

- Theoretical Neuroscience Research, LLC, Assistant Editor-in-Chief, Surgical Neurology International, 315 Rolling Meadows Rd, Ridgeland, MS 39157, USA
Correspondence Address:
Russell L. Blaylock
Theoretical Neuroscience Research, LLC, Assistant Editor-in-Chief, Surgical Neurology International, 315 Rolling Meadows Rd, Ridgeland, MS 39157, USA
DOI:10.4103/2152-7806.157890
Copyright: © 2015 Blaylock RL. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.How to cite this article: Blaylock RL. Cancer microenvironment, inflammation and cancer stem cells: A hypothesis for a paradigm change and new targets in cancer control. Surg Neurol Int 29-May-2015;6:92
How to cite this URL: Blaylock RL. Cancer microenvironment, inflammation and cancer stem cells: A hypothesis for a paradigm change and new targets in cancer control. Surg Neurol Int 29-May-2015;6:92. Available from: http://surgicalneurologyint.com/surgicalint_articles/cancer-microenvironment-inflammation-cancer-stem-cells/
Abstract
Since President Nixon officially declared a war on cancer with the National Cancer Act, billions of dollars have been spent on research in hopes of finding a cure for cancer. Recent reviews have pointed out that over the ensuing 42 years, cancer death rates have barely changed for the major cancers. Recently, several researchers have questioned the prevailing cancer paradigm based on recent discoveries concerning the mechanism of carcinogenesis and the origins of cancer. Over the past decade we have learned a great deal concerning both of these central issues. Cell signaling has taken center stage, particularly as regards the links between chronic inflammation and cancer development. It is now evident that the common factor among a great number of carcinogenic agents is activation of genes controlling inflammation cell-signaling pathways and that these signals control all aspects of the cancer process. Of these pathways, the most important and common to all cancers is the NFκB and STAT3 pathways. The second discovery of critical importance is that mutated stem cells appear to be in charge of the cancer process. Most chemotherapy agents and radiotherapy kill daughter cells of the cancer stem cell, many of which are not tumorigenic themselves. Most cancer stem cells are completely resistant to conventional treatments, which explain dormancy and the poor cure rate with metastatic tumors. A growing number of studies are finding that several polyphenol extracts can kill cancer stem cells as well as daughter cells and can enhance the effectiveness and safety of conventional treatments. These new discoveries provide the clinician with a whole new set of targets for cancer control and cure.
Keywords: Cancer stem cell, cell signaling, inflammatory oncogenes, stemness, tumor microenvironment
INTRODUCTION
Oncogene activation leading to the overstimulation of cell growth as a cause of Cancer.
We often hear it said that all the billions spent on the “war on cancer” was essentially wasted, as death rates from metastatic cancer have changed little since the war was declared 42 years ago under President Nixon's National Cancer Act. It is accepted that long-term survival, once a cancer metastasizes, is no more than 5–10% despite intensive chemotherapy and radiotherapy – a pretty dismal conclusion to a 40-year war.[
Much of the research was directed at cancer cell biology, in particular genetics and cell-signaling mechanisms. Based on early research, it was assumed that most cells in the body, under particular conditions, could transform into immortalized cancer cells through a specific gene-directed process. Oncogenes, as the paradigm concluded, were either mutated or overexpressed leading to excessive stimulation of cell cycling and growth signals and/or suppression of cancer suppressor signals – the bottom-line being that somatic cells had lost growth restraint signals and were transformed into cancer cells.
Further, it was assumed that carcinogenic agents affected cell signaling and their carcinogenicity was based on their effects on oncogenes, which could occur by a number of mechanisms. Based on this theory of carcinogenesis, chemotherapeutic treatments were mostly directed at controlling cell cycling, induction of apoptosis and reducing cell growth signaling.
WILL CANCER TREATMENT UNDERGO A PARADIGM SHIFT?
Consideration of the role of inflammation in cancer
Sarah Crawford in a series of important papers asks this critical question based on a considerable amount of research that indicates conventional treatments have failed to live up to early promises and that new discoveries suggest that we may have been following an incorrect paradigm.[
In this paper, I have reviewed some of these studies. What we have learned is that central to all cancers is inflammation and that the cell processes involved in inflammation not only are responsible for initiation of the cancer, but also persist during its growth and play a central role throughout every phase of the cancer's existence, including progression, invasion, angiogenesis, and metastasis [
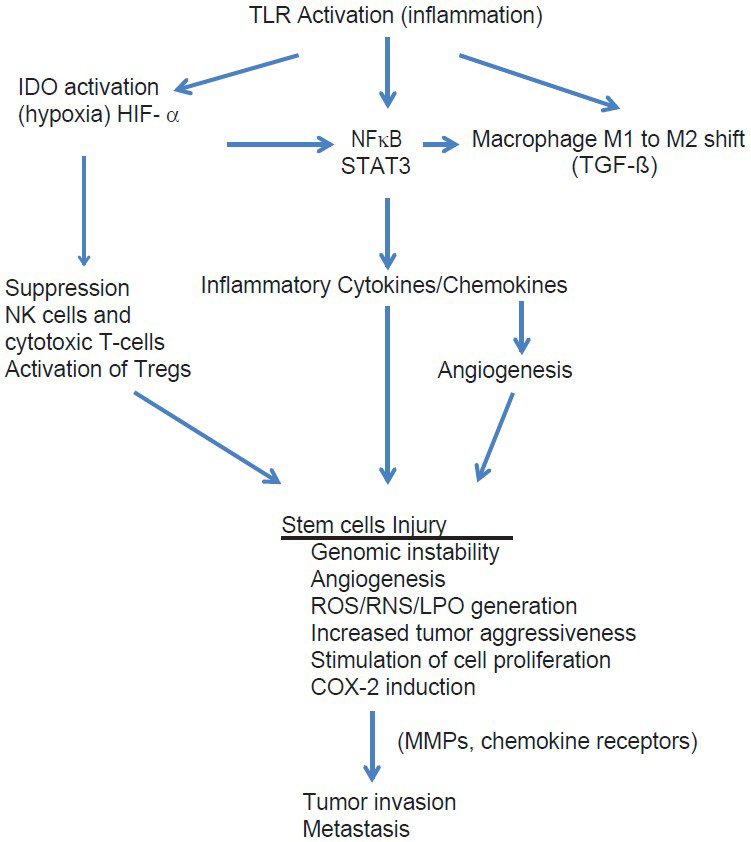
The role of short term and chronic inflammation in cancer and disease
While short-term inflammation is rarely associated with cancer induction, chronic smoldering inflammation, as seen with a large number of disorders, is almost always linked to carcinogenesis.[
Common molecular signaling pathways in cancer
What all of these carcinogenic events have in common is that they activate two main cell-signaling molecules – nuclear factor kappa (NFκB) and signal transducer and activator of transcription-3 (STAT3).[
The NFκB and STAT3 pathways are central pathways in both inflammation and tumorigenesis. Both are activated by a wide assortment of tumor-associated events, such as growth factors (epidermal growth factor [EGF]), hypoxia, acidic microenvironment, hyperglycemia (diabetes and insulin resistance), and proinflammatory cytokines (TNF-α). In fact, TNF-α is one of the most powerful activators of NFκB, which explain the strong association found between high levels of TNF-α and the aggressive behavior of several cancers, such as glioblastomas, head and neck squamous cell cancer, mantle cell lymphoma and acute myeloid leukemia, and others.[
Growth factors, such as EGF and growth receptors, such as HER2 and EGFR, are universally activated in a variety of cancers and they also activate NFκB.[
The proinflammatory cytokine IL-6, a major growth factor in prostate and other cancers, activates both NFκB and STAT3. NFκB is also a major controller of IL-6 production, a major cancer growth factor.[
An explanation for resistance of cancer to radiation and chemotherapy
Of major interest is that activation of NFκB plays a major role in resistance to chemotherapy and radiation therapy.[
