
Abstract
Cyclic adenosine 3, 5′-monophosphate (cAMP) is a ubiquitous second messenger regulating many biological processes, such as cell migration, differentiation, proliferation and apoptosis. cAMP signaling functions not only on the plasma membrane, but also in the nucleus and in organelles such as mitochondria. Mitochondrial cAMP signaling is an indispensable part of the cytoplasm-mitochondrion crosstalk that maintains mitochondrial homeostasis, regulates mitochondrial dynamics, and modulates cellular stress responses and other signaling pathways. Recently, the compartmentalization of mitochondrial cAMP signaling has attracted great attentions. This new input should be carefully taken into account when we interpret the findings of mitochondrial cAMP signaling. In this review, we summarize previous and recent progress in our understanding of mitochondrial cAMP signaling, including the components of the signaling cascade, and the function and regulation of this signaling pathway in different mitochondrial compartments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria, unique organelles enclosed by a two-layered membrane, produce the majority of cellular ATP in the eukaryotic cells through oxidative phosphorylation (OXPHOS). They also house the biosynthetic processes for many of the cell’s building blocks, including lipids, nucleotides, amino acids, and heme. Besides their well-recognized roles in energy and intermediate metabolism, mitochondria are now accepted as an important nexus for signaling cascades involved in cell growth, proliferation, differentiation and death [1].
Mitochondria originated from the endosymbiosis of ancestral bacteria inside primitive eukaryotic cells, approximately two billion years ago [1, 2]. Over their long evolutionary history, the engulfed bacteria ceased to function as free-living organisms, becoming instead semi-autonomous organelles. The majority of the mitochondrial ancestor’s genetic content was either lost or transferred to the host nuclear genome [3], and only a small fraction was retained in mitochondria. A typical animal cell contains hundreds to thousands copies of mitochondrial DNA (mtDNA), each encoding 13 essential subunits of the electron transport chain (ETC), 2 ribosomal RNAs and 22 tRNAs required for protein synthesis inside the organelle. The majority of the approximately 1500 mitochondrial proteins are encoded by the nuclear genome, synthesized in the cytoplasm and imported into mitochondria after translation [4, 5].
Mitochondria cannot be produced de novo. Instead, their proliferation requires mtDNA replication and the addition of lipids and proteins to the existing organelles [6, 7]. Mitochondria also undergo constant fusion and fission to adjust their shape and numbers in cells [8, 9]. Fusion and fission are also critical for the positioning, movement and even destruction of mitochondria. Mitochondrial biogenesis (the growth and division of pre-existing mitochondria) and dynamics (movement and morphological changes) are highly plastic in response to the cell’s energy demand, to developmental cues and to environmental stimuli. The success of such symbiosis requires carefully orchestrated communications between the eukaryocyte host and the prokaryocyte organelle. In the past a few years, it has become apparent that these communications are mediated in great part by cAMP signaling, a universal pathway conserved in all cellular organisms.
In this review, we briefly describe the compartmentalized landscape of mitochondrial cAMP signaling and discuss its regulation and diverse functions. We summarize the established roles of cAMP-PKA signaling the outer mitochondrial membrane (OMM) in protein import, mitochondrial fission and apoptosis. We also discuss the recent evidence for cAMP signaling in the mitochondrial matrix, as well as its effectors in the regulation of OXPHOS and mitochondrial biogenesis. In addition, we present unresolved questions about intra-mitochondrial cAMP signaling that need to be addressed in the future. Finally, we speculate on the therapeutic potential of managing mitochondrial cAMP signaling for diseases linked to mitochondrial deficiencies.
Cellular cAMP signaling and compartmentalization (Fig. 1)
cAMP, one of the first identified and most versatile second messengers, mediates diverse cellular responses to extracellular signals. Over the past decades, various mechanisms triggering the production of cAMP have been identified, as well as its downstream effectors. Upon the activation of membrane receptors, downstream transducers, mainly G proteins, activate adenylyl cyclases on the plasma membrane (TmACs) to convert ATP to cAMP [10, 11]. Intracellular cAMP can also be produced by soluble adenylyl cyclases (sACs) in response to bicarbonate, calcium and the change of ATP level [12–15]. The major downstream effector of cAMP signaling is protein kinase A (PKA), a heterotetramer consisting of two catalytic subunits and two regulatory subunits. cAMP binds to the regulatory subunits, which releases and activates the catalytic subunits [16, 17]. Activated PKA phosphorylates and activates cAMP response element (CRE)-binding protein (CREB), a transcriptional co-factor that initiates an array of transcriptional cascades involved in immune response, cellular metabolism and mitochondrial biogenesis [18–20]. Besides its role in transcriptional regulation, PKA phosphorylates and modulates the activity of ion channels [21–23], cellular motor proteins [24, 25] and many enzymes involved in intermediate metabolism [26]. Many signaling and regulatory proteins, such as phospholipase C (PLC) [27], protein kinase C (PKC) [28], phosphoinositide 3-kinase (PI3K) [29, 30] and inositol trisphosphate (IP3) receptors [31], are also regulated by PKA-dependent phosphorylation. These phosphorylation events intertwine cAMP-PKA signaling with other cellular messengers and signaling cascades, and provide multiple feedback loops further modulating cAMP signaling [32, 33]. Intracellular cAMP level is regulated by the balanced act of ACs and cyclic nucleotide phosphodiesterases (PDEs), which terminate cAMP-PKA signaling by hydrolyzing cAMP to AMP [34, 35]. Besides the main effector PKA, cAMP can also directly activate the exchange protein Epac [36], the cyclic nucleotide-gated channels [37] and the Popeye domain-containing proteins [38, 39]. Within a single cAMP cascade, ACs, PKA, other downstream effectors and PDEs are often tethered together by scaffold proteins, the A-kinase anchoring proteins (AKAPs), at distinct subcellular locations [16, 40]. The compartmentalization of these signaling proteins [41–44] not only promotes the efficiency of cAMP signaling transduction, but also allows the same second messenger, cAMP, to mediate diverse physiological responses.

General cAMP signaling pathways. Intracellular cAMPs are generated by two classes of ACs, the transmembrane AC (TmAC) and the soluble AC (sAC). Both TmAC and sAC convert ATP to cAMP upon stimulus. TmAC can be activated by G proteins when an extracellular signal is received by the G protein-coupled membrane receptor. sAC is insensitive to G-proteins but can be activated by bicarbonate and calcium. Elevated cAMP level activates protein kinase A (PKA), the major effector of cAMP signaling, by releasing the two catalytic subunits (Cs) from the two regulatory subunits (Rs). Activated PKA in turn phosphorylates and activates numerous downstream protein targets, including the cAMP response element binding protein (CREB), a transcriptional co-factor regulating multiple cellular processes. Cyclic nucleotide phosphodiesterases (PDEs) are the negative regulators that terminate cAMP-PKA signaling by hydrolyzing cAMP to AMP. As a result, cAMP level and signaling activity are determined by the equilibrium between ACs and PDEs. In addition to its main effector PKA, cAMP can also directly activate the exchange protein Epac, the cyclic nucleotide-gated channels (CNGCs) and the Popeye domain-containing proteins (Popdcs). Within a single cAMP cascade, ACs, PKA, other downstream effectors and PDEs are often tethered together by an A-kinase anchoring proteins (AKAPs) at distinct subcellular locations
The compartmentalized structure of mitochondria [45] further shapes the cAMP signaling profile. The cAMP produced by TmAC or sAC in the cytosol can freely diffuse to the outer mitochondrial membrane (OMM) and activate the local PKAs at the mitochondrial surface. Moreover, as the OMM is readily permeable to ions and small molecules of 5 kDa or less [46], cytosolic cAMP might also regulate pathways inside the inter-membrane space (IMS). The inner mitochondrial membrane (IMM), however, is intrinsically impermeable due to a lack of Porins [47] and a high content of cardiolipin [48]. Nevertheless, cAMP is detected in the mitochondrial matrix of metazoans [49], which raises the questions of its origin in the matrix, and of its potential role in the regulation of biochemical pathways in this compartment.
