 100 µL of the same solution to all other wells of the S-plates.
- 13.
- Conduct heat treatment on a 45 µL aliquot of undiluted/neat, vortexed mouse serum in an Eppendorf tube at 57 °C for 30 min using a temperature-controlled water bath.
- 14.
- Transfer 20 µL of each heat-treated serum sample to a designated well in column 1, e.g., well 1H, of the S-plate (Figure 5). Transfer another 20 µL of serum from the same sample to another well of column 1, e.g., well 1D, of the same S-plate (Figure 5). Thus, each serum sample is added twice, to two different wells, of column 1; the second well will serve as a blank ACE2 control.
- 15.
- After serum addition to the S-plates, mix the contents of the column 1 wells by pipetting up and down a 50 µL volume five times. Then, conduct a two-fold serial dilution using a multichannel pipette by transferring 100 µL from column 1, rows A–H, to column 2, rows A–H, and repeat until you reach column 12. Discard the last 100 µL you withdraw from column 12. Repeat Steps 14 and 15 for all S-plates.
- 16.
- Add 100 µL of 0.5% BSA solution to all wells of the CC-plate without further treatment.
- 17.
- Repeat incubation Step 7.
- 19.
- Repeat washing Step 8.
8.3. Addition of hFC-ACE2, 2ry Ab and Substrate (5 h)
- 19.
- Add 100 µL of 2.5 µg/mL hFc-ACE2 solution to all wells of the CC-plate.
- 20.
- Add 100 µL of 2.5 µg/mL hFc-ACE2 solution to the E-H row wells of the S-plates only using a multichannel pipette, i.e., avoid the blank ACE2 control wells in rows A–D of the S-plates (Figure 5).
- 21.
- Repeat incubation Step 7.
- 22.
- Repeat washing Step 8.
- 23.
- Dilute goat anti-human IgG-Fc 2ry Ab to 1/3000 by adding 3.3 µL of 2ry Ab stock to 10 mL of 0.5% BSA solution for each plate (3.3 µL/10 mL 0.5% BSA per plate).
- 24.
- Add 100 µL of diluted 2ry Ab solution to all wells of the S-plates and CC-plate.
- 25.
- Repeat incubation Step 7.
- 26.
- Repeat washing Step 8, and wipe underneath the plates dry using Kimwipes.
- 27.
- Finally, add 100 µL of OPD substrate solution to all wells of the CC-plate and S-plates. Incubate the plates in the dark and allow them to react for 25 min at room temperature, then add a volume of 100 µL of stop solution, 1N sulfuric acid. Determine the absorbance at 450 nm OD450 using a common plate reader for all plates.
8.4. Calculation Procedure (2 h)
- 28.
- Subtract the background OD450 (mean of the first three rows (A–C)) from individual calibration curve OD450 for each of the five rows (D–H) of each dilution (columns 1–9) to yield blank-subtracted calibration curve individual values (five values, rows D–H, for each dilution step, columns 1–9) (Figure 5).
- 29.