Reactive oxygen and nitrogen species as the initiator of the cancer cascade
Within the microenvironment of the stem cells, before conversion to cancer stem cells, one witnesses a transition of the stem cell niche into an area of high concentrations of reactive oxygen species (ROS) and reactive nitrogen species (RNS), lipid peroxidation products (LPPs) and inflammatory cytokines and chemokines.[
The cancer stem cell, its microenvironment and inflammation: effects on cancer biology
As basic research further expanded our understanding of the biology of the cancer process, a different story began to appear. Ironically, it was a story that had been suggested almost 150 years ago by pathologist Rudolph Virchow.[
The frequent association of various tumor types with known chronic inflammatory diseases suggested that inflammation was playing an essential role in cancer biology. For example, colon cancer risk was associated with inflammatory bowel diseases, such as ulcerative colitis and Crohn's disease; pancreatitis with pancreatic cancer; obesity with breast cancer; gastric reflux with esophageal cancer and Schistosmoma infections with bladder cancer. Further support came from the observation that certain antiinflammatory drugs not only reduced the risk of cancer development but also reduced recurrence, metastasis and tumor size.[
CANCER STEM CELLS AND STEMNESS
Early hypotheses on the genesis of tumors from dormant cells, trophoblasts
One of the most important discoveries in cancer biology is one that actually surfaced over 100 years ago. And that is the idea that uncommitted cells lying dormant throughout the body are the source of most cancers.[
Unfortunately, his ideas soon fell into oblivion. I say unfortunately, because so much time was lost examining other theories that did not lead to treatments that could make a significant impact against the major killer cancers.
STEM CELL HYPOTHEIS OF CANCER
Cancerous tumors are said to represent aberrant attempts to produce organs and contain heterogenous populations of cells that differ in their accumulated mutations and degree of differentiation.[
Considerable evidence suggests that cancer stem cells closely resemble stem cells themselves.[
Because stem cells can exist for a lifetime they are vulnerable to varying episodes of attack by ROS/RNS as well as LPPs, such as 4-hydroxynonenal and acrolein.[
Exposure of these stem cells’ DNA to intense or prolonged, unrepaired assaults by ROS/RNS and LPPs can produce varying degrees of genetic mutations that over time can convert a somatic stem cell into a cancer stem cell.[
The literature on cancer stem cells speaks of stemness, indicating that certain influences can alter progenitor cells to revert back to stem cells or cancer stem cells, depending on the conditions. Normally, progenitor cells are less likely to produce tumor formation, as they proliferate for a shorter time before terminally differentiating.[
One of the important findings is that cancer stem cells generally make up only a very small proportion of the cellular structure of the tumor; most cells being daughter cells derived from the cancer stem cells.[
Cancer stem cells are isolated using flow cytometry according to the expression pattern of surface markers such as CD24, CD44, and CD133.[
The first isolation of cancer stem cells from a solid tumor was from breast cancer.[
One of the unsettled questions is whether the daughter cells can at some time dedifferentiate into cancer stems cells, which would create a moving target for cancer treatment and make cures much more difficult. Important in any context is the importance of killing both cancer stem cells and daughter cells of the tumor.
Three studies examined this issue in some detail.[
The strongest evidence of cancer stem cells as the origin of cancers comes from two studies, the Dressens et al. study and the Schepers et al. study mentioned above.[
THE CENTRAL IMPORTANCE OF THE MICROENVIRONMENT OF THE TUMOR
Early history of inflammation in the development of cancer
Rudolph Virchow, over 150 years ago, noted that at its earliest stages, all cancerous tumors were infiltrated with leukocytes of various kinds and that advanced tumors had characteristics of infectious boils.[
More recent studies have confirmed his observations and that leukocyte infiltration occurs even in the precancerous phase of cancer development.[
The link between inflammation and viral and chemical transformation of cells
An example of the central role played by inflammation is seen with malignancies induced by the Rous sarcoma virus. Without inflammation, the virus cannot induce malignant transformation.[
Solid tumors and Inflammation – How it works; other observations
It is becoming evident that inflammation is playing a central role in tumor initiation, progression, invasion and metastasis – that is, in every phase of the carcinogenic process.[
Inflammation is known to induce genomic instability, angiogenesis, alterations in the epigenomic state, stimulation of cell proliferation, increase in cytokine growth factors, generation of reactive oxygen and nitrogen species, induction of chemokine receptors on malignant cells, induction of COX-2 and activation of NFκB and STAT3.[
The degree of inflammation appears to determine the proliferative potential of the tumor as well as its invasive and metastatic aggressiveness.[
Recruitment of macrophages, neutrophils and mast cells increase nitric oxide (NO) levels within the tumor microenvironment and this promotes tumor proliferation.[
Unfortunately, for most malignancies, macrophages are switched to an M2 immune-suppressing phenotype that allows the tumor to escape immune detection and destruction.[
One of the central control elements for immune tolerance under a variety of conditions is the tryptophan metabolizing enzyme indoleamine 2,3-dioxygenase (IDO), an enzyme found in all tissues, including tumor and immune cells.[
Studies have shown that small molecule inhibitors of IDO can cause rapid regression of aggressive tumors that are otherwise known to be treatment resistant.[
Inflammation and oncogene activation within stem-like cells – how cancers develop
It is generally accepted that the trigger for conversion of normal somatic cells into malignant cells involved alteration in their genes controlling cell proliferations and/or tumor suppression/apoptosis. Newer evidence suggests that it is the stem cells in which activation of oncogenes is occurring.[
The MYC oncogene is overexpressed in many human cancers and promotes the first wave of angiogenesis by stimulating the production of the inflammatory cytokine IL-1ß.[
The essential nature of inflammation in the initial stem cell transformation is emphasized by the findings that in the case of pancreatic adenocarcinoma both mutation of the oncogene K-RAS and pancreatitis are necessary for tumor cell development.[
