
Abstract
Distal hereditary motor neuropathy (dHMN) or distal spinal muscular atrophy (OMIM #182960) is a heterogeneous group of disorders characterized by an almost exclusive degeneration of motor nerve fibers, predominantly in the distal part of the limbs1. Silver syndrome (OMIM #270685) is a rare form of hereditary spastic paraparesis mapped to chromosome 11q12–q14 (SPG17) in which spasticity of the legs is accompanied by amyotrophy of the hands and occasionally also the lower limbs2,3. Silver syndrome and most forms of dHMN are autosomal dominantly inherited with incomplete penetrance and a broad variability in clinical expression. A genome-wide scan in an Austrian family with dHMN-V (ref. 4) showed linkage to the locus SPG17, which was confirmed in 16 additional families with a phenotype characteristic of dHMN or Silver syndrome. After refining the critical region to 1 Mb, we sequenced the gene Berardinelli-Seip congenital lipodystrophy (BSCL2) and identified two heterozygous missense mutations resulting in the amino acid substitutions N88S and S90L. Null mutations in BSCL2, which encodes the protein seipin, were previously shown to be associated with autosomal recessive Berardinelli-Seip congenital lipodystrophy5 (OMIM #269700). We show that seipin is an integral membrane protein of the endoplasmic reticulum (ER). The amino acid substitutions N88S and S90L affect glycosylation of seipin and result in aggregate formation leading to neurodegeneration.
Similar content being viewed by others


Main
We previously reported the clinical details of a large Austrian family with dHMN-V in whom linkage to the loci known to be associated with dHMN was excluded4. A whole-genome scan established linkage to chromosome 11q12–q14, and several candidate genes were excluded by sequence analysis6. This chromosomal region overlaps the region involved in Silver syndrome3. The dHMNs and Silver syndrome are characterized by predominant involvement of lower motor neurons. Unlike other forms of dHMN, dHMN-V and Silver syndrome share the unusual characteristic that prominent hand muscle weakness and wasting occur early in the course of the disease and may still predominate later on. Individuals with Silver syndrome also have mild to severe spasticity of the lower limbs, indicating involvement of upper motor neurons as well. The clinical similarities and the shared linkage region suggested that the two diseases might be allelic.
To investigate this possibility, we recruited the following additional families: four Austrian families6 with spastic paraparesis and amyotrophy of the hands (F01–F04), one Italian family with pure motor neuropathy sometimes starting in the hands (F16), seven Austrian families with dHMN-V (F08–F15), one Belgian family with spastic paraparesis and with distal amyotrophy always starting and predominating in the legs (F05), one Brazilian family (F06) and the original English family described by Silver2 (F07) and one English family (F17) with dHMN-V. Fine mapping indicated that the disease in all families could be linked to SPG17 and reduced the candidate gene interval from 13 cM to 2.5 cM, placing the critical region between the markers D11S1765 and D11S1883 (ref. 6). Haplotype analysis further refined the critical region to an interval of ∼1 Mb between the markers WP7 and D11S4205 (Table 1) containing 31 genes.
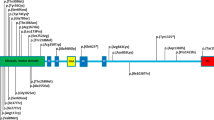
Subsequent sequence analysis identified two distinct missense mutations in the gene BSCL2 (Fig. 1), which encodes the protein seipin whose function was unknown. Mutations in BSCL2 were previously reported to cause autosomal recessive Berardinelli-Seip congenital lipodystrophy type 2, a disorder clinically unrelated to dHMN and Silver syndrome5,7. Affected individuals of the Austrian families (F01–F04, F08–F15), the Italian family (F16) and the English families (F07, F17) were heterozygous with respect to a 263A→G transition mutation leading to the amino acid change N88S, whereas those of the Belgian family (F05) and the Brazilian family (F06) were heterozygous with respect to a 269C→T (S90L) missense mutation. These mutations cosegregated with the disease in 117 affected individuals and in 10 clinically unaffected at-risk individuals who occasionally showed aberrant nerve conduction velocities but no clinical signs, confirming incomplete penetrance and subclinical expression of the mutations. We detected no mutations in a panel of 1,100 control chromosomes (500 Austrian, 200 Belgian and 400 English). Therefore, these sequence changes are probably not single-nucleotide polymorphisms unrelated to the phenotype. Comparison of the haplotypes of the Austrian, Italian and two English families suggested that these families did not share a common ancestry and that the N88S mutation arose independently in each. No mutations were identified in 24 unrelated individuals with the phenotype of dHMN-V and Silver syndrome, confirming genetic heterogeneity3.