cAMP signaling in the outer-mitochondrial compartment (Fig. 2)
Metabolic switch and mitochondrial protein import
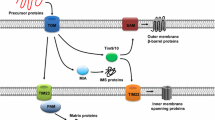
As the interface between mitochondria and cytosol, the OMM is enriched in proteins that respond to intracellular signaling messengers regulating mitochondrial biogenesis, morphology and removal. Mitochondrial biogenesis entails the synthesis of approximately 1500 nucleus-encoded polypeptides in the cytoplasm and their import to mitochondria [4, 5]. The translocase of the outer membrane (TOM) complex represents the predominant pathway for importing proteins across the OMM [4, 5]. It is also one of the main downstream effectors of cAMP-PKA signaling tuning mitochondrial biogenesis to metabolism [50]. Three core components of the TOM complex, TOM70, TOM22 and TOM40, are phosphorylated by PKA in response to the glucose-induced cAMP increase triggered by a metabolic switch from respiratory to fermentable conditions [51–53]. Phosphorylation by PKA impairs the interaction between TOM70 and metabolite carrier/chaperone complexes (e.g., AAC/Hsp70) [51], inhibits the translocation of TOM22 to mitochondria [52] and prevents the integration of TOM40 into the OMM [53]. Thus, elevated cAMP-PKA signaling slows down the import of mitochondrial proteins, and fosters the metabolic switch from OXPHOS to glycolysis in conditions of increased glucose or reduced oxygen availability.

cAMP signaling in the outer-mitochondrial compartment. cAMP from the cytosol or produced by sAC on the OMM can activate the local PKA which in turn phosphorylates different targets associated with the OMM. a PKA phosphorylation impairs the receptor activity of TOM70 and its interaction with the metabolite carrier/chaperone, prevents TOM22 translocation and TOM40 integration into the OMM, and eventually slows down the import of mitochondrial proteins. b PKA phosphorylation can also block Drp1’s translocation to the OMM surface and thus lead to reduced mitochondrial fission. c In mammals, PKA phosphorylation inhibits Bad’s apoptotic activity but promotes Bim’s by increasing its stability against proteasome-dependent degradation. PKA phosphorylation of Bax promotes its translocation to mitochondria and triggers the release of cytochrome c (CC) and the maturation of the apoptosome, which eventually leads to apoptosis. AKAPs tether PKA and other proteins, e.g., Bad, on the OMM to facilitate the cAMP-PKA targeting. They also promote different signaling specificities under the same environmental context by providing a dynamic platform of proteins complex in multiple combinations
Stress response and regulation of mitochondrial fission and fusion
Mitochondrial fission and fusion are indispensable for mitochondrial homeostasis [8, 9]. In a pool of overall healthy mitochondria, fusion mingles proteins, lipids and metabolites among individual mitochondria, thereby mitigating the defects of dysfunctional ones. On the other hand, fission enables the segregation of damaged mitochondria from a healthy population for their eventual removal, possibly through autophagy and mitochondrion-derived vesicles (MDV) [54–56]. Unbalanced fusion and fission can impair mitochondrial biogenesis, cause excessive mitochondrial fragmentation and trigger apoptosis [8, 9]. Elevated cAMP signaling upon bioenergetic stresses like starvation can inhibit mitochondrial fission, suspend unnecessary biogenesis and promote survival by sharing metabolites and boosting energy metabolism [57]. Mitochondrion fission is mediated by the dynamin-like GTPase, Drp1, which is recruited to the mitochondrial surface and assembled into a multimeric ring-like structure wrapping around the constriction points of the dividing mitochondria [58]. Phosphorylation of Drp1 by PKA blocks its translocation to the mitochondrial surface, leading to mitochondrial elongation rather than fission, which promotes cell survival [57, 59–61]. Reciprocally, Drp1 de-phosphorylation facilitates Drp1 recruitment to mitochondria and promotes fission, autophagy and apoptosis [54, 59, 62].
Under conditions of increased autophagy, a feedback response can promote fusion and survival by activating cAMP-PKA-dependent Drp1 phosphorylation and mitochondria elongation [57]. In addition to Drp1, PKA can also phosphorylate mitofusin 2 (Mfn2) [63] and bind to optic atrophy 1 (OPA1) [64, 65]. Both Mfn2 and OPA1 are involved in mitochondrial fusion [9]. However, the physiological significance of cAMP-PKA signaling in regulating mitochondrial fusion remains elusive.
Apoptosis
Several apoptosis-related proteins are substrates of PKA [66–73]. In mammals, the intrinsic apoptotic pathway is regulated by the concerted action of anti-apoptotic Bcl-2 like proteins and pro-apoptotic BH3-only proteins [74, 75]. The pro-apoptotic proteins Bax [68, 69], Bad [66, 67, 70, 71] and Bim [72] can all be phosphorylated by PKA. However PKA phosphorylation on these proteins have completely opposite consequences: IL3-induced PKA phosphorylation of Bad inhibits its apoptotic activity [66], and the effect is mediated through the formation of PKA-Bad complex on the OMM [67, 70, 71]. By contrast, PKA phosphorylation of Bax promotes its translocation to mitochondria and triggers cytochrome c release and apoptosis [68, 69]. In addition, PKA phosphorylation of an isoform of Bim increases its stability against proteasome-dependent degradation and promotes its apoptotic effect [72].
AKAPs as mediators of cAMP signaling specificity on the OMM
It is perplexing that a common messenger, cAMP, can elicit such diverse and distinct responses through different effectors on the OMM. It is now recognized that a family of PKA-anchoring proteins, the AKAPs, bind to and target PKA to distinct subcellular locations including the mitochondrial surface, the plasma membrane and the nucleus [16, 40]. AKAPs also act as scaffold proteins that tether PKA, PDEs, phosphatases, cytoskeleton proteins as well as other signaling molecules, such as PKC, MAPK, GSK-3β together, to form multi-protein signaling complexes [16, 40, 76–79]. Multiple AKAP isoforms have been found localized to mitochondria in metazoan [76, 78–86], and most AKAPs localize to the OMM. Specifically, D-AKAP1 has been proved directly siting in the OMM with its C-terminus protruding into the cytoplasm [87, 88]. The OMM-bound AKAPs tether PKA and other signaling molecules on the OMM [66, 78, 79, 84, 89–92]. This spatial organization allows the signaling specificities of different PKA pathways with a shared common messenger, cAMP, and thereby supports versatile combinations of mitochondrion related regulations. The sphingosine kinase interacting protein (SKIP) is an exceptional AKAP that recruits PKA to the IMS and the matrix of murine heart mitochondria [86].
cAMP signaling inside the matrix (Fig. 3)
Mitochondrial biogenesis relies on the coordination between mitochondrial and nuclear genomes [6, 7]. In particular, the assembly of ETC complexes and mitochondrial ribosomes require the proper stoichiometry of nucleus- and mitochondria-encoded components. Mitochondria originated from bacteria, and bacteria use cAMP signaling to regulate motility, metabolism and DNA replication [93]. One can therefore speculate that a local cAMP signaling might have been retained in the mitochondrial matrix during evolution even though most mitochondrial components became encoded in the nuclear genome. Considering the emerging roles of cAMP signaling in the regulation of nuclear transcriptional activity and protein import into mitochondria, it is conceivable that cAMP signaling might act in parallel inside the matrix to modulate mitochondrial activities locally. Indeed, accumulating evidence supports a role for cAMP signaling inside the matrix as a necessary complement to the nuclear regulation.