According to the present hypothesis, activation of oncogenes results from damage to DNA by high levels of ROS/RNS and these are generated by smoldering inflammation, either systemically or locally at the site of tumor development. The tumor-initiating inflammation can results from a number of insults, such as trauma, chronic, smoldering infections, latent viruses, parasitic infections, chemical carcinogens, or autoimmune disorders. Most types of cancer are found to have high levels of ROS/RNS.[
Cell signaling and control of cancer stem cell behavior
In general, adult stem cells are normally quiescent and this state is dependent on the microenvironment of the stem cell niche. This quiescence requires interaction with various cell types within and surrounding the niche or tumor bed.[
Quiescence is controlled by a number of cell signaling pathways, including p53, FoxO, HIF-1α, nuclear factor of activated T cells c1 (NFATc1), Phosphatase and tensin homolog (PTEN), mammalian Target of Rapamycin (mTOR), bone morphagenic proteins (BMPs), transforming growth factor beta (TGF-β), thrombopoietin, angiopoietin-1 (ang-1), and Wnt/B-caterin signaling.[
Granulocyte colony stimulating factor (G-CSF), interferon-α, and the chemokine CXCL12 can all mobilize dormant cancer stem cells into the circulation.[
Nanog: A master controller of cancer behavior
Newer studies are finding that the transcription factor nanog plays a major and central role in regulating pluipotency and tumorigenesis of cancer stem cells.[
Nanog is expressed in a number of cancers including cancer of the breast, cervix, kidney, prostate, lung, brain, ovary, gastric carcinoma, and oral cancers.[
Another way overexpression of nanog promotes tumor aggressiveness, invasion, and metastasis is by activating Wnt signaling, which allows the cancer cells to adapt to the immune system, that is, it leads to immune escape. Both the cancer cells and surrounding stromal cells can express high levels of nanog.[
Sonic hedgehog, another cell signaling mechanism, also promotes cancer stem cell survival, tumor growth, and invasion in human glioma cells.[
Micro RNA: New guys on the block
These cell signaling pathways, in conjunction with the previously described quiescence cell signaling, play a major role in controlling tumor behavior, especially as regards invasiveness and metastatic potential. A great deal of attention is now being paid to another regulator of stem cells and this includes microRNA, short, noncoding fragments of RNA. MicroRNAs appear to control a great number of processes in cells and are especially important in regulation of stem cells. This is well documented both in embryogenesis and in cancers.[
During brain development, CD133+ stem cells regulate cell differentiation and orientation.[
As controllers of a number of cell processes, microRNA dysfunction can result in tumor progression and aggressiveness by inhibiting the normal microRNA functions that control tumor suppressor genes and by overexpression of microRNAs that promote stemness and stem cell self-renewal.[
Studies have shown abundant levels of the microRNAs miR-9, miR-9± in cancer stem cells of glioblastomas.[
Downregulation of the microRNA miR-199b-5p is associated with metastatic spread of medulloblastoma cells.[
THE CENTRAL ROLE OF NUCLEAR FACTOR KAPPAB AND STAT3 IN TUMOR INFLAMMATION AND BIOLOGY
The central activating molecular processes in tumor initiation, invasion, and metastasis
Activation of NFκB is central to regulation of the inflammatory state of the tumor cells themselves and plays a major role in tumor biology.[
Most cancers demonstrate increased NFκB activation.[
Hypoxia link to inflammation and cancer
Hypoxia, which plays a major role in tumor induction as well as maintenance, activates hypoxia inducible factor-1α (HIF-1α), which in turn activates NFκB.[
The role played by STAT3
STAT3, another inflammation controlling transcription factor, also plays an essential role in the tumor inflammatory microenvironment and therefore tumor behavior.[
Once a condition of protumor immunity is activated, the tumor, by switching Th1 cytotoxic immunity to Th2 type immunity (an immune suppressing phenotype) in the immune cells, allows the cancer to grow unimpeded by the immune cytotoxic system. A key element in this immune suppression is the generation of large numbers of interleukin-10 (IL-10) producing immune cells.[
Proinflammatory cytokines, such as IL-1ß, IL-2, IL-6, IL-17, and IL-23, can act through the STAT3 signaling system. For example, IL-6 is a growth stimulating cytokine that is associated with rapid growth and invasion of a number of cancers, including ovarian cancer, prostate cancer, nonsmall cell lung cancer, squamous cell carcinoma of the head and neck, lymphomas and gastric carcinomas induced by H. pylori.[
INVASION AND METASTASIS: MOLECULAR BASIS OF METASTASIS
Chemokines
Of particular interest is the strong link between inflammation and invasion and metastasis of cancers. In some animal studies, inflammation was necessary for a cancer to metastasize.[
A strong relationship also exists between the presence of chemokine receptors and metastasis.[
One of the better-studied chemokine receptors includes CXCR4 and its ligand CXCL12, which is frequently expressed by malignant cells. Studies have shown that the amount of CXCR4 receptor expressed by primary tumors correlates with the extent to which metastasis to regional lymph nodes occurs. This is has been demonstrated for breast, colorectal, liver, and esophageal cancers.[
Other chemokine receptors expressed by malignant cells include CX3CR1, CCR1, CCR7, CCR9, CCR10, CXCR1, CXCR2, CXCR3, CXCR5, and CXCR7. Interestingly, malignant melanomas express a number of chemokine receptors and may explain its high propensity to metastasize to a number of sites.[
Normally, tissues such as epithelial cells and mesenchymal cells do not express chemokine receptors but the appearance of these chemokine receptors occurs early with malignant transformation.[
Suppression of chemokines and effect on tumor cell invasion
Suppressing inflammatory cell signaling has been shown to significantly reduce metastatic spread in animal models of prostate cancer, for example.[
MACROPHAGE CONVERSION AIDING TUMOR CELL INVASION
Switching from the antitumor M1 phenotype macrophage to the M2 protumor mode is accomplished by activation of NFκB and this promotes proliferations, invasion, and metastasis of the tumor.[
Important in the switching process of macrophages from M1 to M2 phenotype is TGF-ß, an inflammation-triggered mediator of immune suppression as well as the generation and release of MMPs enzymes by cancer cells. MMPs promote tumor invasion and high levels are an independent risk factor for a poor prognosis.[
Tumor microenvironment: Special characteristics