(a) Graphical representation of the exons comprising BSCL2, predicted protein domains (I, inside; TM, transmembrane; O, outside), consensus sequence and position of the N-glycosylation site in the protein. (b) Partial electropherograms after direct DNA sequencing of PCR products for the missense mutation 263A→G (resulting in the amino acid substitution N88S; left), control DNA from an unaffected person (middle) and the missense mutation 269C→T (resulting in the amino acid substitution S90L; right), destroying the two conserved amino acids for seipin N-glycosylation (heterozygosity indicated by an arrow).
Northern-blot analysis with a 5′-specific probe detected the expression of two BSCL2 variants (Fig. 2a). We detected a brain-specific transcript of ∼1.8 kb and a ubiquitously expressed 2.2-kb mRNA, both encoding a predicted protein with 398 amino acids. An alternative start codon, potentially encoding a protein of 464 amino acids, seems improbable based on the statistical predictions of the Kozak's consensus sequence. A brain-specific northern blot showed the expression of the 1.8-kb mRNA in all regions analyzed (Fig. 2b). Analysis of the mouse homolog of BSCL2 showed expression in ventral horn and dorsal root ganglia and adult mouse brain (Supplementary Fig. 1 online).

(a) Hybridization with a 481-bp probe specific to the 5′ untranslated region on a multiple-tissue northern blot (Clontech) detected a brain-specific transcript of ∼1.8 kb and ubiquitous expression of a 2.2-kb BSCL2 transcript in all lanes except heart. (b) Brain-specific northern blot gave signals in all brain regions analyzed. (c) Western blots. Lane S, molecular weight standard; lane 1, cell lysate of COS-7 cells transiently transfected with wild-type seipin–EGFP; lane 2, cell lysate of COS-7 cells transiently transfected with N88S seipin–EGFP; lane 3, cell lysate of COS-7 cells transiently transfected with S90L seipin–EGFP; lane 4, cell lysate of untransfected COS-7 cells; lane 5, pellet fraction of untransfected COS-7 cells; lane 6, pellet fraction of COS-7 cells transiently transfected with wild-type seipin–EGFP; lane 7, pellet fraction of COS-7 cells transiently transfected with N88S seipin–EGFP; lane 8, pellet fraction of COS-7 cells transiently transfected with S90L seipin–EGFP; lane 9, positive control cell lysate of COS-7 cells transfected with rat hormone sensitive lipase-EGFP (molecular weight of ∼115 kDa). (d) Deglycosylation assay of wild-type seipin–EGFP (lanes 1, 2) and N88S seipin–EGFP (lanes 3, 4) fusion protein. Seipin-EGFP in pellet fractions was either kept on ice untreated (lanes 1, 3) or treated with N-glycosidase F, hydrolyzing all classes of asparagine-linked glycans (lanes 2, 4). Markers to the left of lane 1 indicate the molecular mass: >, 90 kDa; *, 85 kDa.
Both mutations are located in exon 3 of BSCL2 and destroy a predicted N-glycosylation site of the protein, affecting the highly conserved residues of the consensus sequence N-X-S/T at positions 88–90 (Fig. 1a). Comparison of protein masses after western blotting showed a shift in the apparent molecular weight of both mutant proteins (Fig. 2c). The wild-type fusion protein migrated at an apparent molecular weight of ∼90 kDa, whereas both mutant proteins had a molecular weight of ∼85 kDa. This migration shift cannot be explained by the amino acid exchanges, and so we analyzed N-glycosylation of seipin. After treatment of isolated protein pellets with N-glycosidase F, wild-type seipin tagged with enhanced green fluorescent protein (EGFP) and N88S seipin–EGFP had identical molecular masses after deglycosylation, comparable to that of untreated N88S seipin–EGFP (Fig. 2d), confirming the predicted glycosylation site.
The addition of N-linked oligosaccharides to newly synthesized polypeptides occurs cotranslationally in the ER, and its most important function in eukaryotes is to promote proper protein folding8. Inhibition of glycosylation most commonly leads to the generation of misfolded and aggregated proteins that are nonfunctional. To investigate the cellular localization of seipin, we transiently transfected immortalized neuroblastoma–spinal cord (NSC34) cells and visualized the subcellular localization of seipin–green fluorescent protein (GFP) by fluorescence microscopy. After transfection, most cells expressing the wild-type protein showed staining throughout the cytoplasm (Fig. 3a–c). In contrast, most NSC34 cells expressing the mutant proteins had a variable number (typically two) of brightly fluorescent spots (Fig. 3d–i). We obtained similar results in studies using Chinese hamster ovary (CHO) cells, COS-7 cells and human umbilical vein endothelial cells (EA.hy926). Mutant seipin seems to become instantly concentrated in brightly fluorescent spots resembling aggresomes9,10. With increasing time after transfection, aggregation of the expressed protein developed in cells containing the wild-type seipin–EGFP protein (data not shown). This may simply reflect overexpression of the protein11.