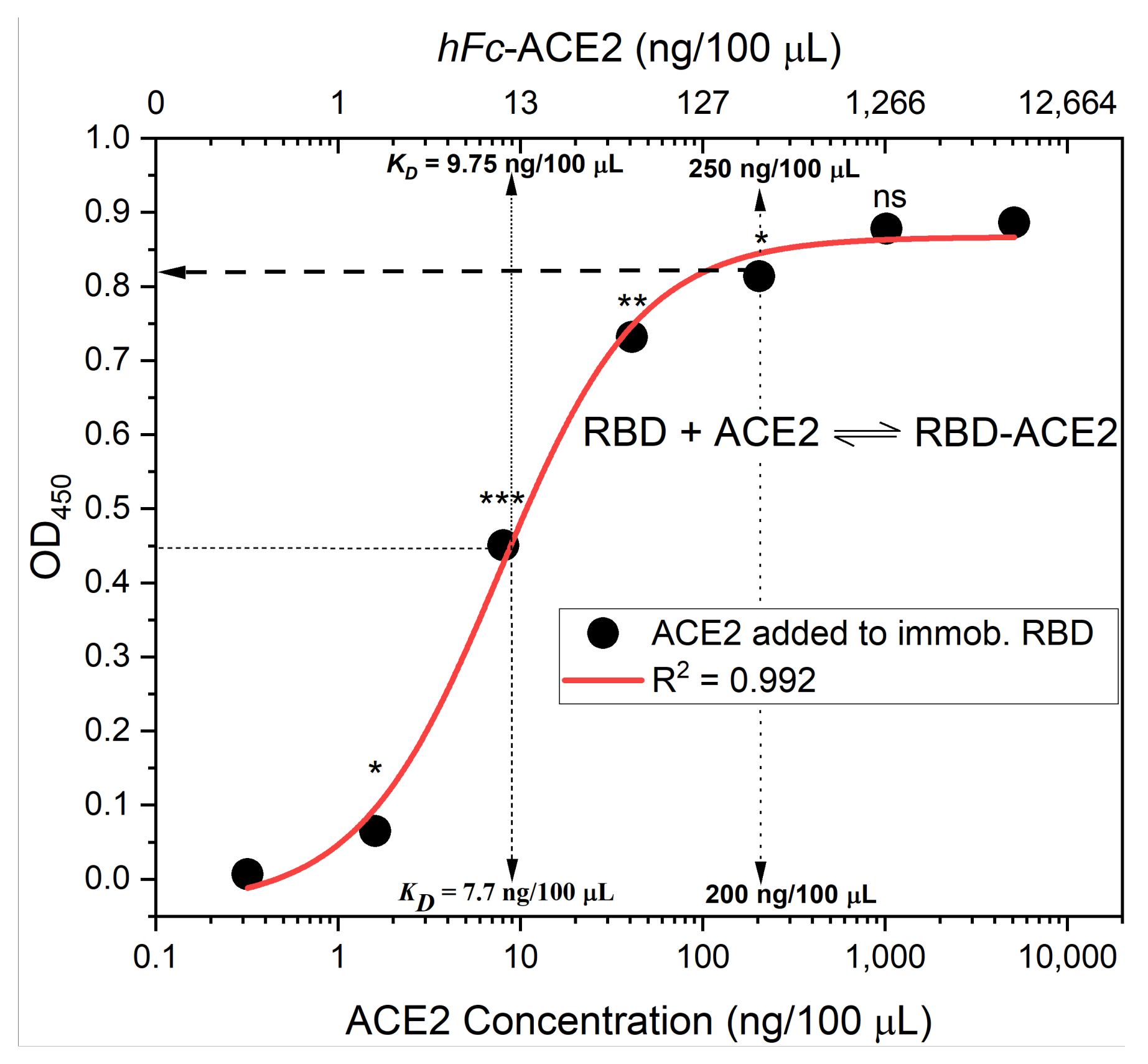
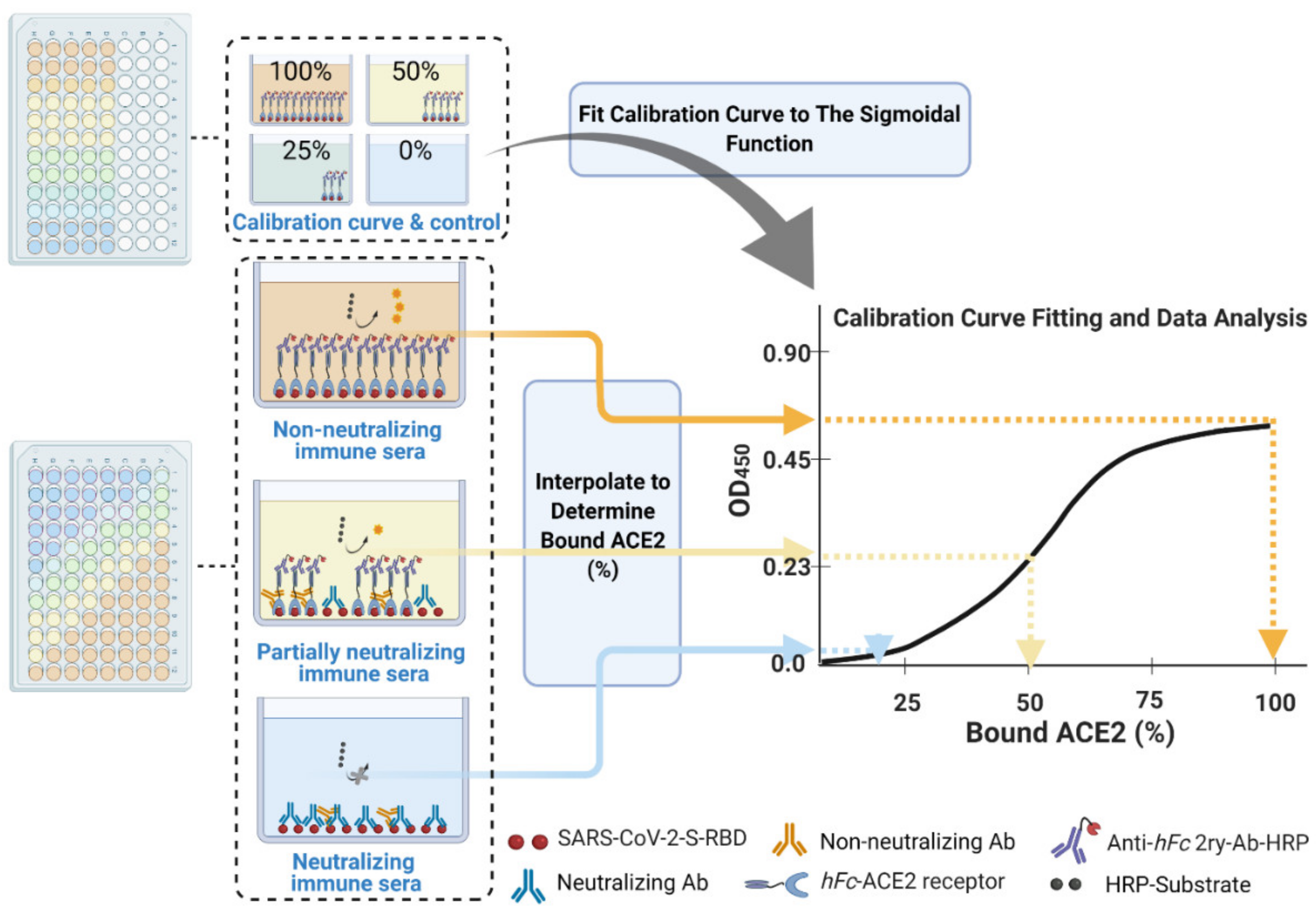
- Calculate the mean value (±standard deviation) of blank-subtracted calibration curve data and plot these against their associated concentration expressed in terms of bound ACE2 (%) (Figure 6), i.e., the OD reading average of the second and third columns correspond to the percent of available RBDs (37.5 and 25 ng, respectively) compared to the total RBD amount in column 1 (50 ng), 75% and 50% respectively.Critical: Check whether each point on the calibration curve (mean ± standard deviation) fits the following criteria:
- Each point is significantly different from its neighboring points—this means a tight standard deviation is required. The mean and standard deviation OD450 values for 25%, 50%, 75%, and 100% bound ACE2 should all be significantly different (p < 0.05).
- The lowest mean OD450 value is significantly higher than the background.
- The highest mean OD450 value should be in the range of 0.5 to 1.0.
- 30.
