
Abstract
Intercellular communication plays an important role in cancer initiation and progression through secretory molecules, including growth factors and cytokines. Recent advances have revealed that small membrane vesicles, termed extracellular vesicles (EVs), served as a regulatory agent in the intercellular communication of cancer. EVs enable the transfer of functional molecules, including proteins, mRNA and microRNAs (miRNAs), into recipient cells. Cancer cells utilize EVs to dictate the unique phenotype of surrounding cells, thereby promoting cancer progression. Against such “education” by cancer cells, non-tumoral cells suppress cancer initiation and progression via EVs. Therefore, researchers consider EVs to be important cues to clarify the molecular mechanisms of cancer biology. Understanding the functions of EVs in cancer progression is an important aspect of cancer biology that has not been previously elucidated. In this review, we summarize experimental data that indicate the pivotal roles of EVs in cancer progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The malignant phenotypes of tumors not only are determined by cancer cells themselves but also depend on their surrounding tumor microenvironments [1, 2]. These microenvironments consist of various cell types, such as fibroblasts, lymphocyte, inflammatory cells, epithelial cells, endothelial cells, and mesenchymal stem cells. These cells within the tumor microenvironment and cancer cells interact with each other and form the intrinsic communication networks that affect several cancer hallmarks, as described by Hanahan and Weinberg [3]. Several reports documented that such intercellular communications were modulated by various humoral factors, such as growth factors, cytokines, and chemokines. Similar to these molecules, recent advances in cancer biology revealed that extracellular vesicles (EVs) also served as a regulatory agent in such communications.
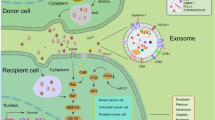
EVs have a heterogenetic population and are generally categorized as exosome, microvesicles or ectosomes, and apoptotic bodies [4–6]. These vesicles originate from different subcellular compartments [4–6]. Exosomes are small membrane vesicles, ranging from 50 to 150 nm in diameter, that have a lipid bilayer membrane and originate from the exocytosis of multivesicular bodies (MVBs) containing intraluminal vesicles [6]. Exosome biogenesis and release are modulated by the endosomal sorting complex that is required for transport (ESCRT) machinery and the ceramide-dependent pathway [6]. Researchers in EV biology have identified several types of exosome markers, including tetraspanins (CD9, CD63, CD81), heat shock proteins (HSP60, 70, and 90), membrane transporters and fusion proteins (Annexins and flotillin), and MVB synthesis proteins (Alix and TSG101) [7]. Microvesicles are 100–1000 nm in diameter and are produced directly from the plasma membrane via budding [8]. Microvesicles are enriched in some lipid components and phosphatidylserine [9]. The biogenesis of microvesicles is modulated by the interaction between phospholipid redistribution and the contraction of cytoskeletal structures [10]. Apoptotic bodies (500–4000 nm in diameter) are formed during the apoptotic process and contain organelles and nuclear fragments [6, 10, 11]. Apoptotic bodies also contain DNA fragments and RNA. Macrophages subsequently clear apoptotic bodies by phagocytosis [11]. However, these apoptotic bodies may participate in the intercellular communication of the cancer microenvironment. Indeed, H-rasV12- and human c-myc-transfected to rat fibroblasts could transfer their DNA to other fibroblasts by apoptotic bodies, thereby inducing tumorigenic phenotypes [12].
EVs contain functional cellular components such as proteins, mRNAs, and microRNAs (miRNAs) that enable the transfer of these principal factors to various cell types [13]. These components of EVs are also functional in the recipient cells and are highly variable depending on the origin cells [6]. As shown in Figs. 1 and 2, this EV-mediated interaction between cancer cells and their surrounding cells within tumor microenvironment confers advantages for cancer initiation and progression. Non-tumoral cells also utilize EVs to transfer the tumor-suppressive molecules that affect cancer initiation and progression (Fig. 2). Therefore, researchers consider EVs to be important cues for understanding the molecular mechanisms underlying the intercellular communication in the tumor microenvironment. In this review, we will summarize the current knowledge regarding the functional role of EV components on intercellular communication between cancer cells and each cell type within the tumor microenvironment.