An inflammatory microenvironment plays a key role in this conversion of stem cells into cancer stem cells, and newer research is finding that cells in the stroma have a major influence on stem cell behavior. For example, Rao et al. found that endothelial cells play a critical role in the development and behavior of glioblastoma multiforme tumors by regulating the release of the chemokine CXCL12.[
A critically important aspect of tumor microenvironment is hypoxia, as mentioned above, especially cyclic hypoxia. It has been shown that hypoxia can predict the likelihood of tumor aggressiveness, invasion, metastasis, tumor recurrence, resistance to chemotherapy and radiotherapy, and patient survival.[
Hypoxia, by increasing the release of HIF-1α in the microenvironment, induces the expression of the chemokine receptor CXCR4 on the membrane surface of stem cells, which is responsible for migration and metastasis of cancer stem cells.[
Another characteristic of cancer stem cells is their resistance to chemotherapy and radiotherapy. Currently used chemotherapeutic drugs can often dramatically shrink metastatic tumors, but these effects are usually quite transient and do not significantly extend the life of the patient. In essence, the chemotherapy drugs are killing only daughter cells and not cancer stem cells.[
NATURAL MOLECULAR AGENTS AND THEIR POTENTIAL EFFECT ON CANCER
Natural molecular agents
For example, resveratrol, curcumin, quercetin, hesperidin, luteolin, apigenin, naringenin, urolic acid, and silymarin have all been shown to have powerful inhibitory effects on tumor mechanism without toxicity to normal cells.[
The natural compounds, including flavonoids, special molecules, and certain vitamins and minerals, have also been shown to reverse MDR and radioresistance in tumors.[
The alteration of the immune system in the response to cancer by natural molecular agents
Increasing evidence indicates that the immune system, especially cellular immunity, is a major barrier to successful tumor growth and persistence.[
Natural molecular agents and their influence in reversing the resistance to radiation and chemotherapy
Several studies have shown that a number of natural products can enhance the cancer cell killing effects of conventional treatments, including radiotherapy, and at the same time protect normal cells from damage by these treatments – the best of all worlds.[
A number of natural products have shown an ability to reverse chemotherapy drug resistance, including an ability to restore apoptotic mechanisms such as p53 activity.[
Low toxicity of natural molecular agents
When I practiced neurosurgery, I gave all of my cancer patients selected anticancer natural products, primarily curcumin, quercetin, mixed tocopherols, vitamin C, and resveratrol and have never observed interference with conventional treatments. Dr Jerome Block, former chief of the Division of Medical Oncology/Hematology at Harbor UCLA Medical Center not only used complementary nutraceuticals in his cancer patients, but also taught visiting doctors on their use.[
Why the traditional Western diet may be proinflammatory and procarcinogenic
Physicians treating cancer should be aware of the fact that most of the omega-6 oils, such as corn, safflower, sunflower, peanut, and soybean oils, promote tumor proliferation, invasion, and metastasis. The main reason is that these oils are proinflammatory. Americans eating the typical Western diet consume 50-fold higher levels of the oils than are needed for good health. High sugar intake also promotes inflammation and cancer growth and invasion. Finally, glutamate and the other excitatory amino acids (aspartate, homocysteic acid, and cysteic acid) also promote tumor proliferation, invasion, and metastasis. Many hospital feeding formula contain high levels of glutamate and rarely are cancer patients told to avoid glutamate additives, aspartame, or foods naturally high in glutamate.
CONCLUSIONS
In this Hypothesis on the Genesis of Cancer paper, what we have learned after 43 years of the war on cancer is that we managed to overlook a critical mechanism that was correctly recognized over 150 years ago, mainly that inflammation is at the center of the cancer process. Our investment also allowed us to correct our having overlooked another important piece of the puzzle – that not all cells can become cancer, rather stem cells appear to be the major cell type involved.
These two changes in our thinking may well lead to a dramatic reduction in cancer development and may change the way we treat established cancers by changing our targets. Over the past 40 years, we have learned an enormous amount about cell signaling and how it is altered in cancer cells. Two of the most important systems are transcription control mechanisms of gene activation known as NFκB and STAT3. It is through these transcription pathways that cancers are formed, proliferate, develop a blood supply (angiogenesis), invade surrounding tissues (including blood and lymphatic vessels), and metastasized to distant sites.
Nonsteroidal antiinflammatory medications and aspirin have shown a significant ability to suppress inflammation and thereby alter the cancer process. A growing number of natural molecular products and their extracts are showing an ability to suppress multiple pathways involved in the cancer process, including suppression of NFκB and STAT3 in the plant and animal kingdom.
Presently, human trials and responses to these natural substances have been hampered by poor absorption and bioavailability of these extracts but newer techniques, such as nanosizing and microencapsulation with phospholipids, have greatly improved both gut absorption and bioavailability. When combined with dietary programs designed to utilize what we now know about the anticancer effects of various foods can greatly improve the prevention and treatment of cancers.
The vast majority of the natural products found to have powerful and versatile anticancer effects have shown a very wide margin of safety. Curcumin, for example, in extremely high doses is nontoxic to normal cells and tissues. One can appreciate the careful testing of manufactured drugs, as most have extremely high toxicity and treatment concentrations are often close to fatal systemic toxic effects.
The fact that these natural compounds are powerful anticancer agents when used alone and significantly improve conventional chemotherapy and radiation therapy treatments, plus the fact that they protect normal tissues and cells, is reason enough to begin use of these valuable agents now, both as cancer preventatives and in the treatment of established cancers. The patients with advanced cancers or high aggressive cancers cannot afford to wait another 10 years while these safe compounds are tested for years as if they were dangerous drugs.