cAMP signaling in the mitochondrial matrix. The intra-mitochondrial cAMP-PKA pathway has been proposed to fine-tune metabolism by directly regulating the TCA cycle and respiration. sACs have been found in the mitochondrial matrix and produce cAMP locally in response to the CO2/HCO3 − generated by the TCA cycle. The matrix cAMP-PKA cascades are then activated, leading to the phosphorylation of ETC proteins such as Complex I and Complex IV subunits, modulating the OXPHOS and ATP production. In addition, PKA phosphorylation of the ATPase inhibitory factor 1 (IF1) abolishes its ability to bind to and inhibit Complex V. The sACs can also be activated by mitochondrial uptake of Ca2+. Active mechanisms for transporting cAMP into the matrix remain to be identified. To coordinate the OXPHOS and energy needs, the cAMP-PKA pathways outside mitochondria can activate the nuclear CREBs and the downstream transcription factors (PGC-1α, NRF) to promote TFAM (mtTFA) production, mtDNA replication and eventually mitochondrial biogenesis. On the other hand, the matrix cAMP signaling could exert a negative regulation on this process by increasing the PKA phosphorylation-dependent degradation of TFAM
Origin of cAMP in the mitochondrial matrix
Although cAMP per se cannot freely diffuse across the IMM from the cytosol, both biochemical assays with isolated mitochondria [94, 95] and live-cell experiments using genetic reporters [49, 96, 97] have demonstrated the presence of cAMP inside mammalian mitochondria. However, the origin of matrix cAMP is still debated. After a brief incubation with purified mitochondria in vitro, cAMP readily accumulates in the mitochondrial matrix, suggesting that it could be actively transported into the matrix [94]. But recent studies using genetic cAMP reporters in cultured human cell lines (Hela, HEK293T) and primary rat cardiomyocytes argue that mitochondrial inner membrane is impermeable to cAMP [96, 97]. A model for generating cAMP locally has hence been proposed. It involves a mitochondrially localized sAC that can be directly activated by HCO3 − [12] and was shown to synthesize cAMP locally in response to the CO2/HCO3 − produced in the TCA cycle in mammalian cultured cells [95–100]. The matrix CO2-sAC-cAMP-PKA cascade is also involved in the allosteric regulation of COX activity by ATP in the S. cerevisiae, demonstrating a conserved role of cAMP signaling in fine-tuning energy metabolism [101]. However, the Drosophila and C. elegans genomes do not encode any known sAC [102], and the prevalence of intra-mitochondrial cAMP signaling in metazoans has been called into question.
We recently constructed a matrix-localized cAMP sensor by fusing the new generation of cAMP reporter, ICUE3, with SOD2, a bona fide mitochondrial matrix protein [103]. Using this construct, we demonstrated the existence of cAMP in the mitochondrial matrix of Drosophila cultured cells. Given the lack of sAC in the fly genome, our finding suggests that an unidentified mechanism allows the rapid entry of cytosolic cAMP into the mitochondrial matrix, which is consistent with a previous biochemical study [94]. Still, the Drosophila genome contains 14 genes that encode 38 different AC isoforms (http://flybase.org/). We thus cannot rule out the possibility that a matrix AC might be among them and produce cAMP locally. Although the sources of matrix cAMP may differ in different organisms and cell types, the presence of cAMP in the mitochondrial matrix now appears universal.
PKA as the main effector of cAMP signaling in the mitochondrial matrix
Protein kinase A has long been considered as the main effector of intra-mitochondrial cAMP to regulate mitochondrial energy metabolism [95, 96, 104–107]. Large-scale phosphoproteome analyses revealed that many enzymes in the TCA cycle and ETC complex subunits were phosphorylated at the PKA consensus sites [108, 109]. These observations suggest that PKA is one of the most active kinases in the matrix [108, 109]. In support of this idea, 85 % of PKA activity associated with purified mitochondria is derived from the mitochondrial matrix fraction [110]. However, FRET-based PKA activity reporters that are targeted to the mitochondrial matrix do not respond to the addition of membrane-permeable cAMP analogs or bicarbonate [97]. It has been argued that the phosphorylation of matrix proteins at PKA consensus sites might take place before their import to the matrix [97]. We have developed a bimolecular fluorescence complementation (BiFC) assay for assessing the submitochondrial localization of proteins [103]. The assay is based on the fact that two halves of a GFP molecule will reconstitute into a whole, fluorescent GFP molecule only when they are in physical proximity [111]. By applying this sensitive assay, we demonstrated that PKA indeed locates to the mitochondrial matrix [103], substantiating the presence of local cAMP-PKA signaling.
Other members of the cAMP signaling pathway in the matrix
Besides the sACs and PKA, other components of cAMP signaling cascade have also been identified in the matrix, including cAMP PDE activity proteins such as PDE2A2 [100] and Prune [103]. Interestingly, PDEs can be inhibited by hydrogen sulfide (H2S), which is produced in the matrix by enzymes such as 3-mercaptopyruvate sulfurtransferase (3-MST), cystathionine β-synthase (CBS) and cystathionine g-lyase (CSE) in response to environmental stresses like hypoxia [112]. These findings suggest a potential link between the environmental stresses and cAMP signaling in the mitochondrial matrix. In summary, almost a full complement of key components of the cAMP signaling cascade (CO2/HCO3 −, Ca2+, sAC, cAMP, PKA, PDE and H2S) has been identified in the matrix, strongly supporting the existence of a distinct cAMP signaling in the mitochondrial matrix, which may have specific physiological and pathological implications.
Intra-mitochondrial cAMP signaling regulates OXPHOS
A rich body of literature has documented the phosphorylation of ETC complexes and enzymes in the TCA cycle by PKA [95, 104–109, 113–116]. This observation suggests that the cAMP-PKA pathway in the matrix may fine-tune metabolism by directly regulating the TCA cycle and respiration. A rise in CO2 or bicarbonate concentration is thought to activate a matrix-localized sAC. The sAC produces cAMP locally, which in turn activates PKA and leads to phosphorylation of cytochrome c oxidase subunit IV isoform 1 (COXIV-1), resulting in enhanced cytochrome c oxidase (COX) activity and OXPHOS [95]. In yeast, a similar phosphorylation-dependent regulation on Cox5a, the homolog of COXIV-1, has also been reported [101]. Evidences also suggest that PKA can phosphorylate subunits of Complex I, which increases Complex I activity and stability, and hence enhances respiration [107, 113, 117–122]. Given that CO2 is the end product of the TCA cycle, the cAMP-PKA pathway provides a feed-forward regulation on two modules of energy metabolism: TCA cycle and ETC complexes. Recently, Complex V activity has also been found under the regulation of matrix cAMP-PKA pathway [123–125]. Complex V (mitochondrial ATP synthase) catalyzes the synthesis of ATP through the proton gradient generated by the ETC complexes and executes the reverse hydrolysis when the membrane potential falls below a threshold (pH of ~6.7 or below) [126]. An ATPase inhibitory factor 1 (IF1) is thought to act as a “reverse rotation brake” for the ATP synthase motor, preventing the “wasteful” ATP hydrolysis under the anaerobic condition [126, 127]. A recent study demonstrated that IF1 binds to Complex V and inhibits not only its hydrolase but also its ATP synthase activity [123]. IF1 can be phosphorylated by PKA, which prevents it from binding to Complex V, and thereby relieves its inhibitory effects on both the hydrolytic and synthetic activities of Complex V [123]. Thus the phosphorylation status of IF1 appears to be key in regulating the flux of glycolysis and OXPHOS corresponding to certain physiological context [124]. Furthermore, De Rasmo and colleagues recently have demonstrated the importance of the sAC-cAMP signaling for the organization and activity of Complex V in isolated rat mitochondria and myoblast cultures [125].
The impact of PKA on ETC complexes appears to be multifaceted depending on the environment cues. Under hypoxia/ischemia condition, excess of reactive oxygen species (ROS) induces the sequestration of PKA catalytic α subunit into the matrix, and leads to the hyper-phosphorylation of Complex IV and reduced COX activity [114, 128]. PKA phosphorylation on several Complex IV subunits during heart failure may also inhibit OXPHOS by either disrupting the complex assembly or reducing its stability [129]. Taken together, the intra-mitochondrial cAMP-PKA signaling may serve as an acute and local mechanism allowing mitochondria to rapidly adjust energy output in response to environmental stress, and to oxygen and nutrient availability. Under the aerobic condition, the steady stream of CO2 produced from TCA cycle activates sAC-cAMP-PKA cascade, which in turn tunes up the activities of the ETC complexes by coordinating TCA cycle and ETC complexes. While CO2 production is diminished under metabolic or environmental stress, ROS can directly activate PKA to inhibit ETC complexes to avoid excessive ROS production. Of note, Ca2+ can also activate sAC, the principal source of matrix cAMP [96], highlighting a potential crosstalk and synergy between these two common second messengers (cAMP and Ca2+) in regulating energy metabolism.