Cancer cell-derived EVs modify the characters of cancer surrounding microenvironment. Several kinds of cell types, such as cancer cells, fibroblasts, immune cells, endothelial cells, epithelial cells, and mesenchymal stem cells, comprise unique microenvironment for cancer progression. Cancer cells utilize EVs to modify surrounding cells within tumor microenvironment. Cancer-derived EVs have multiple functions that depend on component molecules of EVs. To induce cancer-associated fibroblast (CaF)-like phenotypes in cancer surrounding fibroblasts and mesenchymal stem cells, cancer cells secrete EVs and transfer growth factors and microRNAs (miRNAs), including transforming growth factor-beta (TGF-β) and miR-155, respectively. To escape from immune surveillance, cancer cells transfer several types of immunoregulatory molecules into immune cells. However, these cancer-derived EVs also stimulate cancer immunity to kill tumor cells because tumor antigens were packaged in EVs and stimulated cancer immunity. Cancer-derived EVs also contain angiogenic proteins and miRNAs that promote migration and proangiogenic activity of endothelial cells. In addition, miR-105 and miR-181c in EVs are capable of rupturing the vascular system to increase the permeability that supports cancer metastasis. Cancer-derived EVs confer malignant phenotypes in other cancer cells and epithelial cells by transferring oncogenic proteins and miRNAs, such as EGFRvIII, miR-200, and tissue transglutaminase (tTG). Taken together, cancer cells “dictate” the characters of their surrounding stromal cells and create a convenient microenvironment to support cancer progression via EVs

The functional role of non-tumoral cell-derived EVs in cancer initiation and progression. Non-tumoral cells utilize EVs to affect cancer initiation and progression. Cancer-associated fibroblasts secrete EVs and affect invasion, proliferation, chemoresistance, and metabolic properties of cancer cells by transferring CD81, miR-21, miR-409, and metabolites. Macrophage-derived EVs contain miR-223, which stimulates the invasive activity of cancer cells. Monocytes transfer miR-155 to neuroblastoma via EVs and induce chemoresistance in neuroblastoma. “Non-educated” mesenchymal stem cells (MSCs) by cancer cells secrete EVs containing miR-16 to suppress tumor growth and angiogenesis. However, interestingly, “educated” MSCs by cancer cells enable to secrete EVs that contain tumor promotive proteins such as plakoglobin and CCL2. MSC-derived EVs also transfer miR-23b, which induce dormant state of cancer cells to survive in a quiescent state while waiting for the appropriate environmental conditions to begin proliferation again. Non-aberrant epithelial cells secreted EVs to transfer miR-143 into cancer cells and suppress tumor growth
Interaction between cancer cells and surrounding stromal fibroblasts via EVs
The fibroblasts within tumor stroma, which are also termed cancer-associated fibroblasts (CaFs), have heterogeneous populations and include myofibroblasts that are similar to fibroblasts associated with wound healing [14]. CaFs enable the formation of a unique microenvironment that plays a pivotal role in cancer development and progression. Although the origins of CaFs and the signaling that mediates CaF induction remain controversial, several types of factors, including transforming growth factor-beta (TGF-β), are required for the induction and maintenance of CaFs [14–18]. In addition to these factors, the EVs derived from cancer cells induce such CaF-like phenotypes in cancer surrounding stromal cells (Table 1). Webber et al. showed that TGF-β was loaded on the surface of EVs derived from prostate cancer and mesothelioma cell lines [19, 20]. These cancer-derived EVs could trigger the TGF-β/SMAD3 signaling pathway in fibroblasts and induce the myofibroblast-like phenotypes, including the induction of α-smooth muscle action (α-SMA) expression and the production of fibroblasts growth factor 2 (FGF2) [19]. These data suggest that cancer cells could dictate the characters of their surrounding stromal cells via EVs and create a convenient microenvironment to support cancer cell survival and progression.
In contrast, stromal fibroblasts that are “educated” by cancer cells also secreted EVs and established the communication networks that provide the ultimate benefit for cancer progression. Luga et al. demonstrated that CD81-positive CaF-derived EVs enhanced breast cancer cell motility and metastasis by activating the Wnt-planar cell polarity (PCP) signaling pathway [21]. Endocytosis and internalization of the Wnt-receptor complex from the plasma membrane are required for signal transduction [22]. Wnt11 and CD81 are localized on EVs, and the internalization of CD81 molecules on the EVs in breast cancer cell may support the endocytic trafficking of autocrine Wnt ligand to activate the Wnt-PCP pathway [21]. Because CD81 is a well-known marker of EVs [7], it is possible that CD81-positive EVs derived from other cell types within the tumor microenvironment may contribute to the activation of the Wnt-PCP pathway in cancer cells. Boelens et al. demonstrated that resistance to chemotherapy and radiation in breast cancer is mediated by CaF-derived EVs [23]. These CaF-derived EVs contain non-coding or transposable RNAs that stimulate RIG-I recognition to activate STAT1. Activated STAT1 cooperates with juxtacrine-activated NOTCH3 to mediate NOTCH target gene transcriptions. NOTCH and its target genes support the maintainance of tumor-initiating cells that are known to be resistant to chemotherapy and radiation [24]. Therefore, NOTCH target gene induction via EVs results in the expansion of therapy-resistant tumor-initiating cells [23]. Interestingly, CaF-derived EVs preferentially affect breast cancer cells with the basal-like subtype that is associated with aggressive phenotype of breast cancer [23, 25]. Although the mechanism by which CaF-derived EVs could preferentially affect basal-like subtype breast cancer cells has not been addressed, CaF-derived EVs potentially have a tropism to influence treatment-resistant cancer cells.