References
1. Acikalin MF, Oner U, Topeu I, Yasar B, Kiper H, Colak E. Tumor angiogenesis and mast cell density in the prognostic assessment of colorectal carcinomas. Dig Live Dis. 2005. 37: 162-9
2. Alberti C, Pinciroli P, Valeri B, Ferri R, Ditto A, Umezawa K. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFκB pathway in advanced-stage epithelial ovarian cancer. Oncogene. 2012. 31: 4139-49
3. Alexandrow MG, Song LJ, Altiok S, Gray J, Haura EB, Kumar NB. Curcumin: A novel STAT3 pathway inhibitor for chemoprevention of lung cancer. Eur J Cancer Prev. 2012. 21: 407-12
4. Al-Hajj M, Wicha MS, Benito-Hernendez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003. 100: 3983-8
5. Allavena P, Garlanda C, Borrello MG, Sica A, Mantovani A. Pathways connecting inflammation and cancer. Curr Opin Genet Dev. 2008. 18: 3-10
6. Ambs S, Glynn SA. Candidate pathways linking inducible nitric oxide synthase to a basal-like transcription pattern and tumor progression in human breast cancer. Cell Cycle. 2011. 10: 619-24
7. Annemijn M, Algra B, Rothwell P. effects of regular aspirin n long-term cancer incidence and metastasis: A systematic comparison if evidence from observational studies versus randomized trails. Lancet. 2012. 13: 518-27
8. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004. 4: 540-50
9. Balkwill F. Tumor necrosis factor and cancer. Nat Rev Cancer. 2009. 9: 361-71
10. Balkwill F, Charles KA, Montovani A. Smoldering and polarizing inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005. 7: 211-7
11. Balkwill F, Mantovani A. Inflammation and cancer: Back to Virchow?. Lancet. 2001. 357: 539-45
12. Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R. A randomized trail of aspirin to prevent colorectal adenomas. N Engl J Med. 2003. 348: 891-9
13. Block JB. Interview with Jerome B Block, MD. J Am Nutraceutical Assoc. 1999. 2: 57-
14. Boccardo E, Lepique AP, Vilia LL. The role if inflammation in HPV carcinogenesis. Carcinogenesis. 2010. 31: 1905-12
15. Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP. Human melanoma-initiating cells express neuronal crest nerve growth factor receptor CD271. Nature. 2010. 466: 133-7
16. Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech Aging Dev. 2004. 125: 811-26
17. Bourillot PY, Aksoy I, Schreiber V, Wianny F, Schultz H, Hummel O. Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells. 2009. 27: 1760-71
18. Bruning A. Inhibition of mTOR signaling by quercetin in cancer treatment and prevention. Anticancer Agents Med Chem. 2013. 13: 1025-31
19. Burleigh AR. Of germs cells, trophoblast, and cancer stem cells. 2008. 7: 276-81
20. Cabrera MC, Hollingsworth RE, Hurt EM. Cancer stem cell plasticity and tumor hierarchy. World J Stem Cells. 2015. 7: 27-36
21. Cao HH, Tse AK, Kwan HY, Yu H, Chen CY, Su T. Quercetin exerts anti-melanoma activities and inhibits STAT3 signaling. Biochem Pharmacol. 2014. 87: 424-34
22. Carocho M, Ferreira IC. The role of phenolic compounds in the fight against cancer - a review. Anticancer Agents Med Chem. 2013. 13: 1236-58
23. Chan A, Ogino S, Fuch C. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009. 302: 649-58
24. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012. 488: 522-6
25. Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000. 60: 2132-5
26. Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res. 2014. 2: 1124-31
27. Clement V, Sanchez P, de Tribolet N, Taobovanic I, Ruizialtaba A. Hedgehog-GL13 signaling regulates human glioma growth, cancer stem cells self-renewal, and tumorigenicity. Curr Biol. 2007. 17: 165-72
28. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis. 2009. 30: 1073-81
29. Condeelis J, Pollard JW. Macrophages: Obligate partners for tumor cell migration, invasion and metastasis. Cell. 2006. 124: 263-6
30. Correa GT, Bandeira GA, Cavalcanti BG, de Carvalho Fraga CA, dos Santos EP, Silva TF. Association of – 308 TNF-alpha promoter polymorphism with clinical aggressiveness in patients with head and neck squamous cell carcinoma. Oral Oncol. 2011. 47: 888-94
31. Crawford S. Anti-inflammatory/antioxidant use in long-term maintenance cancer therapy: A new therapeutic approach to disease progression and recurrence. Ther Adv Med Oncol. 2014. 6: 52-68
32. Crawford S. Is it time for a new paradigm for systemic cancer treatment? Lessons from a century of cancer chemotherapy. Front Pharmacol. 2012. 4: 68-
33. Dannenmann SR, Thielicke J, Stokli M, Matter C, von Boehmer L, Cecconi V. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunology. 2013. 2: e23562-
34. Das M, Sahoo SK. Folate decorated dual drug loaded nanoparticle: role of curcumin in enhancing therapeutic potential of nutlin-3a by reversing multidrug resistance. PloS One. 2012. 7: e32920-
35. Demaria S, Pikarsky E, Karin M, Coussens LM, Chen YC, El-Omar EM. Cancer and inflammation: Promise for biological therapy. J Immunother. 2010. 33: 335-51
36. Dennis KL, Blatner NR, Gounari F, Khazaie K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr Opin Oncol. 2013. 25: 637-45
37. Dhandapani KM, Mahesh VB, Brann DW. Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFkappaB transcription factors. J Neurochem. 2007. 102: 522-38
38. Dressens G, Beck B, Caawe A, Simons BD, Blanpain C. Defining the mode of tumor growth by clonal analysis. Nature. 2012. 488: 527-30
39. Ebrahimi B, Tucker SL, Li D, Abbruzzese JL, Kurzrock R. Cytokines in pancreatic carcinoma: Correlation with phenotypic characteristics and prognosis. Cancer. 2004. 101: 2727-36
40. Elgert K, Alleva D, Mullens D. Tumor-induced immune dysfunction: The macrophage connection. J Leukoc Biol. 1998. 64: 275-90
41. Enewold L, Mechanic LE, Bowman ED, Zheng YL, Yu Z, Trivers G. Serum concentrations of cytokines and lung cancer survival in African Americans and Caucasians. Cancer Epidemiol Biomarkers Prov. 2009. 18: 215-22
42. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S. Isolation and characterization of tumorigenic, stem cell-like neuronal precursors from human glioblastoma. Cancer Res. 2004. 64: 7011-21
43. Garg AK, Buchholz TA, Aggarwal BB. Chemosensitization and radiosensitization of tumors by plant polyphenols. Antioxid Redox Signal. 2005. 7: 1630-47
44. Garzia L, Andolfo I, Cusanelli E, Marino N, Petrpsino G, De Martino D. MicroRNA-199b-5p impairs cancer stem cells through negative regulation if HES1 in medulloblastoma. PLoS One. 2009. 4: e4998-
45. Gat U, Das Gupta R, Degenstein L, Fuchs E. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell. 1998. 95: 605-14
46. Gilmore TD. The Rel/NF-κB/IκB signal transduction pathway and cancer. Cancer Treat Res. 2003. 115: 241-65
47. Gu Q, Hu C, Chen Q, Xia Y. Tea polyphenols prevent lung from preneoplastic lesions and effect p53 and bcl-2 expression in rat lung tissues. Int J Clin Exp Pathol. 2013. 6: 1523-31
48. Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007. 11: 291-302
49. Gurchot C. The trophoblastic theory of cancer (John Beard, 1857-1924) revisted. Oncology. 1975. 31: 310-33