Phase contrast pictures are shown on the left (a,d,g), fluorescence pictures are in the middle column (b,e,h) and merged images are on the right (c,f,i). Expression of wild-type seipin fusion protein (a–c), N88S seipin–GFP (d–f) and S90L seipin–GFP (g–i) in NSC34 cells. Wild-type seipin was distributed in the cytoplasm, whereas the mutant proteins were localized in bright fluorescent spherical bodies (arrowheads).
We quantified the EGFP fluorescence signals on the single-cell level in transfected CHO cells (Fig. 4). Fluorescence intensities per μm2 of the ER-targeted wild-type seipin–EGFP fusion protein were significantly higher than those of the ER-located EGFP fusion proteins of both mutant seipins (wild-type, 26.49 ± 6.64, n = 23; N88S, 9.08 ± 3.54, n = 14 and S90L, 4.76 ± 0.82, n = 13). When calculating the fluorescence intensities per μm2 in the protein accumulated outside the ER, the findings were reversed. Both mutants had higher fluorescence intensities per μm2 than wild-type seipin (wild-type, 1,964 ± 308, n = 35; N88S, 2,908 ± 268, n = 26 and S90L, 3,298 ± 595, n = 25). Intracellular localization of wild-type seipin fusion protein in the ER was shown in the EA.hy926 cell line by confocal microscopy using calreticulin protein as marker (Fig. 5).

Fluorescence signals of single cells (n = 13–36) were analyzed and statistical significance was evaluated using Scheffé's post hoc F test. (a,b) Comparison of fluorescence (F) intensities of wild-type seipin, N88S mutant seipin and S90L mutant seipin showing differences in the distribution between aggregates and ER. (c,d) Fluorescence intensities per μm2 determined for protein accumulations and for the ER showed significant differences in cellular location between wild-type and mutant seipin-EGFP fusion proteins (statistical significance indicated by an asterisk).

(a) Fluorescence micrograph indicates distribution of wild-type seipin–EGFP 16 h after transfection. (b) ER distribution depicted by calreticulin staining for the same subcellular region shown in red. (c) Merged analysis showing colocalization of wild-type seipin and calreticulin in the ER.
We showed that heterozygous missense mutations in BSCL2 lead to dHMN-V and Silver syndrome, and we established that Silver syndrome and some forms of dHMN are, in fact, extreme phenotypes that share the same genetic etiology. In addition, we showed that both conditions are genetically heterogeneous, as some individuals with each phenotype do not carry a BSCL2 mutation. Also, the originally described dHMN-V phenotype12 (also called distal spinal muscular atrophy V and CMT 2D), which has already been associated with mutations in the gene glycyl-tRNA synthetase13 (GARS; OMIM #600794), supports the presence of genetic heterogeneity. Further genotype-phenotype studies are needed to find out whether the distinct phenotypes correlate with specific mutations and to establish whether there are phenotypically different forms of dHMN-V caused by mutations in BSCL2 versus GARS.
How BSCL2 mutations lead to motor neuron degeneration remains enigmatic. But we determined that seipin is glycosylated and that both N88S and S90L affect the consensus sequence for N-glycosylation. GFP-labeled mutant seipin also results in aggregates, which may share some features in common with aggresomes, a well known characteristic of a steadily increasing number of neurodegenerative disorders14,15,16. The fact that mutations in the N-glycosylation sequence of seipin seem to be specifically required to cause dHMN-V and Silver syndrome, whereas null mutations result in Berardinelli-Seip congenital lipodystrophy type 2, supports the hypothesis that a gain-of-function mechanism underlies the motor neuron degeneration in these conditions. As glycosylation is a fundamental modification for normal protein processing, a gain-of-function mechanism may result from seipin misfolding.