Recently, an increasing number of reports have demonstrated that secreted miRNAs also enable to compose the communication networks between cancer cells and CaFs to modulate cancer progression. Pang et al. showed that miR-155 was secreted through cancer-derived EVs and dictated CaF-like phenotypes in fibroblasts by repressing TP53INP1 [26]. This finding suggests that cancer-derived EVs also affect stromal fibroblasts through transferring miRNAs. However, Josson et al. demonstrated that CaF-derived EVs contain miR-409, which promotes the epithelial-to-mesenchymal transition (EMT) [27]. Moreover, Yeung et al. demonstrated that CaFs and cancer-associated adipocytes secreted miR-21 via EVs and conferred chemo-resistance to cancer cells by regulating apoptotic peptidase activating factor 1 (APAF1) expression [28]. Because miRNA expression changes involve regulating the tumor-promoting function of CaFs [29], the miRNA expression profile in CaFs and their EVs reflect the disease status of cancer. Understanding EV communication networks in tumor-stromal interaction will enable the development of therapeutic strategies to target EVs and may provide novel functional marker of CaFs.
Cancer cells can reprogram their energy metabolism to be distinct from that of normal cells [3, 30]. Interestingly, CaFs within tumor stroma also support the metabolic properties of cancer cells [31, 32]. Zhao et al. showed that CaF-derived EVs alter the metabolic properties of prostate cancer cells [33]. In the presence of CaF-derived EVs, oxidative phosphorylation is inhibited, but glycolysis and lactate levels are increased in prostate cancer cell lines. This finding suggests that EVs induce the “Warburg effect” [30, 34]. These CaF-derived EVs also transfer TCA cycle metabolites, amino acids, and lipids into cancer cells. Interestingly, breast cancer-derived EVs also modify the glucose consumption of fibroblasts in metastatic sites by targeting pyruvate kinases [35]. Therefore, cancer-derived EVs enable to educate stromal cells within distant niche to promote cancer metastasis. Similarly, several reports also showed the important aspects of cancer-derived EV on the cancer progression through remodeling the stromal cells within pre-metastatic niche [36, 37]. Although further studies are required to clarify the advantages of metabolic change in cancer progression, these findings suggest that the communication networks of EVs contribute to cancer proliferation and survival in the surrounding environment with limited oxygen and nutrient supplies.
Cancer-derived EVs in the immune system
The major function of the immune system is not only to protect against several infectious pathogens but also to eradicate the abnormal cells [40]. Therefore, to promote cancer initiation and progression, evasion from immune surveillance is one of the key hallmarks of cancer cells [3]. However, increasing evidences indicate that the immune systems play a dual role in cancer progression and can both suppress and support the tumor cells. In addition, several reports suggested that macrophages and neutrophils within tumor tissue were associated with cancer progression [40–44]. These macrophages and neutrophils also exhibit polarity on either a tumor-suppressive or a tumor-promoting phenotype, depending on the surrounding microenvironment [43, 44]. Hence, cancer cells and immune cells form intricate communication networks that affect the risk of cancer development.