50. Hamberger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977. 197: 461-3
51. Hashimoto O, Shimizu K, Semba S, Ciba S, Ku Y, Yokozaki H. Hypoxia induces tumor aggressiveness and the expansion of CD133-positive cells in a hypoxia-inducible factor-1α-dependent manner in pancreatic cancer cells. Pathobiology. 2011. 76: 181-92
52. Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010. 101: 1913-9
53. Hellegass JM, Shukla A, Lathrop SA, MacPherson MB, Beuschel SL, Butnor KJ. Inflammation precedes the development f human malignant mesotheliomas in a SCID mouse xenograft model. Ann NY Acad Sci. 2010. p. 1203-14
54. Holmes M, Chen W, Li L, Hertzmark E, Spiegelman D, Hankinson S. Aspirin intake and survival after breast cancer. J Clin Oncol. 2010. 28: 1467-72
55. Hsieh CH, Lee CH, Liang JA, Yu CY, Shyu WC. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signaling. Oncol Rep. 2010. 24: 1629-36
56. Hsieh CH, Shyu WC, Chiang CY, Kuo JW, Shen WC, Liu RS. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS One. 2011. 6: e23945-
57. Huber MA, Azoitel N, Baumann B, Grunert S, Sommer A, Pehamberger H. NF-kappaB is essential for epithelial-mesenchyme transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004. 114: 569-81
58. Ibrahim EE, Babael-Jadidi R, Saadeddin A, Spencer-Dene B, Houssaini S, Abuzinadah M. Embryonic NANOG activity define colorectal cancer stem cells and modulates through AP1-and TCF-dependent mechanisms. Stem Cells. 2012. 30: 2076-87
59. Jensen RL. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg Focus. 2006. 20: E24-
60. Ji RC. Macrophages are important mediators of either tumor-or-inflammation-induced lymphangiogenesis. Cell Mol Life Sci. 2012. 69: 897-914
61. Johnson TS, Munn DH. Host indolamine 2,3 dioxygenase: contribution to systemic acquired tumor tolerance. Immunol Invest. 2012. 41: 765-97
62. Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S. Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J Clin Invest. 2014. 124: 528-42
63. Kaifi JT, Yekebas EF, Schurr P, Obonyo D, Wachowiak R, Busch P. Tumor-cell homing lymph nodes and bone marrow and CXCR4 expression in esophageal cancer. J Nat Cancer Inst. 2005. 97: 1840-7
64. Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006. 441: 431-6
65. Katz JB, Muller AJ, Pendergast GC. Indolamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2008. 222: 206-21
66. Keibel A, Singh V, Sharma MC. Inflammation, microevironment, and the immune system in cancer progression. Curr Pharm Des. 2009. 15: 1949-55
67. Kim YS, Farrar W, Colburn NH, Milner JA. Cancer stem cells: potential target for bioactive food components. J Nutr Biochem. 2012. 23: 691-8
68. Kisseleva T, Song L, Voromtchikhina M, Feirt N, Kitajewski J, Schindler C. NF-kappaB regulation of endothelial cell function during LPS-induced toxemia and cancer. J Clin Invest. 2006. 116: 2955-63
69. Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008. 216: 15-24
70. Kono Y, Kawakami S, Higuchi Y, Yasashita F, Hashida M. In vitro evaluation of inhibitory effects of nuclear factor-kappaB activity by small interfering RNA on pro-tumor characteristics of M2-like macrophages. Biol Pharm Bull. 2014. 37: 137-44
71. Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005. 24: 315-27
72. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S. Inhibiting STAT3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005. 11: 1314-21
73. Krstic J, Santibanez JF. Transforming growth factor-beta and matric metalloprotenases: Functional interactions in tumor stroma-infiltrating myeloid cells. Scientific World Journal 2014. 2014. p.
74. Kulbe H, Hagemann T, Szlosarek PW, Balkwill FR, Wilson JL. The inflammatory cytokine tumor necrosis factor-alpha regulates chemokine receptor expression on ovarian cancer cells. Cancer Res. 2005. 65: 10355-62
75. Kunnummakara AB, Guha S, Krishnan S, Diagaradjiane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res. 2007. 67: 3853-61
76. Lee M, Nam EJ, Kim SW, Kim S, Kim JH, Kim YT. Prognostic impact of the cancer stem cell-related marker NANOG in ovarian serious carcinoma. Int J Gynecol Cancer. 2012. 22: 1489-96
77. Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 2011. 17: 4936-41
78. Li Q, Withoff S, Verma IM. Inflammation-associated cancer: NF-κB is the lynchpin. Trends Immunol. 2005. 26: 318-25
79. Liao WC, Lin JT, Wu CY, Huang SP, Lin MT, Wu As. Serum interleukin-6 level but not genotype predicts survival after resection in stages II and III gastric carcinoma. Clin Cancer Res. 2008. 14: 428-34
80. Liao Y, Shen W, Kong G, Lv H, Tao W, Bo P. Apigenin induces apoptosis via extrinsic pathway, inducing p53 and inhibiting STAT3 and NFκB signaling in HER2-overexpressing breast cancer cells. Mol Cell Biochem. 2012. 366: 319-34
81. Lin L, Hutzen B, Li PK, Ball S, Zuo M, DeAngelis S. A novel small molecule, LLL12 inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia. 2010. 12: 39-50
82. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010. 44:
83. Lu H, Quyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res. 2006. 4: 1-33
84. Luo JL, Tan W, Ricino JM, Korchynskyi O, Zhang M, Gonias SL. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007. 446: 690-4
85. Malhotra GK, Zhao X, Band H, Band V. Shared signaling pathways in normal and breast cancer stem cells. J Carcinog. 2011. 10: 38-
86. Malianna SK, Rizzino A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev Biol. 2010. 344: 16-25
87. Mantovani A, Allaventa P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008. 454: 436-44
88. Martin-Green M, Boudreau N, Bissel MJ. Inflammation is responsible for the development of wound-induced tumors in chickens infected with Rous sarcoma virus. Cancer Res. 1994. 54: 4334-41
89. Medema JP, Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature. 2011. 474: 318-26
90. Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 2012. 12: 306-
91. Menegazzi M, Mariotto S, Dal Bosco M, Darra E, Vaiana N, Shiji K. Direct interaction of natural and synthetic catechins with signal transducer activator of transcription 1 affects both its phosphorylation and activity. FEBS J. 2014. p. 281724-38
92. Moss RW. An annotated bibliography of works by John Beard. Integ Cancer Ther. 2008. 7: 317-21
93. Munn DH. Indolamine 2,3-dioxygenase, tumor-induced tolerance and counter-regulation. Curr Opin Immunol. 2006. 18: 220-5
94. Murley JS, Kataoka Y, Cao D, Li JJ, Oberley LW, Grdina DJ. Delayed radioprotection by Nf kappaB-mediated induction of Sod2 (MnSOD) in Sa-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res. 2004. 162: 536-46
95. Nagata T, Shimada Y, Sekine S, Hori R, Matsui K, Okumura T. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2012. 21: 96-101
96. Oh YS, Kim HY, Song IC, Yun HJ, Jo DY, Kim S. Hypoxia induces CXCR4 expression and biological activity in gastric cancer cells through activation of hypoxia-inducible factor-1α. Oncol Rep. 2012. 28: 2239-46
97. Ohnishi S, Ma N, Thanan R, Pinlaor S, Hammam O, Murata M. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxid Med Cell Longev 2013. 2013. p.