The presence of aggresomes may have additional deleterious effects. In autosomal dominant retinitis pigmentosa, the presence of the opsin aggresome can recruit wild-type protein17 and eventually leads to apoptosis. Protein folding is a complex process that can be disturbed by genetic causes or environmental factors11. Alterations of seipin function, either as a causative defect or a disease-modulating factor, should therefore be further investigated in other inherited and acquired lower and upper motor neuron disorders, including amyotrophic lateral sclerosis. The unrelated lipodystrophy and neurodegenerative phenotypes produced by BSCL2 mutations, which seem to arise from either null mutations or protein misfolding, highlight the fact that any molecule can be considered for mutation screening in other HMNs.
Methods
Clinical studies.
We included 7 families with Silver syndrome (F01–F07) and 11 families with dHMN-V (F08–F17) in our linkage studies. We extended the previously described4 Austrian family with dHMN-V (F15) with four new branches (F01–F04) showing typical signs of Silver syndrome and seven branches (F08–F14) with dHMN-V. We also included the English family (F07) in whom the Silver syndrome locus on 11q12–q14 was originally mapped, a Belgian family (F05), a Brazilian family (F06) with Silver syndrome, an Italian family (F16) and an additional English family with dHMN-V (F17). We also studied 24 unrelated individuals with a phenotype characteristic of dHMN or Silver syndrome. All individuals had a detailed neurological examination. We analyzed nerve conduction velocities in several affected and at-risk individuals. At-risk individuals were diagnosed as affected if they had either spasticity of the lower extremities or wasting of the small hand muscles, or if there were signs of a motor neuropathy in the upper and lower limbs on electrophysiological testing. There was no clinical evidence of lipodystrophy or abnormal body fat distribution in these individuals.
The local Ethical Committee of each University and Institution approved the study, and we obtained written informed consent from all participants according to the Declaration of Helsinki.
Molecular genetics.
We isolated genomic DNA from total blood samples of family members and control individuals using a standard extraction protocol. We carried out a whole-genome scan in the Austrian family with dHMN-V (F15) using the ABI human Linkage Mapping Set v2.5-MD10 according to the instructions of the manufacturer (Applied Biosystems).
From sequences of human high-throughput genomic sequence clones localized in the Silver syndrome region, we selected known short tandem repeat (STR) sequences and identified nine probable polymorphic STR markers by BLAST searches (WP1–WP9). Four of these newly designed markers were polymorphic in the families in our study. To genotype STRs, we designed primer pairs with the Primer3 program. We carried out PCR amplification with dye-labeled primers on a MJ Research thermocycler. We carried out fragment analysis on an ABI Prism 3100 DNA Analyzer and processed data with the ABI GENESCAN 3.5 and GENOTYPER 3.6 software (Perkin-Elmer, Applied Biosystems). We computed genetic linkage with the LINKAGE program package considering the disease-causing mutation as a rare allele (1%). To calculate two-point lod scores, we assumed complete penetrance as electrophysiological measurements allows clinical diagnosis in asymptomatic individuals.
Mutation analysis.
We used the National Center for Biotechnology Information Entrez Genome Map Viewer, Ensembl Human Genome Server and GenBank databases to find known genes, expressed-sequence tags and putative new genes in the region associated with Silver syndrome. We determined the exon-intron boundaries of the candidate sequences by BLAST searches against the human genome sequence database at National Center for Biotechnology Information. We amplified all exons of the functional candidate genes by PCR using intronic primers (primer sequences are available on request). We sequenced PCR products using the BigDYEv3 ET Terminator Cycle Sequencing Kit (Perkin-Elmer, Applied Biosystems). We loaded the sequencing reactions on the ABI Prism 3100 DNA Analyzer (Perkin-Elmer, Applied Biosystems) and collected and analyzed data using the ABI DATA COLLECTION version 1.1 and DNA SEQUENCING ANALYSIS version 3.6 software, respectively.
Expression analysis.
We probed a multiple-tissue northern blot (Clontech) and a brain-specific northern blot using a cDNA probe for the 5′ untranslated region of BSCL2 (NM_032667) of 481 bp in size generated by PCR (primer sequences are available on request) following the manufacturer's recommendations. The probe was labeled with αP32dCTP by random priming and hybridized for 1 h at 65 °C in ExpressHybrid solution (Clontech). After washing, we detected and quantified signals with a Storm 840 phosphorimager.
We isolated ventral horn and dorsal root ganglia from 13-d-old mouse embryos. Brain tissue was from an adult mouse. We extracted total RNA using the Totally RNA Kit (Ambion) and carried out RT-PCR using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). We amplified exons 1–5 of mouse Bscl2, resulting in a 665-bp fragment, and exons 4–10, yielding a 697-bp PCR product (primer sequences are available on request).