EVs also modulate the evasion of cancer cells from immune surveillance (Table 2). For example, cancer-derived EVs induce apoptosis of CD8-positive T-cells and promote regulatory T-cell expansion to suppress anticancer immunity [45, 46]. Additionally, cancer-derived EVs included several types of immunoregulatory molecules, such as FasL [47], TGF-β [48], NKG2D ligands [49], galectin-9 [50], and HSP72 [51], to support the immune escape of cancer cells. These EVs also induce the differentiation of monocytes into immunosuppressive macrophages [52]. Interestingly, cancer-derived EVs increase the survival rate of monocytes [53, 54]. Consequently, cancer-derived EVs facilitate the formation of macrophages from monocytes in the tumor microenvironment. These macrophages are also capable of producing the EVs that enhance the metastatic properties of cancer cells [55]. Moreover, cancer-derived EVs internalize into macrophages and induce pro-inflammatory cytokines, including IL-6, TNF-α, and CCL2, through activating NF-κB signaling [56]. Thus, EV communications between cancer cells and immune cells seem to provide the advantage of evasion from immune surveillance and cancer progression. However, several researchers suggested that tumor antigens were packaged in EVs and stimulated cancer immunity to kill tumor cells [57, 58]. Therefore, cancer-derived EVs are also utilized as anti-tumoral vaccines [59, 60]. In addition, dendritic cells (DC)-derived EVs induce CD8-positive T-cell activation in both a CD4-positive T-cell and B-cell-dependent manner [61]. The availability of these DC-derived EVs as a tool of vaccine immunotherapy was also tested [62–64]. Taken together, EVs within the tumor microenvironment also participate in such complex interactions between cancer cells and immune cells.
Among these communication networks of the EVs involved in cancer immunity, the miRNAs secreted via EVs are also involved in the regulation of cancer immunity (Fig. 1; Table 2). Indeed, pancreatic cancer-derived EVs containing miR-203 regulate the expression of the immune response-relative genes, including toll-like recepter-4, and may contribute to suppress anticancer immunity [65]. In addition, macrophage-derived EVs can transfer miR-223, which stimulates the invasive activity of breast cancer cells, by regulating the Mef2c-β-catenin pathway [55]. Furthermore, miR-21 is transferred into monocytes via neuroblastoma-derived EVs and can bind to TLR8 to stimulate NF-κB pathway and induce miR-155 expression in monocytes [66]. Interestingly, miR-155 is also transferred from monocytes to neuroblastoma via EVs and induces chemoresistance in neuroblastoma through targeting telomeric repeat binding factor 1 (TERF1). Such communication networks of secreted miRNAs via EVs are likely associated with programmed cell death protein ligand-1 (PD-L1) in mediating the immune response. PD-L1 is well known as a ligand of programmed cell death protein-1 (PD-1) that plays a pivotal role in T-cell inhibition and exhaustion. PD-L1 is up-regulated in several cancers to suppress the cytotoxic activity of T-cells and escape from the immune system. Instability of the PD-L1 3′-untranslated region (UTR) was recently reported to be associated with aberrant PD-L1 expression in cancer cells [67]. Importantly, most miRNA targeting sites are located in the 3′-UTR region of target genes [68]. Several reports have demonstrated the functional role of miRNAs in immune checkpoints by regulating PD-L1 expression [69, 70]. Although it is unclear that miRNAs targeting PD-L1 are packaged in EVs, it is conceivable that the genetic status of PD-L1 may affect the functional role of the secreted miRNAs that are included in EVs in the evasion of cancer cells from immune surveillance. Further understanding of the molecular mechanisms underlying these complex communication networks via EVs will provide several avenues for cancer immunotherapy and diagnosis.
The communication network of EVs between cancer cells and endothelial cells
Angiogenesis within tumor tissue is required to provide nutrients and oxygen to cancer cells and allows entry to the blood circulation system [3, 77, 78]. Angiogenesis can trigger the formation of metastatic foci in secondary sites. Tumor vascularization is regulated by a number of potential mechanisms, and vascular endothelial cells are one of the origins of the tumor vessel [79]. Cancer-derived EVs support the vascularization by affecting endothelial cells within the tumor microenvironment and may confer the aggressive phenotypes to cancer cells (Table 3). For instance, prostate and ovarian cancer cells secrete EVs to transfer sphingomyelin and CD147 into endothelial cells. These EV molecules promote migration and proangiogenic activity of endothelial cells [80, 81]. Similarly, nasopharyngeal cancer-derived EVs contained intercellular adhesion molecule-1 (ICAM-1) and CD44 variant isoform 5 (CD44v5), which significantly increase the migration, invasion, and tubulogenesis of endothelial cells [82]. Although these reports did not clarify the contribution of these EV components in cancer progression, cancer-derived EV-induced angiogenesis within the tumor microenvironment probably supports cancer survival and metastasis.