98. Okada F. Inflammation and free radicals tumor development and progression. Redox Rep. 2002. 7: 375-68
99. Orr WS, Denbo JW, Saab KR, Ng CY, Wu J, Li K. Curcumin potentiates rhabdomyosarcoma radiosensitivity by suppressing NF-κB activity. PloS One. 2013. 8: e51309-
100. Ott G. Impact of MYC on malignant behavior. Hematology Am Soc Hematol Educ Program 2014. 2014. p. 100-6
101. Park CH, Bergaagel DE, McCulloch EA. Mouse myloma stem cells: A primary culture assay. J Natl Cancer Inst. 1971. 46: 411-22
102. Patel N, Joseph C, Corcoran GB, Ray SD. Silymarin modulates doxorubicin-induced oxidative stress, Bcl-xL and p53 expression while preventing apoptotic and necrotic cell death in the liver. Toxicol Appl Pharmacol. 2010. 245: 143-52
103. Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. 2002. 118: 915-22
104. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S. NF-kappaB functions as a tumor promoter in inflammation-associated cancer. Nature. 2004. 431: 461-6
105. Polakis P. Wnt signaling and cancer. Genes Dev. 2000. 14: 1837-51
106. Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007. 104: 973-8
107. Rao S, Sengupta R, Choe EJ, Woerner M, Jackson E, Sun T. CXCL12 mediates trophic interactions between endothelial and tumor cells in glioblastoma. PLoS One. 2012. 7: e33005-
108. Ravishankar D, Rajora AK, Greco F, Osborn HM. Flavonoids as prospective compounds for anti-cancer therapy. In J Biochem Cell Biol. 2013. 45: 2821-31
109. Read SA, Douglas MW. Virus induced inflammation and cancer development. Cancer Lett. 2014. 345: 174-81
110. Reya T, Morrison SJ, Clarke MF, Weissman IK. Stem cells, cancer, and cancer stem cells. Nature. 2001. 414: 105-11
111. Rothman N, Skibola CF, Wang SS, Morgan G, Lan Q, Smith MT. Genetic variation in TNF and IL10 and risk of non-Hodgkin lymphoma: A report from the InterLymph Consortium. Lancet Oncol. 2006. 7: 27-38
112. Rutkowski P, Kaminska J, Kowalska M, Ruka W, Steffen J. Cytokine and cytokine receptor serum levels in adult bone sarcoma patients: correlations with local tumor extent and prognosis. J Surg Oncol. 2003. 84: 151-9
113. Sakuma S, Sawamura Y, Tada M, Aida T, Abe H, Suzuki K. Responses of human glioblastoma cells to human tumor necrosis factor-alpha: Susceptibility, mechanism of resistance and cytokine production studies. J Neurooncol. 1993. 15: 197-208
114. Salsbury AJ. The significance of the circulating cancer cells. Cancer Treat Rev. 1975. 2: 55-72
115. Salvucci O, Bouchard A, Baccarelli A, Deschenes J, Sauter G, Simon R. The role of CXCR4 receptor in breast cancer: a large tissue microarray study. Breast Cancer Res Treat. 2006. 97: 275-83
116. Sarvalya PJ, Guo D, Ulasov I, Gabikian P, Lesniak MS. Chemokines in tumor progression and metastasis. Oncotarget. 2013. 4: 2171-85
117. Sato A, Okada M, Shibuya K, Watanabe E, Seino S, Narita Y. Pivotal role for ROS activation of p38 MAPK in control of differentiation and tumor initiating capacity of glioma initiating cells. Stem Cell Res. 2014. 12: 119-31
118. Sato Y, Goto Y, Narita N, Hoon DS. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron. 2009. 2: 205-14
119. Schepers AG, Snippert HJ, Strange DE, van den Born M, van Es JH, van de Wetering M. Linage tracing reveals Lgr5+stem cell activity in mouse intestinal adenomas. Science. 2012. 337: 730-5
120. Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010. 31: 37-49
121. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A. Regulation of chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003. 198: 1391-402
122. Schraivogel D, Weinmann L, Beier D, Tabatabai G, Eichner A, Zhu JY. CAMTA1 is a novel tumor suppressor regulated by miR-9/9 in glioblastoma stem cells. EMBRO J. 2011. 30: 4309-22
123. Schrocksnadel K, Wirleitner B, Winkler C, Fuchs D. Monitoring tryptophan metabolism in chronic immune activation. Clin Chim Acta. 2006. 364: 82-90
124. Senggunprai L, Kuongviriyapan V, Prawan A, Kukongviriyapan U. Quercetin and EGCG exhibit chemopreventative effects in cholangiocarcinoma cells via suppression of JAK/STAT signaling pathway. Phytother Res. 2013. 28: 841-8
125. Seo HS, Choi HS, Kim SR, Choi YK, Woo SM, Park SY, Wung BS, Hsu MC, Wu CC, Hsieh CW. Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: Effects on inhibition of STAT3 phosphorylation. Life Sci. 2005. 78: 389-97
126. Sethi S, Sarkar FH. Regulating miRNA by natural agents as a new strategy for cancer treatment. Curr Drug Targets. 2013. 14: 1167-74
127. Shchors K, Shchors E, Rostker F, Lawlor ER, Brown-Swigart L, Evan GI. The myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006. 20: 2527-38
128. Shehzad A, Park JW, Lee J, Lee YS. Curcumin induces radiosensitivity of in vitro and in vivo cancer models by modulating pre-mRNA processing factor 4 (Prp4). Chem Biol Interact. 2013. 206: 394-402
129. Shen HM, Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009. 14: 348-63
130. Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T. Role of tumor-associated macrophages ion the progression of hepatocellular carcinoma. Surg Today. 2012. 42: 1-7
131. Si M, Zhao J, Li X, Tian JG, Li YG, Li JM. Reversion effects of curcumin on multidrug resistance pf MNNG/HOS human osteosarcoma cells in vitro and in vivo through regulation of P-glycoprotein. Chin Med J. 2013. 126: 4116-23
132. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J. Identification of cancer stem cells in human brain tumors. Cancer Res. 2003. 63: 5821-8