We obtained two plasmid clones, IMAGp956M0263 and IMAGp956M2246, containing complete human BSCL2 cDNA sequences from The Resource Center of the German Human Genome Project). We cloned the full-length human BSCL2 cDNA into the pDNR-Dual donor vector (BD Creator cloning kit) after PCR amplification using the in-flash system according to the manufacturer's instructions (BD Biosciences Clontech). We then subcloned the coding region into the pLPS-3′EGFP expression vector, introduced the missense mutations, N88S or S90L, by PCR-mediated site-directed mutagenesis and confirmed the correct DNA sequence. We transiently transfected CHO cells with 2 μg of purified plasmid DNA using CLONfectin (BD Biosciences Clontech). We transiently transfected the human umbilical vein endothelial cell line EA.hy926 with 1.5–3 μg of purified plasmid DNA using TransFast Transfection Reagent (Promega). For analysis of NSC34 cells, we introduced the mutations using Quikchange II XL site-directed mutagenesis kit (Stratagene), cloned this into CT-GFP fusion TOPO TA expression kit (Invitrogen) and transiently transfected our cells with 0.8 μg of DNA using the effectene transfection reagent (Qiagen).
Protein analysis.
We cultured COS-7 cells in 100-mm-diameter dishes until 50–60% confluent cells were reached and transfected them with a complex consisting of 3 μg of plasmid DNA and 5 μl of the liposomal transfection reagent Metafectene (Biontex) for 48 h. We washed cells twice with 10 ml of cold phosphate-buffered saline (PBS) 48 h after transfection and incubated them with 0.5 or 1 ml of hypotonic lysis buffer (1:100 PBS, pH 7.2; 0.5% Triton X-100; protease inhibitors (0.1 mM phenylmethylsulfonyl fluoride, 10 μM leupeptin, 25 μg ml−1 aprotinin or a cocktail available from Sigma (#P8340)). We incubated the cells for 30 min at 4 °C on a shaker and scraped them into 1.5-ml microcentrifuge tubes. We centrifuged them at 14,000g for 30 min at 4 °C. We resuspended pellets in 200 μl of 1:100 PBS, pH 7.2 containing 1% SDS and protease inhibitors. Supernatants were adjusted to 1% SDS and all samples were sonicated in a single burst of 10–15 s at 70 W.
We carried out SDS-PAGE and western blotting according to standard methods. We carried out electroblotting in a Mini Trans-Blot cell (Bio-Rad) in transfer buffer (48 mM Tris-HCl, pH 9.2; 39 mM glycine, 20% methanol and 0.025% SDS) at a constant voltage of 100 V for 2 h. Before immunodetection, we compared the total amounts of protein per lane after staining with Ponceau-S. We carried out immunodetection using standard methods. Dilution of the primary antibody (antibody to GFP ab290; Abcam) was 1:2,500. We visualized bands using a horseradish peroxidase–labeled goat antibody to rabbit IgG and the ECL detection system (Amersham Pharmacia Biotech).
To detect N-glycosylation, we sonicated pellets of transiently transfected COS-7 cells in 100 μl buffer (1:100 PBS, pH 7.2; 0.5% Triton X-100; 0.1% SDS and protease inhibitors) by two bursts of 5 s each at 50 W. We treated aliquots of samples with N-glycosidase F for 30 min at 37 °C according to the kit protocol (#1836552, Roche) or kept them on ice.
Laser scanning microscopy and immunohistochemistry.
We visualized seipin-GFP fusion protein in transfected cells using an array confocal laser scanning microscope (VoxCell Scan, VisiTech) on a Zeiss Axiovert 200M based around the QLC100 laser confocal scanning module (Nipkow disc; VisiTech) controlled by Metamorph 4.0 (Universal Imaging, Visitron Systems). We also carried out experiments using a deconvolution microscope from Nikon (Eclipse 300TE inverted microscope) with a liquid-cooled CCD camera (−30 °C; Quantix KAF 1400G2, Roper Scientific), an epifluorescence system (150 W XBO, Optiquip) and a computer-controlled filter wheel (Ludl Electronic Products).
We used the following antibodies: antibody to calregulin (c-17; 0.4 μg ml−1; Santa Cruz) goat polyclonal antibody to IgG for 3 h and rabbit antibody to goat conjugated to Alexa Fluor 633 (4 μg ml−1; Molecular Probes) for 1 h.
Statistics.