In contrast, cancer cell-derived EVs also transported miRNAs into endothelial cells to modulate neovascularization. Metastatic breast cancer-derived EVs transport miR-210 into endothelial cells [83]. This miRNA secretion is regulated by neutral sphingomyelinase 2 (nSMase2), which is highly expressed in cancer cells compared with non-neoplastic cells [83–85]. Because nSMase2 expression promotes metastatic initiation, miRNA secretion via cancer-derived EVs modulates cancer metastasis through neo- vascularization. The functional role of EVs containing miR-135 and miR-210 in tumor angiogenesis is also observed in multiple myeloma and leukemia, respectively [86, 87]. Interestingly, hypoxic conditions promote the secretion of EVs containing miR-135 and miR-210 in cancer cells [86, 87]. Hypoxic conditions within tumor tissues affect several characteristics of cancer, including angiogenesis [88]. Furthermore, hypoxia promotes EV release and alteration in the EV components [89, 90]. Therefore, EVs are utilized to facilitate the malignant behavior in hypoxic tumor microenvironments. However, the exact contribution of miRNAs and proteins in EVs during angiogenesis and cancer metastasis remains unclear. Understanding these relationships will enable the development of cancer therapy. Indeed, anti-angiogenic strategies to inhibit EVs secretion and miRNA contents were examined [91].
Cancer-derived EVs mediate tumor angiogenesis within proximal microenvironments. However, cancer cells also utilize EVs for endothelial cell modifications within distant microenvironments to form a metastatic niche. For instance, the blood–brain barrier (BBB) tightly regulates the permeability from the vascular system to the central nervous system [88, 92, 93]. Thus, BBB destruction is one of the key steps to allow the extravasation of cancer cells during brain metastasis [88]. Although several types of molecules contributed to the BBB disruption [92, 94], EVs are also associated with brain metastasis of cancer cells by affecting BBB endothelial cells. A recent study indicated that brain metastasis breast cancer-derived EVs containing miR-181c were closely associated with the BBB destruction and brain metastasis [95]. miR-181c is transferred into endothelial cells of the BBB by EVs and causes the delocalization of actin by suppressing the expression of 3-phosphoinoitide-dependent protein kinase-1 (PDPK1). PDPK1 suppression leads to the degradation of the phosphorylated cofilin, which is important for actin dynamics and disrupts the permeability of the BBB. Similar to this report, Zhou et al. reported that metastatic breast cancer-derived EVs also contain miR-105 that directly inhibits tight junction protein 1 (ZO-1) expression in endothelial cells and destroys the tight junction to promote metastasis [96]. It appears that miR-181c is included in the EVs that preferentially accumulate in brain endothelial cells, but EVs that contain miR-105 may affect endothelial cells throughout the body. Therefore, cancer-derived EVs may participate in such organ-specific metastasis of cancer cells through remodeling the distal pro-metastatic niche. Indeed, pancreatic cancer-derived EVs are selectively uptaken by liver Kupffer cells and promote liver metastasis of cancer cells [97]. In addition, cancer-derived EVs had a tropism depending on surface integrin marks [38]. However, it is still controversial whether all of cancer-derived EVs exhibit specificity for the pre-metastatic niche. Clarification of these points is necessary for the use of EVs as novel therapeutic targets of cancer metastasis.
The functional role of EVs on intercellular communication between cancer cells and mesenchymal stem cells
Cancer-derived EVs affect the mesenchymal stem cells (MSCs) as well as fibroblasts and endothelial cells (Table 4). MSCs reside in several types of mesodermal tissues, such as bone marrow (BM), adipose tissue, umbilical cord, and peripheral blood [103–105]. Several lines of evidence indicated that these MSCs are recruited into the tumor microenvironment and could promote tumor growth and metastasis [106, 107]. Cancer-derived EVs control the differentiation of BM-MSCs to a CaF-like state through activating the TGF-β signaling pathway [108]. This differentiated BM-MSC with EVs exhibits proangiogenic properties to enhance tumor proliferation and invasion. Similarly, Paggetti et al. demonstrated that chronic lymphocytic leukemia (CLL) cells also secreted EVs to induce CaF-like phenotypes in BM-MSCs and endothelial cells [109]. BM-MSCs and endothelial cells with CLL-derived EVs support CLL adhesion, survival, and growth in vitro and in vivo. The origins of CaFs are still controversial, and cancer-derived EVs also induce CaF-like phenotypes in cancer surrounding fibroblasts [14]. Therefore, cancer-derived EVs can convert several types of stromal cells into CaF-like cells. These data suggest that EVs are associated with CaF population diversity.