- Fit the OD450 mean (±standard deviation) versus bound ACE2 (%) plot with a four-parameter logistic function (Figure 6) using Graphpad Prism or Originpro 2020 software using (Equation (1)), where y is the mean OD reading, A1 and A2 are the minimum and maximum mean OD450 readings, x is bound ACE2 (%), xo is bound ACE2 (%) at 50% OD450 signal, and p is the power exponent.
- 31.
- Similarly, for the S-plates, subtract the OD450 of each serum’s blank ACE2 (from rows A–D) from the OD450 of the main serum (rows E–H) to yield corrected main serum OD450 data (the main serum placed in row H has its blank ACE2 serum sample in row D, see experimental procedure Steps 14–15), then subtract well D1 from H1, and D2 from H2 then D3 from H3, etc. This step aims to eliminate any 2ry Ab cross-reactivity with murine sera or any non-specific interaction that positively contributed to the OD450 signal, including background.Critical: Check the following criteria:
- Do any corrected main serum OD450 data points from the S-plate main sera exceed the sum of the 100% bound ACE2 calibration curve point? If several such points exist, then the heat treatment was not effective.
- Does the OD450 of any of the blank ACE2 wells exceed 10% of the calibration curve’s 100% bound ACE2 OD450 value after blank subtraction? If high OD450 values are consistent across several wells, then the 2ry Ab has high cross-reactivity to the serum employed. In this case, either switch to a less murine-cross-reactive 2ry Ab, or switch to a biotinylated-ACE2 and streptavidin-HRP system. If the high values are found only on a few plate edges or sporadic wells, then it is more likely that the washing step was ineffective.
- 32.
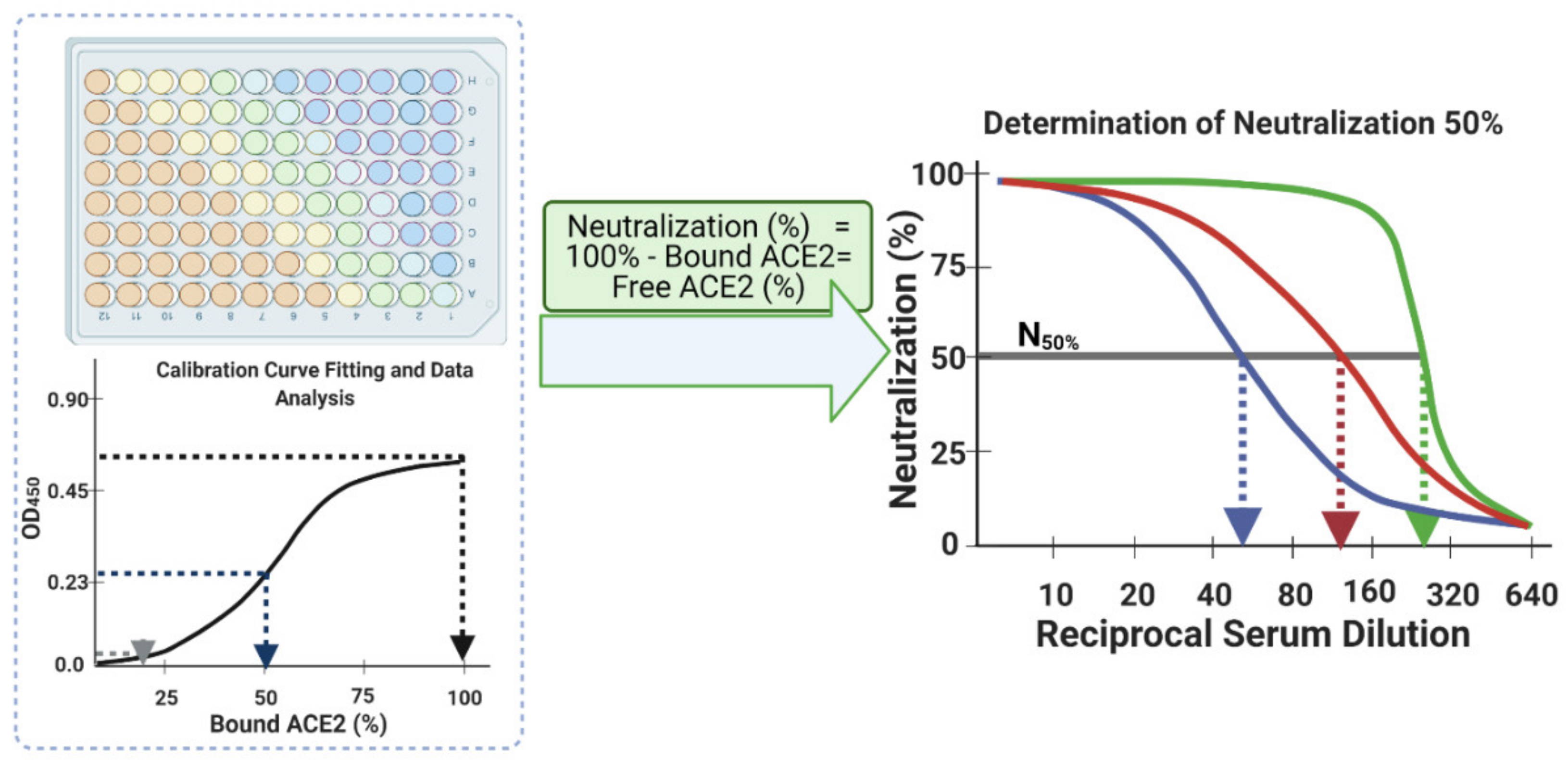