133. Sinkovics JG. Molecular biology of oncogenic inflammatory processes. I. Non-oncogenic and oncogenic pathogens, intrinsic inflammatory reactions without pathogens, and microRNA/DNA interactions (review). Int J Oncol. 2012. 40: 305-49
134. Siu MK, Wong ES, Kong DS, Chan HY, Jiang L, Wong OG. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene. 2013. 32: 3500-9
135. Smirnova L, Grafe A, Seiler A, Schumacher S, Nitsh R, Wulczyn FG. Regulation of miRNA expression during neural cell specification. Eur J Neurosci. 2005. 21: 1469-77
136. Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Fick HE. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2012. 2: 722-35
137. Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007. 13: 1211-8
138. Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004. 6: 447-58
139. Spradling A, Drummond-Barbosa D, Kai T. Stem cells find their niche. Nature. 2001. 414: 98-104
140. Stockler M, Wilcken NR, Ghersi D, Simes RJ. Systematic reviews of chemotherapy and endocrine therapy in metastatic breast cancer. Cancer Treat Rev. 2000. 26: 151-68
141. Takahashi RU, Miyazaki H, Ochiya T. The role of microRNAs in the regulation of cancer stem cells. Front Genet. 2014. 4: 295-
142. Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nature Med. 2002. 8: 1089-97
143. Tian F, Fan T, Zhang Y, Jiang Y, Zhang X. Curcumin potentiates the antitumor effects of 5-FU in treatment of esophageal squamous cells through downregulating the activation of NF-κB signaling pathway in vitro and in vivo. Acta Biochim Biophys Sin (Shanghai). 2012. 44: 847-55
144. Todd R, Wong DT. Wong. Oncogenes. Anticancer Res. 1999. 19: 4729-46
145. Trabanelli S, Ocadlikova D, Evangelisti C, Parisi S, Curti A. Induction of regulatory T cells by dendritic cells through indolamine 2,3-dioxygenase: A potent mechanism of acquired peripheral tolerance. Curr Med Chem. 2011. 18: 2234-9
146. Tu SP, Jin H, Shi JD, Zhu LM, Suo Y, Lu G. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev Res. 2012. 5: 205-15
147. Visvader JE. Cells of origin in cancer. Nature. 2011. 469: 314-22
148. Wang CY, Cusack JC, Liu R, Baldwin AS. Control of inducible chemoresistance enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999. 5: 412-7
149. Wang G, Zhang J, Sharma S, Dong O. Quercetin potentiates doxorubicin mediated antitumor effects against liver cancer through p53/Bcl-xl. PloS One. 2012. 7: e51764-
150. Wang J, Pae M, Meydani SN. Green tea epigallocatechin-3 gallate modulates differentiation of naïve CD4 T cells into specific lineage effector cells. J Mol Med (Berl). 2013. 91: 485-95
151. Wang ML, Chiou SH, Wu CW. Targeting cancer stem cells: Emerging role of nanog transcription factor. OncoTargets Ther. 2013. 6: 1207-20
152. Wang Q, Fan H, Liu Y, Yin Z, Cai H, Liu J. Curcumin enhances the radiosensitivity in nasopharyngeal carcinoma cells involving the reversal of differentially expressed long non-coding TNAs. In J Oncol. 2014. 44: 858-64
153. Wang TT, Wang SK, Huang GL, Sun GJ. Luteolin induced-growth inhibition and apoptosis of human esophageal squamous carcinoma cell line Eca109 cells in vitro. Asian Pac J Cancer Prev. 2012. 13: 5455-61
154. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 2006. 66: 1883-90
155. Wiejak J, Dunlop J, Mackay SP, Yarwood SJ. Flavonoids induce expression of the suppressor of cytokine signaling 3 (SOCS3) gene and suppress TL-6-activated signal transducer and activator of transcription 3 (STAT3) activation in vascular endothelial cells. Biochem J. 2013. 454: 283-93
156. Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. 2010. 102: 639-44
157. Yang CL, Liu YY, Ma YG, Xue YX, Liu DG, Ren Y. Curcumin blocks small cell lung cancer cells migration, invasion, angiogenesis, cell cycle and neoplasia through janus kinase-STAT3 signaling pathway. PloS One. 2012. 7: e37960-
158. Ye MX, Zhao YL, Li Y, Miao Q, Li ZK, Ren XL. Curcumin reverses cis-platin resistance and promotes human lung adenocarcinoma A549/DDP cell apoptosis through HIF-1α and caspase-3 mechanisms. Phytomedicine. 2012. 15: 779-87
159. Yiannakopoulou ECh. Effect of green tea catechins on breast carcinogenesis: A systematic review of in-vitro and in-vivo experimental studies. Eur J Cancer Prev. 2014. 23: 84-9
160. Yu L, Baxter PA, Voicu H, Gurusiddappa S, Zhao Y, Adesina A. A clinically relevant orthoptic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo . Neuro Oncol. 2010. 12: 580-94
161. Yu L, Chen C, Wang LF, Kuang X, Liu K, Zhang H. Neuroprotective effect of kaempferol glycosides against brain injury and neuroinflammation by inhibiting the activation of NFκB and STAT3 in transient focal stroke. PloS One. 2013. 8: e55839-
162. Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer Stem Cells. Int J Biochem Cell Biol. 2012. 44: 2144-51
163. Zhai K, Ding J, Zhou Y. Different role of tumor necrosis factor-alpha polymorphism in non-Hodgkin lymphomas among Caucasion and Asian populations: A meta-analysis. Int J Mol Sci. 2014. 15: 7684-98
164. Zhang M, He Y, Sun X, Li G, Wang W, Zhao A. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. 2014. 7: 19-
165. Zhang Y, Sime W, Juhas M, Sjolander A. Crosstalk between colon cancer cells and macrophages via inflammatory mediators and CD47 promotes tumor cell migration. Eur J Cancer. 2013. 49: 3320-34
166. Zhou D, Bigarella CL, Liang R, GhaffRI S. Stem cells and the impact of ROS signaling. Development. 2014. 141: 4206-18
167. Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001. 7: 1028-34