Intra-mitochondrial cAMP signaling regulates mitochondrial biogenesis
Regulating OXPHOS is not the only way through which mitochondria respond to the cellular and environmental cues. A long-term adaptation of energy homeostasis can be achieved by modulating mitochondrial biogenesis. As previously described, during stresses such as hypoxia and starvation, the cAMP-PKA pathway can suppress mitochondrial biogenesis by inhibiting protein import and mitochondrial fission. On the other hand, the cytosolic cAMP-PKA pathway can also activate the nuclear CREBs and the downstream transcription factors (PGC-1α, NRF), which in turn activates the transcription of mitochondrial transcription factor A (mtTFA, also abbreviated as TFAM) and mitochondrial biogenesis [6, 130]. CREBs are also found inside mitochondria, binding to the CREs on the mtDNA D-loop, and directly regulating mtDNA-encoded gene expression [131–134]. The translocation of CREBs into mitochondria may be facilitated by chaperones like mtHsp70 [132] or by a process that depends on both membrane potential and TOM complex [134]. Both nuclear and mitochondrial CREB pathways promote neuronal survival in the brain [132, 133, 135], which is consistent with their positive roles in mitochondrial biogenesis.
Matrix cAMP signaling could also exert negative regulations on mitochondrial biogenesis. Human mtTFA can be phosphorylated by PKA, leading to decreased mtTFA-mtDNA interaction and increased mtTFA degradation [136]. Recently, we confirmed such phosphorylation in Drosophila and found that Prune, a mitochondrial PDE, stabilizes mtTFA by down-regulating cAMP levels in the mitochondrial matrix, thereby promotes mtDNA replication [103]. It thus appears the cAMP-PKA signaling acts as a “double agent” to fine-tune the mtTFA level. It promotes the expression of nuclear-encoded mtTFA, while counterbalances this action by negatively modulating mtTFA protein level through PKA-dependent degradation in the mitochondrial matrix. Our finding demonstrates the prevalence of mitochondrial matrix cAMP signaling and provides a new insight into its role in coordinating nuclear and mitochondrial regulation on mitochondrial biogenesis.
Future perspectives
Remaining mysteries in the mitochondrial cAMP signaling pathways
The repertoire of cAMP-PKA signaling components identified both on the OMM and in the mitochondrial matrix is growing. New regulations on mitochondrial functions by cAMP-PKA signaling keep emerging. We now recognize that cAMP signaling can respond to extracellular stimuli, intracellular cues or even environmental stresses to modulate a variety of mitochondrial behaviors including energy homeostasis, mitochondrial biogenesis and cell death. The literature also demonstrates the great complexity of mitochondrial cAMP signaling. The compartmentalization of mitochondrial cAMP signaling ensures its specificity. PKA phosphorylation on the same protein under different environmental or physiological context can lead to distinct, even opposite functional consequences, further demonstrating the complexity of mitochondrial cAMP signaling. Meanwhile, cAMP signaling pathway in different mitochondrial compartments (the OMM and the matrix) can work synergistically, and may interact with cytosolic and nuclear cAMP signaling to achieve coordinated and balanced regulations on mitochondrial functions.
The discovery of intra-mitochondrial cAMP signaling in Drosophila not only demonstrates the prevalence of mitochondrial cAMP signaling in metazoan, but also reveals an unknown regulation on mitochondrial biogenesis. However the lack of sACs in Drosophila and C. elegans genomes [102] presents an unsettled issue regarding the source of mitochondrial cAMP in these organisms. It is worth exploring whether there are unidentified AC isoforms that localize to mitochondria in these species. Meanwhile it remains to be determined whether there is indeed an active transport mechanism for cAMP into the matrix.
It is noteworthy that ATP-binding cassette (ABC) transporters have been found to work synergistically with PDEs regulating both local and global cAMP level [137, 138]. Recently one of the ABC B subfamily members has been shown to export cAMP in D. discoideum [139]. Several evolutionarily related ABC B subfamily proteins have been found localized on the IMM [140, 141]. It would be interesting to test whether any of these ABC transporters might contribute to the matrix cAMP transport.
sACs can be activated by direct Ca2+ binding in a dose-dependent manner [13], demonstrating a potential crosstalk between these two most common second messengers [32, 142, 143]. In cells that lack the active transporting mechanism for cAMP into the matrix, Ca2+ may relay the cytosolic cAMP signaling to the matrix and activate sAC wherein [96]. Thus the intra-mitochondrial Ca2+ and cAMP signaling may act synergistically in regulating energy metabolism [15, 32, 96, 144]. Whether Ca2+, or other messengers, affords coordination between cytosolic and intra-mitochondrial cAMP signaling, awaits further investigation. And to what extent a crosstalk between cAMP and other signaling is achieved to regulate mitochondrial behaviors remains to be explored.
Potential applications to the treatment of mitochondrial dysfunction diseases
Given the essential role of mitochondrial cAMP signaling in mitochondrial dynamics, biogenesis and metabolism, it is not surprising that its misregulation can cause various disorders, particularly in cells and tissues with high-energy demand. For instance, displacement of AKAP121 increases ROS production, induces apoptosis and triggers cardiac hypertrophy in transgenic rodents [145]. Loss of AKAP and decline of mitochondrial PKA signaling are believed to contribute to the etiology of several brain degenerative disease models [146]. In Drosophila, the misregulation of the intra-mitochondrial cAMP signaling impairs mitochondrial biogenesis and triggers neurodegeneration [103]. On the other hand, manipulating mitochondrial cAMP signaling might provide a handle to modulate energy metabolism and control cell death, thereby offering potential avenues for managing mitochondrial diseases and neuromuscular diseases associated with mitochondrial dysfunctions. Several compounds that modulate cAMP-PKA signaling globally, including PDE inhibitors and PKA inhibitors (H89), have shown promise in treating inflammation, diabetes and cardiovascular disorders [35, 147, 148]. However, their application to modulating mitochondrial cAMP signaling locally has to be cautioned. In particular, specific delivery of drugs into mitochondrial compartments is necessary to avoid broad activation or inhibition of cellular cAMP signaling. A lipophilic cation, triphenylphosphonium (TPP+), which preferentially accumulates in the mitochondrial matrix can effectively target antioxidants and metabolic-modulating compounds to mitochondria [149]. Peptides containing a unique aromatic-cationic sequence motif also concentrate on the mitochondria inner membrane [149, 150]. It would be interesting to test whether these molecules can be used to deliver cAMP signaling-modulating compounds to specific mitochondrial compartments, and thereby to improve mitochondrial functions without interfering with other cAMP signaling processes.