- Interpolate each corrected main serum OD450 data point using the established calibration curve logistic fit equation. As the four parameters of the function have already been established (Step 30), substitute ‘y’ with each corrected main serum OD450 data point to determine the amount of bound ACE2 (%) at each dilution down the row of that serum, i.e., the corrected main serum OD450 data points from rows E–H and columns 1–12. This provides an accurate transformation of OD450 data into bound ACE2 (%) for each serum sample at each dilution step.
- 33.
- Finally, plot the neutralization (%) value for each serum sample in each well against its corresponding reciprocal serum dilution, as in Figure 5. The reciprocal serum dilutions of columns 1, 2, 3, 4, and 5 are 10, 20, 40, 80, 160, etc. Each curve represents the serum neutralization profile of different dilutions (Figure 7), and fit neutralization (%) versus corresponding reciprocal serum data (i.e., 1/serum dilution value, so 1/20 dilution becomes 20), again using the four-parameter logistic function (Figure 7).
- 34.
- Interpolate each serum curve at 50% neutralization value (Figure 7) to determine the neutralization 50% reciprocal serum dilution (N50 or IC50), other common determinant/endpoint of neutralization efficacy. The common endpoint/determinant of neutralization parameters (nAb titer, N95) can also be determined from the plot by interpolation at 95% neutralization; it is the maximum serum dilution value that results in 95% neutralization of ACE2 binding to coated RBD (Figure 7).
- Standard nAbs can also be included and treated as serum, but without heat inactivation (Step 13). For comparison purposes, nAbs can be serially diluted from their stock solution until N50/IC50 is achieved. This also provides an additional external standard, as cell-based assay procedures often employ standard nAbs.
- Interpolation at 80% or 90% neutralization yields N80 and N90 values, which are also commonly used neutralization efficacy endpoints.
- If the results are not as expected, refer to troubleshooting section (Table 1).