We carried out analysis of variance and evaluated statistical significance using Scheffé's post hoc F test. We defined the level of significance as P < 0.05.
URLs.
The Resource Center of the German Human Genome Project is available at http://www.rzpd.de/. Primer3 is available at http://www.broad.mit.edu/cgi-bin/primer/ primer3_www.cgi. LINKAGE is available at http://linkage.rockefeller.edu/. Glycosylation prediction is available at http://www.cbs.dtu.dk/services/NetNGlyc/.
Accession numbers.
BSCL2 mRNAs: AF052149, BC009866, NM_032667 (GenBank). Seipin: NP_116056, AAH09866 (GenBank protein database). Genomic sequence clone containing BSCL2: NT_033903 (GenBank).
Note: Supplementary information is available on the Nature Genetics website.
References
Harding, A.E. Hereditary spastic paraplegias. Semin. Neurol. 13, 333–336 (1993).
Silver, J.R. Familial spastic paraplegia with amyotrophy of the hands. J. Neurol. Neurosurg. Psychiatry 29, 135–144 (1966).
Patel, H. et al. The Silver syndrome variant of hereditary spastic paraplegia maps to chromosome 11q12-q14, with evidence for genetic heterogeneity within this subtype. Am. J. Hum. Genet. 69, 209–215 (2001).
Auer-Grumbach, M. et al. Phenotypic and genotypic heterogeneity in hereditary motor neuronopathy type V: a clinical, electrophysiological and genetic study. Brain 123, 1612–1623 (2000).
Magré, J. et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 28, 3665–370 (2001).
Windpassinger, C. et al. Refinement of the “Silver syndrome locus” on chromosome 11q12-q14 in four families and exclusion of eight candidate genes. Hum. Genet. 114, 99–109 (2003).
Van Maldergem, L. et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J. Med. Genet. 39, 722–33 (2002).
Helenius, A. & Aebi, M. Intracellular functions of N-linked glycans. Science 291, 2364–2369 (2001).
Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 (2000).
Garcia-Mata, R., Gao, Y.-S. & Sztul, E. Hassels with taking out the garbage: Aggravatine aggresomes. Traffic 3, 388–396 (2002).
Johnston, J.A., Ward, C.L. & Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898 (1998).
Christodoulou, K. et al. Mapping of a distal form of spinal muscular atrophy with upper limb predominance to chromosome 7p. Hum. Mol. Genet. 4, 1629–1632 (1995).
Antonellis, A. et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 72, 1293–1299 (2003).
Ryan, M.C., Shooter, E.M. & Notterpeck, L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol. Dis. 10, 109–118 (2002).
Lee, M.J. et al. Hereditary sensory neuropathy is caused by a mutation in the delta subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct4) gene. Hum. Mol. Genet. 12, 1917–1925 (2003).
Johnston, J.A., Dalton, M.J., Gurney, M.E. & Kopito, R.R. Formation of high molecular weight complexes of mutant Cu, ZN-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 97, 12571–12576 (2000).
Saliba, R.S., Munro, P.M., Luthert, P.J. & Cheetham, M.E. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 115, 2907–2918 (2002).
Acknowledgements
We thank the affected individuals and their family members for their participation; H. Offenbacher, A. Irmler and R. Fischer for their contribution in recruiting the families; A. Krenn and A. Legenstein for technical assistance; and W. Graier and E. Steyrer for critical discussion of the manuscript. This research project was supported by the Muscular Dystrophy Association, USA; the Fonds zur Förderung der wissenschaftlichen Forschung, Austria; the Tom-Wahlig-Stiftung Jena, Germany; the Fachabteilung 6A - Wissenschaft und Forschung of the Land Steiermark; the Birth Defects Foundation, UK; the University of Antwerp, the Fund for Scientific Research - Flanders; the Medical Foundation Queen Elisabeth; the Association Belge contre les Maladies Neuromusculaires, and the Interuniversity Attraction Poles programme of the Belgian Federal Science Policy Office, Belgium. I.D. and N.V. are supported by Ph.D. fellowships of the Institute for Science and Technology, Belgium. The Institutes of Medical Biology and Human Genetics and of Medical Biochemistry & Medical Molecular Biology are members of the Institutes of Basic Medical Sciences at the University of Graz and were supported by the infrastructure program of the Austrian ministry of education, science and culture.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Windpassinger, C., Auer-Grumbach, M., Irobi, J. et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet 36, 271–276 (2004). https://doi.org/10.1038/ng1313
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1313