Abbreviations
- cAMP:
-
Cyclic adenosine 3′,5′-monophosphate
- ATP:
-
Adenosine triphosphate
- OXPHOS:
-
Oxidative phosphorylation
- mtDNA:
-
Mitochondrial DNA
- ETC:
-
Electron transport chain
- OMM:
-
Outer mitochondrial membrane
- AC:
-
Adenylyl cyclase
- TmAC:
-
Transmembrane adenylyl cyclase
- sAC:
-
Soluble adenylyl cyclase
- PKA:
-
Protein kinase A
- CREB:
-
cAMP response element binding protein
- PLC:
-
Phospholipase C
- PKC:
-
Protein kinase C
- PI3K:
-
Phosphoinositide 3-kinase
- IP3:
-
Inositol trisphosphate
- PDE:
-
Cyclic nucleotide phosphodiesterase
- AKAP:
-
A-kinase anchoring protein
- IMS:
-
Intermembrane space
- IMM:
-
Inner mitochondrial membrane
- TOM:
-
Translocase of the outer membrane
- AAC:
-
ADP/ATP carrier
- MDV:
-
Mitochondrion-derived vesicles
- Drp1:
-
Dynamin-related protein 1
- Mfn2:
-
Mitofusin 2
- OPA1:
-
Optic atrophy 1
- MAPK:
-
Mitogen-activated protein kinases
- GSK-3β:
-
Glycogen synthase kinase 3β
- SKIP:
-
Sphingosine kinase interacting protein
- FRET:
-
Fluorescence resonance energy transfer
- TCA:
-
Tricarboxylic acid
- BiFC:
-
Bimolecular fluorescence complementation
- 3-MST:
-
3-Mercaptopyruvate sulfurtransferase
- CBS:
-
cystathionine β-synthase
- CSE:
-
Cystathionine g-lyase
- H2S:
-
Hydrogen sulfide
- COXIV-1:
-
Cytochrome c oxidase subunit IV isoform 1
- COX:
-
Cytochrome c oxidase
- IF1:
-
ATPase inhibitory factor 1
- ROS:
-
Reactive oxygen species
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma co-activator 1α
- NRF:
-
Nuclear respiratory factor
- mtTFA:
-
Mitochondrial transcription factor A
- ABC:
-
ATP-binding cassette
- TPP+ :
-
Triphenylphosphonium cation
References
Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407. doi:10.1146/annurev.genet.39.110304.095751
Pallen MJ (2011) Time to recognise that mitochondria are bacteria? Trends Microbiol 19(2):58–64. doi:10.1016/j.tim.2010.11.001
Vafai SB, Mootha VK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491(7424):374–383. doi:10.1038/nature11707
Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138(4):628–644. doi:10.1016/j.cell.2009.08.005
Schmidt O, Pfanner N, Meisinger C (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11(9):655–667. doi:10.1038/nrm2959
Ventura-Clapier R, Garnier A, Veksler V (2008) Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79(2):208–217. doi:10.1093/cvr/cvn098
Vega RB, Horton JL, Kelly DP (2015) Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circ Res 116(11):1820–1834. doi:10.1161/CIRCRESAHA.116.305420
Cerveny KL, Tamura Y, Zhang Z, Jensen RE, Sesaki H (2007) Regulation of mitochondrial fusion and division. Trends Cell Biol 17(11):563–569. doi:10.1016/j.tcb.2007.08.006
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11(12):872–884. doi:10.1038/nrm3013
Cooper DM (2003) Regulation and organization of adenylyl cyclases and cAMP. Biochem J 375(Pt 3):517–529. doi:10.1042/BJ20031061
Tresguerres M, Levin LR, Buck J (2011) Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int 79(12):1277–1288. doi:10.1038/ki.2011.95
Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J (2000) Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 289(5479):625–628
Jaiswal BS, Conti M (2003) Calcium regulation of the soluble adenylyl cyclase expressed in mammalian spermatozoa. Proc Natl Acad Sci USA 100(19):10676–10681. doi:10.1073/pnas.1831008100
Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR (2003) Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem 278(18):15922–15926. doi:10.1074/jbc.M212475200
Zippin JH, Chen Y, Straub SG, Hess KC, Diaz A, Lee D, Tso P, Holz GG, Sharp GW, Levin LR, Buck J (2013) CO2/HCO3(−)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J Biol Chem 288(46):33283–33291. doi:10.1074/jbc.M113.510073
Wong W, Scott JD (2004) AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5(12):959–970. doi:10.1038/nrm1527
Walsh DA, Van Patten SM (1994) Multiple pathway signal transduction by the cAMP-dependent protein kinase. FASEB J 8(15):1227–1236
Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68:821–861. doi:10.1146/annurev.biochem.68.1.821
Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB (2010) Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J Biol Chem 285(1):142–152. doi:10.1074/jbc.M109.030130
Wu Z, Huang X, Feng Y, Handschin C, Feng Y, Gullicksen PS, Bare O, Labow M, Spiegelman B, Stevenson SC (2006) Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci USA 103(39):14379–14384. doi:10.1073/pnas.0606714103
Anderson MP, Berger HA, Rich DP, Gregory RJ, Smith AE, Welsh MJ (1991) Nucleoside triphosphates are required to open the CFTR chloride channel. Cell 67(4):775–784
Catterall WA (2015) Regulation of cardiac calcium channels in the fight-or-flight response. Curr Mol Pharmacol 8(1):12–21
Stival C, La Spina FA, Baro Graf C, Arcelay E, Arranz SE, Ferreira JJ, Le Grand S, Dzikunu VA, Santi CM, Visconti PE, Buffone MG, Krapf D (2015) Src kinase is the connecting player between protein kinase A (PKA) activation and hyperpolarization through SLO3 potassium channel regulation in mouse sperm. J Biol Chem 290(30):18855–18864. doi:10.1074/jbc.M115.640326
Scherer J, Yi J, Vallee RB (2014) PKA-dependent dynein switching from lysosomes to adenovirus: a novel form of host-virus competition. J Cell Biol 205(2):163–177. doi:10.1083/jcb.201307116
Kashina AS, Semenova IV, Ivanov PA, Potekhina ES, Zaliapin I, Rodionov VI (2004) Protein kinase A, which regulates intracellular transport, forms complexes with molecular motors on organelles. Curr Biol 14(20):1877–1881. doi:10.1016/j.cub.2004.10.003
Tripodi F, Nicastro R, Reghellin V, Coccetti P (2015) Post-translational modifications on yeast carbon metabolism: regulatory mechanisms beyond transcriptional control. Biochim Biophys Acta 1850(4):620–627. doi:10.1016/j.bbagen.2014.12.010
Liu M, Simon MI (1996) Regulation by cAMP-dependent protein kinease of a G-protein-mediated phospholipase C. Nature 382(6586):83–87. doi:10.1038/382083a0
Yoon YM, Oh CD, Kang SS, Chun JS (2000) Protein kinase A regulates chondrogenesis of mesenchymal cells at the post-precartilage condensation stage via protein kinase C-alpha signaling. J Bone Miner Res 15(11):2197–2205. doi:10.1359/jbmr.2000.15.11.2197
Ciullo I, Diez-Roux G, Di Domenico M, Migliaccio A, Avvedimento EV (2001) cAMP signaling selectively influences Ras effectors pathways. Oncogene 20(10):1186–1192. doi:10.1038/sj.onc.1204219
Cosentino C, Di Domenico M, Porcellini A, Cuozzo C, De Gregorio G, Santillo MR, Agnese S, Di Stasio R, Feliciello A, Migliaccio A, Avvedimento EV (2007) p85 regulatory subunit of PI3K mediates cAMP-PKA and estrogens biological effects on growth and survival. Oncogene 26(14):2095–2103. doi:10.1038/sj.onc.1210027
Wagner LE 2nd, Li WH, Yule DI (2003) Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2− splice variants. J Biol Chem 278(46):45811–45817. doi:10.1074/jbc.M306270200
Ahuja M, Jha A, Maleth J, Park S, Muallem S (2014) cAMP and Ca(2)(+) signaling in secretory epithelia: crosstalk and synergism. Cell Calcium 55(6):385–393. doi:10.1016/j.ceca.2014.01.006
Bjorgo E, Solheim SA, Abrahamsen H, Baillie GS, Brown KM, Berge T, Okkenhaug K, Houslay MD, Tasken K (2010) Cross talk between phosphatidylinositol 3-kinase and cyclic AMP (cAMP)-protein kinase a signaling pathways at the level of a protein kinase B/beta-arrestin/cAMP phosphodiesterase 4 complex. Mol Cell Biol 30(7):1660–1672. doi:10.1128/MCB.00696-09