9. Anticipated Results
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, W.; Su, W.; Tang, H.; Le, W.; Zhang, X.; Zheng, Y.; Liu, X.; Xie, L.; Li, J.; Ye, J.; et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020, 6, 31. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalash, A.O.; Hussein, W.M.; Skwarczynski, M.; Toth, I. Key Considerations for the Development of Safe and Effective SARS-CoV-2 Subunit Vaccine: A Peptide-Based Vaccine Alternative. Adv. Sci. 2021, 8, 2100985. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.T.; Li, Z.; Samson, R.; Samavarchi-Tehrani, P.; Valcourt, E.J.; Wood, H. A simple protein-based surrogate neutralization assay for SARS-CoV-2. JCI Insight 2020, 5, e142362. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, B.; Sun, P.; Wang, W.; Luo, S.; Zhang, W.; Yang, Y.; Wang, Z.; Lin, J.; Chen, P.R. Severe Acute Respiratory Syndrome Coronavirus-2 Spike Protein Nanogel as a Pro-Antigen Strategy with Enhanced Protective Immune Responses. Small 2020, 16, e2004237. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Ozberk, V.; Eskandari, S.; Shalash, A.O.; Joyce, M.A.; Saffran, H.A.; Day, C.J.; Lepletier, A.; Spillings, B.L.; Mills, J.L.; et al. Antibodies to neutralising epitopes synergistically block the interaction of the receptor-binding domain of SARS-CoV-2 to ACE 2. Clin. Transl. Immunol. 2021, 10, e1260. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, R. New test for AIDS. IDRC Rep. 1991, 19, 22–23. [Google Scholar]
- Butler, J.E. The behavior of antigens and antibodies immobilized on a solid phase. Struct. Antigens 1992, 1, 208–259. [Google Scholar]
- Crowther, J.R. Systems in ELISA. In The ELISA Guidebook; Humana Press: Louisville, KY, USA, 2000; pp. 9–42. [Google Scholar]
- Padoan, A.; Bonfante, F.; Pagliari, M.; Bortolami, A.; Negrini, D.; Zuin, S.; Bozzato, D.; Cosma, C.; Sciacovelli, L.; Plebani, M. Analytical and clinical performances of five immunoassays for the detection of SARS-CoV-2 antibodies in comparison with neutralization activity. EBioMedicine 2020, 62, 103101. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.B.; Zahn, R.; Wegmann, F.; Loos, C.; Chandrashekar, A.; Yu, J.; Liu, J.; Peter, L.; McMahan, K.; Tostanoski, L.H.; et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature 2020, 586, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Koishi, A.C.; Suzukawa, A.A.; Zanluca, C.; Camacho, D.E.; Comach, G.; Dos Santos, C.N.D. Development and evaluation of a novel high-throughput image-based fluorescent neutralization test for detection of Zika virus infection. PLoS Negl. Trop. Dis. 2018, 12, e0006342. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.C.; Bogardus, L.; Giacone, D.G.; Rubinstein, L.J.; Antonello, J.M.; Sun, D.; Daijogo, S.; Gurney, K.B. Virus Reduction Neutralization Test: A Single-Cell Imaging High-Throughput Virus Neutralization Assay for Dengue. Am. J. Trop. Med. Hyg. 2018, 99, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Von Rhein, C.; Scholz, T.; Henss, L.; Kronstein-Wiedemann, R.; Schwarz, T.; Rodionov, R.N.; Corman, V.M.; Tonn, T.; Schnierle, B.S. Comparison of potency assays to assess SARS-CoV-2 neutralizing antibody capacity in COVID-19 convalescent plasma. J. Virol. Methods 2021, 288, 114031. [Google Scholar] [CrossRef]
- Tan, C.W.; Bogardus, L.; Giacone, D.G.; Rubinstein, L.J.; Antonello, J.M.; Sun, D.; Daijogo, S.; Gurney, K.B. A SARS-CoV-2 surrogate virus neutralization test based on antibody-mediated blockage of ACE2–spike protein–protein interaction. Nat. Biotechnol. 2020, 38, 1073–1078. [Google Scholar] [CrossRef]