Conti M, Beavo J (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76:481–511. doi:10.1146/annurev.biochem.76.060305.150444
Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discovery 13(4):290–314. doi:10.1038/nrd4228
Bos JL (2006) Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci 31(12):680–686. doi:10.1016/j.tibs.2006.10.002
Kaupp UB, Seifert R (2002) Cyclic nucleotide-gated ion channels. Physiol Rev 82(3):769–824. doi:10.1152/physrev.00008.2002
Simrick S, Schindler RF, Poon KL, Brand T (2013) Popeye domain-containing proteins and stress-mediated modulation of cardiac pacemaking. Trends Cardiovasc Med 23(7):257–263. doi:10.1016/j.tcm.2013.02.002
Froese A, Breher SS, Waldeyer C, Schindler RF, Nikolaev VO, Rinne S, Wischmeyer E, Schlueter J, Becher J, Simrick S, Vauti F, Kuhtz J, Meister P, Kreissl S, Torlopp A, Liebig SK, Laakmann S, Muller TD, Neumann J, Stieber J, Ludwig A, Maier SK, Decher N, Arnold HH, Kirchhof P, Fabritz L, Brand T (2012) Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J Clin Investig 122(3):1119–1130. doi:10.1172/JCI59410
Scott JD, Dessauer CW, Tasken K (2013) Creating order from chaos: cellular regulation by kinase anchoring. Annu Rev Pharmacol Toxicol 53:187–210. doi:10.1146/annurev-pharmtox-011112-140204
Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J (2003) Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J 17(1):82–84. doi:10.1096/fj.02-0598fje
Sim AT, Scott JD (1999) Targeting of PKA, PKC and protein phosphatases to cellular microdomains. Cell Calcium 26(5):209–217. doi:10.1054/ceca.1999.0072
Stangherlin A, Zaccolo M (2012) Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am J Physiol Heart Circ Physiol 302(2):H379–H390. doi:10.1152/ajpheart.00766.2011
Houslay MD (2010) Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci 35(2):91–100. doi:10.1016/j.tibs.2009.09.007
Logan DC (2006) The mitochondrial compartment. J Exp Bot 57(6):1225–1243. doi:10.1093/jxb/erj151
Zalman LS, Nikaido H, Kagawa Y (1980) Mitochondrial outer membrane contains a protein producing nonspecific diffusion channels. J Biol Chem 255(5):1771–1774
Linden M, Andersson G, Gellerfors P, Nelson BD (1984) Subcellular distribution of rat liver porin. Biochim Biophys Acta 770(1):93–96
Ferreira FM, Seica R, Oliveira PJ, Coxito PM, Moreno AJ, Palmeira CM, Santos MS (2003) Diabetes induces metabolic adaptations in rat liver mitochondria: role of coenzyme Q and cardiolipin contents. Biochim Biophys Acta 1639(2):113–120
DiPilato LM, Cheng X, Zhang J (2004) Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci USA 101(47):16513–16518. doi:10.1073/pnas.0405973101
Opalinska M, Meisinger C (2015) Metabolic control via the mitochondrial protein import machinery. Curr Opin Cell Biol 33:42–48. doi:10.1016/j.ceb.2014.11.001
Schmidt O, Harbauer AB, Rao S, Eyrich B, Zahedi RP, Stojanovski D, Schonfisch B, Guiard B, Sickmann A, Pfanner N, Meisinger C (2011) Regulation of mitochondrial protein import by cytosolic kinases. Cell 144(2):227–239. doi:10.1016/j.cell.2010.12.015
Gerbeth C, Schmidt O, Rao S, Harbauer AB, Mikropoulou D, Opalinska M, Guiard B, Pfanner N, Meisinger C (2013) Glucose-induced regulation of protein import receptor Tom22 by cytosolic and mitochondria-bound kinases. Cell Metab 18(4):578–587. doi:10.1016/j.cmet.2013.09.006
Rao S, Schmidt O, Harbauer AB, Schonfisch B, Guiard B, Pfanner N, Meisinger C (2012) Biogenesis of the preprotein translocase of the outer mitochondrial membrane: protein kinase A phosphorylates the precursor of Tom40 and impairs its import. Mol Biol Cell 23(9):1618–1627. doi:10.1091/mbc.E11-11-0933
Gomes LC, Scorrano L (2008) High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta 1777(7–8):860–866. doi:10.1016/j.bbabio.2008.05.442
Ashrafi G, Schwarz TL (2012) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. doi:10.1038/cdd.2012.81
Sugiura A, McLelland GL, Fon EA, McBride HM (2014) A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33(19):2142–2156. doi:10.15252/embj.201488104
Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13(5):589–598. doi:10.1038/ncb2220
Hoppins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76:751–780. doi:10.1146/annurev.biochem.76.071905.090048
Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8(10):939–944. doi:10.1038/sj.embor.7401062
Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282(30):21583–21587. doi:10.1074/jbc.C700083200
Merrill RA, Dagda RK, Dickey AS, Cribbs JT, Green SH, Usachev YM, Strack S (2011) Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol 9(4):e1000612. doi:10.1371/journal.pbio.1000612
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105(41):15803–15808. doi:10.1073/pnas.0808249105
Zhou W, Chen KH, Cao W, Zeng J, Liao H, Zhao L, Guo X (2010) Mutation of the protein kinase A phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis 211(1):216–223. doi:10.1016/j.atherosclerosis.2010.02.012
Rogne M, Tasken K (2014) Compartmentalization of cAMP signaling in adipogenesis, lipogenesis, and lipolysis. Horm Metab Res 46(12):833–840. doi:10.1055/s-0034-1389955
Pidoux G, Witczak O, Jarnaess E, Myrvold L, Urlaub H, Stokka AJ, Kuntziger T, Tasken K (2011) Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J 30(21):4371–4386. doi:10.1038/emboj.2011.365
Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ (1999) Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell 3(4):413–422
Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP, Korsmeyer SJ (2003) BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424(6951):952–956. doi:10.1038/nature01825
Kumar S, Kostin S, Flacke JP, Reusch HP, Ladilov Y (2009) Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells. J Biol Chem 284(22):14760–14768. doi:10.1074/jbc.M900925200
Appukuttan A, Kasseckert SA, Micoogullari M, Flacke JP, Kumar S, Woste A, Abdallah Y, Pott L, Reusch HP, Ladilov Y (2012) Type 10 adenylyl cyclase mediates mitochondrial Bax translocation and apoptosis of adult rat cardiomyocytes under simulated ischaemia/reperfusion. Cardiovasc Res 93(2):340–349. doi:10.1093/cvr/cvr306
Yang J, Li JH, Wang J, Zhang CY (2010) Molecular modeling of BAD complex resided in a mitochondrion integrating glycolysis and apoptosis. J Theor Biol 266(2):231–241. doi:10.1016/j.jtbi.2010.06.009
Bui M, Gilady SY, Fitzsimmons RE, Benson MD, Lynes EM, Gesson K, Alto NM, Strack S, Scott JD, Simmen T (2010) Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J Biol Chem 285(41):31590–31602. doi:10.1074/jbc.M110.101584
Moujalled D, Weston R, Anderton H, Ninnis R, Goel P, Coley A, Huang DC, Wu L, Strasser A, Puthalakath H (2011) Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep 12(1):77–83. doi:10.1038/embor.2010.190
Dohi T, Xia F, Altieri DC (2007) Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol Cell 27(1):17–28. doi:10.1016/j.molcel.2007.06.004
Strasser A (2005) The role of BH3-only proteins in the immune system. Nat Rev Immunol 5(3):189–200. doi:10.1038/nri1568
Fuchs Y, Steller H (2015) Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 16(6):329–344. doi:10.1038/nrm3999
Skroblin P, Grossmann S, Schafer G, Rosenthal W, Klussmann E (2010) Mechanisms of protein kinase A anchoring. Int Rev Cell Mol Biol 283:235–330. doi:10.1016/S1937-6448(10)83005-9
Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS (2012) AKAPs: the architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol 52(2):351–358. doi:10.1016/j.yjmcc.2011.05.002