- He, Y.; Zhu, Q.; Liu, S.; Zhou, Y.; Yang, B.; Li, J.; Jiang, S. Identification of a critical neutralization determinant of severe acute respiratory syndrome (SARS)-associated coronavirus: Importance for designing SARS vaccines. Virology 2005, 334, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Stevens, V.; Berry, J.D.; Crameri, G.; McEachern, J.; Tu, C.; Shi, Z.; Liang, G.; Weingartl, H.; Cardosa, J.; et al. Determination and application of immunodominant regions of SARS coronavirus spike and nucleocapsid proteins recognized by sera from different animal species. J. Immunol. Methods 2008, 331, 1–12. [Google Scholar] [CrossRef]
- Zhang, F.; Yu, M.; Weiland, E.; Morrissy, C.; Zhang, N.; Westbury, H.; Wang, L.-F. Characterization of epitopes for neutralizing monoclonal antibodies to classical swine fever virus E2 and Erns using phage-displayed random peptide library. Arch. Virol. 2006, 151, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhao, R.; Luo, J.; Xiong, S.; Shangguan, D.; Zhang, H.; Liu, G.; Chen, Y. Design, synthesis and screening of antisense peptide based combinatorial peptide libraries towards an aromatic region of SARS-CoV. J. Mol. Recognit. 2008, 21, 122–131. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, L.; Kuwahara, K.; Li, L.; Liu, Z.; Li, T.; Zhu, H.; Liu, J.; Xu, Y.; Xie, J.; et al. Immunodominant SARS Coronavirus Epitopes in Humans Elicited both Enhancing and Neutralizing Effects on Infection in Non-human Primates. ACS Infect. Dis. 2016, 2, 361–376. [Google Scholar] [CrossRef]
- Stevens, F.J.; Bobrovnik, S.A. Deconvolution of antibody affinities and concentrations by non-linear regression analysis of competitive ELISA data. J. Immunol. Methods 2007, 328, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sadana, A.; Vo-Dinh, T. Antibody-antigen binding kinetics. A model for multivalency antibodies for large antigen systems. Appl. Biochem. Biotechnol. 1997, 67, 1–22. [Google Scholar] [CrossRef]
- Walker, S.N.; Chokkalingam, N.; Reuschel, E.L.; Purwar, M.; Xu, Z.; Gary, E.N.; Kim, K.Y.; Helble, M.; Schultheis, K.; Walters, J.; et al. SARS-CoV-2 Assays to Detect Functional Antibody Responses That Block ACE2 Recognition in Vaccinated Animals and Infected Patients. J. Clin. Microbiol. 2020, 58, e01533-20. [Google Scholar] [CrossRef] [PubMed]
- Amarilla, A.A.; Modhiran, N.; Setoh, Y.X.; Peng, N.Y.; Sng, J.D.; Liang, B.; McMillan, C.L.; Freney, M.E.; Cheung, S.T.; Chappell, K.J.; et al. An Optimized High-Throughput Immuno-Plaque Assay for SARS-CoV-2. Front. Microbiol. 2021, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Watterson, D.; Wijesundara, D.K.; Modhiran, N.; Mordant, F.L.; Li, Z.; Avumegah, M.S.; McMillan, C.L.; Lackenby, J.; Guilfoyle, K.; van Amerongen, G.; et al. Preclinical development of a molecular clamp-stabilised subunit vaccine for severe acute respiratory syndrome coronavirus 2. Clin. Transl. Immunol. 2021, 10, e1269. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Hussain, S.; Harvey, R.; Martin, S.R.; Roustan, C.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Antibody-mediated disruption of the SARS-CoV-2 spike glycoprotein. Nat. Commun. 2020, 11, 5337. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Issue | Potential Reason(s) | Solution |
|---|---|---|
| Neutralizing extent exceeded the plate dilution down the rows | Very highly neutralizing serum, N50 exceeding 2 × 104. | Repeat the test starting with a lower serum dilution, e.g., 1/100, or conduct three-fold serum serial dilutions instead of two-fold. |
| Neutralization unapparent at the highest serum concentration | Irrelevant or non-neutralizing serum sample. Excessive heat treatment. | Repeat the assay with careful monitoring of heat treatment conditions (time and water bath temperature) and include a known standard murine neutralizing antibody (nAb) as an external standard to check assay integrity. |
| OD450 of blank ACE2 wells consistently exceeds 10% of the calibration curve’s 100% bound OD450 signal | Cross-reactivity of secondary antibody (2ry Ab) with murine sera. | Employ a more species-specific 2ry Ab with lower cross-reactivity or switch to the biotinylated ACE2 and streptavidin-HRP system |
| High blank ACE2 well OD450 in plate-edge or sporadic wells | Ineffective washing step. | Ensure you thoroughly wash every well in the plate three times with deionized water and three times with wash buffer. If feasible, use an automatic plate washer. |
| S-plate column 1, main serum well OD450 exceed the sum of blank ACE2 OD450 and 100% bound ACE2 OD450 combined | Heat treatment was not effective. Serum proteins are non-specifically interacting with ACE2 and 2ry Abs at high serum concertation. | Repeat the assay with careful monitoring of heat treatment conditions (time and water bath temperature). Highly neutralizing serum data points (above 2% serum concentrations; S-plates, columns 1–3) can be excluded and neutralization efficacy calculations can still be conducted. |
| Non-significant differences between calibration curve OD450 values for 25%, 50%, 75%, or 100% bound ACE2 | Variable amounts or coating times between wells of the CC-plate, resulting in high standard deviations of a given bound ACE2 (%) point. OD450 are all too high or too low, refer to reasons mentioned below. | Repeat the assay. Be consistent and quick with CC-plate coating. Incubate overnight at 4 °C for coating RBD, if necessary. Make sure the multichannel pipette is performing well and all seals are intact, or employ auto-ELISA equipment. If OD450 values are too high or too low, refer to solutions below. |
| The lowest calibration curve mean OD450 (0.8% bound ACE2 point) is equal to the background | A different, less sensitive substrate type was used, or the substrate was defective. Low substrate concentration or low 2ry Ab concertation. | Conduct a quick experiment using only the CC-plate to adjust the 2ry Ab and substrate. (1) Concentrate 2ry Ab following the manufacturer’s specifications at a higher value than employed, but within the recommended stated range. (2) Adjust the substrate concentration and reaction time. |
| Highest mean OD450 value is ≫1.0. | A different, more sensitive substrate type, e.g., TMB, was employed. High 2ry Ab concentration. OD450 > 1.0 will further convolute the calibration curve non-linearity. | Conduct a quick experiment using only the CC-plate to adjust 2ry Ab and the substrate. (1) Dilute the 2ry Ab following the manufacturer’s specifications at a lower value than employed, but within the recommended stated range. (2) Adjust the substrate concentration and reaction time. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalash, A.O.; Azuar, A.; Madge, H.Y.R.; Modhiran, N.; Amarilla, A.A.; Liang, B.; Khromykh, A.A.; Watterson, D.; Young, P.R.; Toth, I.; et al. Detection and Quantification of SARS-CoV-2 Receptor Binding Domain Neutralization by a Sensitive Competitive ELISA Assay. Vaccines 2021, 9, 1493. https://doi.org/10.3390/vaccines9121493
Shalash AO, Azuar A, Madge HYR, Modhiran N, Amarilla AA, Liang B, Khromykh AA, Watterson D, Young PR, Toth I, et al. Detection and Quantification of SARS-CoV-2 Receptor Binding Domain Neutralization by a Sensitive Competitive ELISA Assay. Vaccines. 2021; 9(12):1493. https://doi.org/10.3390/vaccines9121493
Chicago/Turabian StyleShalash, Ahmed O., Armira Azuar, Harrison Y. R. Madge, Naphak Modhiran, Alberto A. Amarilla, Benjamin Liang, Alexander A. Khromykh, Daniel Watterson, Paul R. Young, Istvan Toth, and et al. 2021. "Detection and Quantification of SARS-CoV-2 Receptor Binding Domain Neutralization by a Sensitive Competitive ELISA Assay" Vaccines 9, no. 12: 1493. https://doi.org/10.3390/vaccines9121493