Mason TA, Goldenring JR, Kolobova E (2014) AKAP350C targets to mitochondria via a novel amphipathic alpha helical domain. Cell Logist 4(3):e943597. doi:10.4161/21592780.2014.943597
Alto NM, Soderling J, Scott JD (2002) Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J Cell Biol 158(4):659–668. doi:10.1083/jcb.200204081
Lin RY, Moss SB, Rubin CS (1995) Characterization of S-AKAP84, a novel developmentally regulated A kinase anchor protein of male germ cells. J Biol Chem 270(46):27804–27811
Sardanelli AM, Signorile A, Nuzzi R, Rasmo DD, Technikova-Dobrova Z, Drahota Z, Occhiello A, Pica A, Papa S (2006) Occurrence of A-kinase anchor protein and associated cAMP-dependent protein kinase in the inner compartment of mammalian mitochondria. FEBS Lett 580(24):5690–5696. doi:10.1016/j.febslet.2006.09.020
Feliciello A, Rubin CS, Avvedimento EV, Gottesman ME (1998) Expression of a kinase anchor protein 121 is regulated by hormones in thyroid and testicular germ cells. J Biol Chem 273(36):23361–23366
Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS (1997) Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem 272(12):8057–8064
Feliciello A, Gottesman ME, Avvedimento EV (2005) cAMP-PKA signaling to the mitochondria: protein scaffolds, mRNA and phosphatases. Cell Signal 17(3):279–287. doi:10.1016/j.cellsig.2004.09.009
Ma MP, Thomson M (2012) Protein kinase A subunit alpha catalytic and A kinase anchoring protein 79 in human placental mitochondria. Open Biochem J 6:23–30. doi:10.2174/1874091X01206010023
Means CK, Lygren B, Langeberg LK, Jain A, Dixon RE, Vega AL, Gold MG, Petrosyan S, Taylor SS, Murphy AN, Ha T, Santana LF, Tasken K, Scott JD (2011) An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc Natl Acad Sci USA 108(48):E1227–E1235. doi:10.1073/pnas.1107182108
Jun YW, Park H, Lee YK, Kaang BK, Lee JA, Jang DJ (2016) D-AKAP1a is a signal-anchored protein in the mitochondrial outer membrane. FEBS Lett. doi:10.1002/1873-3468.12123
Zhang Y, Chen Y, Gucek M, Xu H (2016) The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J. doi:10.15252/embj.201592994
Carlucci A, Lignitto L, Feliciello A (2008) Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol 18(12):604–613. doi:10.1016/j.tcb.2008.09.006
Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA (2011) Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 44(4):532–544. doi:10.1016/j.molcel.2011.08.045
Dickey AS, Strack S (2011) PKA/AKAP1 and PP2A/Bbeta2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci 31(44):15716–15726. doi:10.1523/JNEUROSCI.3159-11.2011
Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, Van Houten B, Cherra SJ 3rd, Chu CT (2011) Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ 18(12):1914–1923. doi:10.1038/cdd.2011.74
Pesavento C, Hengge R (2009) Bacterial nucleotide-based second messengers. Curr Opin Microbiol 12(2):170–176. doi:10.1016/j.mib.2009.01.007
Kulinskii VI, Zobova NV (1985) Submitochondrial distribution of cAMP during incubation with rat liver mitochondria. Biokhimiia 50(9):1546–1552
Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G (2009) Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 9(3):265–276. doi:10.1016/j.cmet.2009.01.012
Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T (2013) Mitochondrial Ca(2)(+) uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab 17(6):965–975. doi:10.1016/j.cmet.2013.05.003
Lefkimmiatis K, Leronni D, Hofer AM (2013) The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J Cell Biol 202(3):453–462. doi:10.1083/jcb.201303159
Acin-Perez R, Salazar E, Brosel S, Yang H, Schon EA, Manfredi G (2009) Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol Med 1(8–9):392–406. doi:10.1002/emmm.200900046
Acin-Perez R, Gatti DL, Bai Y, Manfredi G (2011) Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: coupled mechanisms of energy metabolism regulation. Cell Metab 13(6):712–719. doi:10.1016/j.cmet.2011.03.024
Acin-Perez R, Russwurm M, Gunnewig K, Gertz M, Zoidl G, Ramos L, Buck J, Levin LR, Rassow J, Manfredi G, Steegborn C (2011) A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J Biol Chem 286(35):30423–30432. doi:10.1074/jbc.M111.266379
Hess KC, Liu J, Manfredi G, Muhlschlegel FA, Buck J, Levin LR, Barrientos A (2014) A mitochondrial CO2-adenylyl cyclase-cAMP signalosome controls yeast normoxic cytochrome c oxidase activity. FASEB J 28(10):4369–4380. doi:10.1096/fj.14-252890
Roelofs J, Van Haastert PJ (2002) Deducing the origin of soluble adenylyl cyclase, a gene lost in multiple lineages. Mol Biol Evol 19(12):2239–2246
Zhang F, Qi Y, Zhou K, Zhang G, Linask K, Xu H (2015) The cAMP phosphodiesterase Prune localizes to the mitochondrial matrix and promotes mtDNA replication by stabilizing TFAM. EMBO Rep 16(4):520–527. doi:10.15252/embr.201439636
Schwoch G, Trinczek B, Bode C (1990) Localization of catalytic and regulatory subunits of cyclic AMP-dependent protein kinases in mitochondria from various rat tissues. Biochem J 270(1):181–188
Papa S, Sardanelli AM, Scacco S, Technikova-Dobrova Z (1999) cAMP-dependent protein kinase and phosphoproteins in mammalian mitochondria. An extension of the cAMP-mediated intracellular signal transduction. FEBS Lett 444(2–3):245–249
Sardanelli AM, Technikova-Dobrova Z, Speranza F, Mazzocca A, Scacco S, Papa S (1996) Topology of the mitochondrial cAMP-dependent protein kinase and its substrates. FEBS Lett 396(2–3):276–278
Chen R, Fearnley IM, Peak-Chew SY, Walker JE (2004) The phosphorylation of subunits of complex I from bovine heart mitochondria. J Biol Chem 279(25):26036–26045. doi:10.1074/jbc.M402710200
Zhao X, Leon IR, Bak S, Mogensen M, Wrzesinski K, Hojlund K, Jensen ON (2011) Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol Cell Proteomics 10(1):M110.000299. doi:10.1074/mcp.M110.000299
Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, Bailey DJ, Jochem A, Stapleton DS, Keller MP, Westphall MS, Yandell BS, Attie AD, Coon JJ, Pagliarini DJ (2012) A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab 16(5):672–683. doi:10.1016/j.cmet.2012.10.004
Agnes RS, Jernigan F, Shell JR, Sharma V, Lawrence DS (2010) Suborganelle sensing of mitochondrial cAMP-dependent protein kinase activity. J Am Chem Soc 132(17):6075–6080. doi:10.1021/ja909652q
Kerppola TK (2006) Visualization of molecular interactions by fluorescence complementation. Nat Rev Mol Cell Biol 7(6):449–456. doi:10.1038/nrm1929
Modis K, Panopoulos P, Coletta C, Papapetropoulos A, Szabo C (2013) Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem Pharmacol 86(9):1311–1319. doi:10.1016/j.bcp.2013.08.064
Papa S, Sardanelli AM, Cocco T, Speranza F, Scacco SC, Technikova-Dobrova Z (1996) The nuclear-encoded 18 kDa (IP) AQDQ subunit of bovine heart complex I is phosphorylated by the mitochondrial cAMP-dependent protein kinase. FEBS Lett 379(3):299–301
Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG (2006) Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem 281(4):2061–2070. doi:10.1074/jbc.M507741200
Fang JK, Prabu SK, Sepuri NB, Raza H, Anandatheerthavarada HK, Galati D, Spear J, Avadhani NG (2007) Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett 581(7):1302–1310. doi:10.1016/j.febslet.2007.02.042
De Rasmo D, Palmisano G, Scacco S, Technikova-Dobrova Z, Panelli D, Cocco T, Sardanelli AM, Gnoni A, Micelli L, Trani A, Di Luccia A, Papa S (2010) Phosphorylation pattern of the NDUFS4 subunit of complex I of the mammalian respiratory chain. Mitochondrion 10(5):464–471. doi:10.1016/j.mito.2010.04.005
Scacco S, Vergari R, Scarpulla RC, Technikova-Dobrova Z, Sardanelli A, Lambo R, Lorusso V, Papa S (2000) cAMP-dependent phosphorylation of the nuclear encoded 18-kDa (IP) subunit of respiratory complex I and activation of the complex in serum-starved mouse fibroblast cultures. J Biol Chem 275(23):17578–17582. doi:10.1074/jbc.M001174200
Papa S, Scacco S, Sardanelli AM, Vergari R, Papa F, Budde S, van den Heuvel L, Smeitink J (2001) Mutation in the NDUFS4 gene of complex I abolishes cAMP-dependent activation of the complex in a child with fatal neurological syndrome. FEBS Lett 489(2–3):259–262
Technikova-Dobrova Z, Sardanelli AM, Speranza F, Scacco S, Signorile A, Lorusso V, Papa S (2001) Cyclic adenosine monophosphate-dependent phosphorylation of mammalian mitochondrial proteins: enzyme and substrate characterization and functional role. Biochemistry 40(46):13941–13947
De Rasmo D, Signorile A, Larizza M, Pacelli C, Cocco T, Papa S (2012) Activation of the cAMP cascade in human fibroblast cultures rescues the activity of oxidatively damaged complex I. Free Radic Biol Med 52(4):757–764. doi:10.1016/j.freeradbiomed.2011.11.030
Papa S, De Rasmo D (2013) Complex I deficiencies in neurological disorders. Trends Mol Med 19(1):61–69. doi:10.1016/j.molmed.2012.11.005
De Rasmo D, Signorile A, Santeramo A, Larizza M, Lattanzio P, Capitanio G, Papa S (2015) Intramitochondrial adenylyl cyclase controls the turnover of nuclear-encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim Biophys Acta 1853(1):183–191. doi:10.1016/j.bbamcr.2014.10.016
Garcia-Bermudez J, Sanchez-Arago M, Soldevilla B, Del Arco A, Nuevo-Tapioles C, Cuezva JM (2015) PKA phosphorylates the ATPase inhibitory factor 1 and inactivates its capacity to bind and inhibit the mitochondrial H(+)-ATP synthase. Cell Rep 12(12):2143–2155. doi:10.1016/j.celrep.2015.08.052
Garcia-Bermudez J, Cuezva JM (2016) The ATPase Inhibitory Factor 1 (IF1): a master regulator of energy metabolism and of cell survival. Biochim Biophys Acta. doi:10.1016/j.bbabio.2016.02.004
De Rasmo D, Micelli L, Santeramo A, Signorile A, Lattanzio P (1857) Papa S (2016) cAMP regulates the functional activity, coupling efficiency and structural organization of mammalian FOF1 ATP synthase. Biochim Biophys Acta 4:350–358. doi:10.1016/j.bbabio.2016.01.006
Walker JE (2013) The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans 41(1):1–16. doi:10.1042/BST20110773
Campanella M, Parker N, Tan CH, Hall AM, Duchen MR (2009) IF(1): setting the pace of the F(1)F(o)-ATP synthase. Trends Biochem Sci 34(7):343–350. doi:10.1016/j.tibs.2009.03.006
Srinivasan S, Spear J, Chandran K, Joseph J, Kalyanaraman B, Avadhani NG (2013) Oxidative stress induced mitochondrial protein kinase A mediates cytochrome c oxidase dysfunction. PLoS One 8(10):e77129. doi:10.1371/journal.pone.0077129
Rosca M, Minkler P (1807) Hoppel CL (2011) Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim Biophys Acta 11:1373–1382. doi:10.1016/j.bbabio.2011.02.003
Scarpulla RC, Vega RB, Kelly DP (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. doi:10.1016/j.tem.2012.06.006
Cammarota M, Paratcha G, Bevilaqua LR, Levi de Stein M, Lopez M, Pellegrino de Iraldi A, Izquierdo I, Medina JH (1999) Cyclic AMP-responsive element binding protein in brain mitochondria. J Neurochem 72(6):2272–2277
Lee J, Kim CH, Simon DK, Aminova LR, Andreyev AY, Kushnareva YE, Murphy AN, Lonze BE, Kim KS, Ginty DD, Ferrante RJ, Ryu H, Ratan RR (2005) Mitochondrial cyclic AMP response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J Biol Chem 280(49):40398–40401. doi:10.1074/jbc.C500140200
Ryu H, Lee J, Impey S, Ratan RR, Ferrante RJ (2005) Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc Natl Acad Sci USA 102(39):13915–13920. doi:10.1073/pnas.0502878102
De Rasmo D, Signorile A, Roca E, Papa S (2009) cAMP response element-binding protein (CREB) is imported into mitochondria and promotes protein synthesis. FEBS J 276(16):4325–4333. doi:10.1111/j.1742-4658.2009.07133.x
Liu D, Zhang Y, Gharavi R, Park HR, Lee J, Siddiqui S, Telljohann R, Nassar MR, Cutler RG, Becker KG, Mattson MP (2015) The mitochondrial uncoupler DNP triggers brain cell mTOR signaling network reprogramming and CREB pathway up-regulation. J Neurochem. doi:10.1111/jnc.13176
Lu B, Lee J, Nie X, Li M, Morozov YI, Venkatesh S, Bogenhagen DF, Temiakov D, Suzuki CK (2013) Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA + Lon protease. Mol Cell 49(1):121–132. doi:10.1016/j.molcel.2012.10.023
Cheepala S, Hulot JS, Morgan JA, Sassi Y, Zhang W, Naren AP, Schuetz JD (2013) Cyclic nucleotide compartmentalization: contributions of phosphodiesterases and ATP-binding cassette transporters. Annu Rev Pharmacol Toxicol 53:231–253. doi:10.1146/annurev-pharmtox-010611-134609
Godinho RO, Duarte T, Pacini ES (2015) New perspectives in signaling mediated by receptors coupled to stimulatory G protein: the emerging significance of cAMP e ffl ux and extracellular cAMP-adenosine pathway. Front Pharmacol 6:58. doi:10.3389/fphar.2015.00058
Miranda ER, Nam EA, Kuspa A, Shaulsky G (2015) The ABC transporter, AbcB3, mediates cAMP export in D. discoideum development. Dev Biol 397(2):203–211. doi:10.1016/j.ydbio.2014.11.006
Liesa M, Qiu W, Shirihai OS (2012) Mitochondrial ABC transporters function: the role of ABCB10 (ABC-me) as a novel player in cellular handling of reactive oxygen species. Biochim Biophys Acta 1823(10):1945–1957. doi:10.1016/j.bbamcr.2012.07.013
Herget M, Tampe R (2007) Intracellular peptide transporters in human–compartmentalization of the “peptidome”. Pflugers Arch 453(5):591–600. doi:10.1007/s00424-006-0083-4
Bruce JI, Straub SV, Yule DI (2003) Crosstalk between cAMP and Ca2+ signaling in non-excitable cells. Cell Calcium 34(6):431–444
Kunzelmann K, Mehta A (2013) CFTR: a hub for kinases and crosstalk of cAMP and Ca2+. FEBS J 280(18):4417–4429. doi:10.1111/febs.12457
Katona D, Rajki A, Di Benedetto G, Pozzan T, Spat A (2015) Calcium-dependent mitochondrial cAMP production enhances aldosterone secretion. Mol Cell Endocrinol. doi:10.1016/j.mce.2015.05.002
Perrino C, Feliciello A, Schiattarella GG, Esposito G, Guerriero R, Zaccaro L, Del Gatto A, Saviano M, Garbi C, Carangi R, Di Lorenzo E, Donato G, Indolfi C, Avvedimento VE, Chiariello M (2010) AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc Res 88(1):101–110. doi:10.1093/cvr/cvq155
Dagda RK, Das Banerjee T (2015) Role of protein kinase A in regulating mitochondrial function and neuronal development: implications to neurodegenerative diseases. Rev Neurosci 26(3):359–370. doi:10.1515/revneuro-2014-0085
Bender AT, Beavo JA (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58(3):488–520. doi:10.1124/pr.58.3.5
Lochner A, Moolman JA (2006) The many faces of H89: a review. Cardiovasc Drug Rev 24(3–4):261–274. doi:10.1111/j.1527-3466.2006.00261.x
Gruber J, Fong S, Chen CB, Yoong S, Pastorin G, Schaffer S, Cheah I, Halliwell B (2013) Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnol Adv 31(5):563–592. doi:10.1016/j.biotechadv.2012.09.005
Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279(33):34682–34690. doi:10.1074/jbc.M402999200
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, F., Zhang, L., Qi, Y. et al. Mitochondrial cAMP signaling. Cell. Mol. Life Sci. 73, 4577–4590 (2016). https://doi.org/10.1007/s00018-016-2282-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2282-2