Breast cancer patients have a protracted risk of recurrence within 5 years and even up to 10–20 years after surgery or adjuvant chemotherapy [110–112]. The phenomenon indicates that breast cancer cells acquire a dormant state and survive for a long time in the patient’s body. Although cancer cells cease dividing under a dormant state, they can survive in a quiescent state while waiting for the appropriate environmental conditions to begin proliferation again. Importantly, BM-MSCs contribute to the maintenance of such cancer cell dormancy via EVs [113]. BM-MSCs transfer miR-23b-containing EVs into breast cancer cells and modulate the dormant state by targeting myristoylated alanine-rich C-kinase substrate (MARCKS), which modulates cell motility and cell cycle progression [114]. Consistent with this report, it was also indicated that BM-MSCs secreted miR-16 via EVs and downregulated vascular endothelial growth factor (VEGF) expression to lead to the inhibition of growth and angiogenesis in breast cancer [115]. In contrast, several reports also showed that these MSC-derived EVs promoted tumor growth in renal cancer [116], gastric cancer, and colorectal cancer [117]. Thus, the functional role of MSC-derived EVs depends on cancer cell types. In addition, it was reported that EVs derived from BM-MSCs of multiple myeloma patients enhance tumor growth but that healthy subject-derived BM-MSCs suppress tumor growth via EVs [118]. Taken together, these different functions of MSC-derived EVs depending on the phenotypes of cancer cells. Understanding these points will provide a novel insight into cancer therapy.
Horizontal propagation of oncogenic molecules by EVs
As mentioned above, cancer-derived EVs controlled stromal cells within the tumor microenvironment to promote cancer progression. However, cancer cells can also transfer their oncogenic properties to other cancer cells via EVs (Table 5). For instance, the epithelial growth factor receptor (EGFR) truncated mutant, EGFR variant III (EGFRvIII), is closely associated with cancer progression and poor patient prognosis of glioblastoma (GBM), which is most common brain malignancy in adults [125]. Al-Nedwi et al. reported that the EGFRvIII could be transferred from EGFRvIII-positive GBM cells to negative GBM cells through EVs [126]. Internalization of EGFRvIII-containing EVs activated the MAPK and Akt signaling pathways in the EGFRvIII-negative GBM cells, resulting in the expression of EGFR target genes, including vascular endothelial growth factor (VEGF). Furthermore, EGFRvIII-containing EVs induce both morphological changes and anchorage-independent growth in recipient cells. Interestingly, EGFRvIII mRNA is also detectable in GBM-derived EVs [127]. Given that some reports demonstrated the heterogeneous distribution of EGFRvIII expression in GBM tissue [128, 129], the horizontal propagation of EGFRvIII by EVs might contribute to intratumoral heterogeneity and the progression of GBM. Such intracellular interactions by EVs between malignant tumor cells and less aggressive tumor cells were also observed in other types of cancer. Le et al. showed that highly metastatic breast cancer cells could transfer the EVs contained miR-200 family miRNAs to non-metastatic breast cancer cells to promote lung metastasis [130]. This miR-200 transfer induces the mesenchymal to epithelial transition (MET) by altering the expression of genes, including zeb2 and sec23a, in non-metastatic breast cancer cell lines. Qu et al. demonstrated that long non-coding RNAs (lncRNAs) were also transferred by EVs and contributed to the chemoresistance of renal cell carcinoma (RCC) cells [131]. This lncRNA, termed lncARSR, acts as a competitor of miR-34/miR-449 and stimulates AXL and c-MET expression to enhance chemoresistance in drug-sensitive RCC cells. From these reports, cancer-derived EVs play a crucial role in the acquisition and transfer of the malignant trait by horizontal propagation of oncogenic molecules.
Interaction between non-tumoral epithelial cells and cancer cells via EVs
Competitive cell interactions represent a basic biological process to maintain homeostasis [137]. In particular, during cancer initiation, aberrant cells bearing genetic or epigenetic mutations will conflict with the surrounding non-aberrant normal cells to eliminate them from the cell population [137, 138]. If these aberrant cells eliminate normal cells from tissue, it may lead to cancer formation and progression. Therefore, it is conceivable that normal cells require an eradication system for aberrant cells to prevent tumor initiation. Normal prostate epithelial cell lines secreted EVs to transfer the tumor suppressor miR-143 into cancer cells [85] (Table 6), resulting in the induction of growth inhibitory signals in prostate cancer cells. These results suggest that EVs may contribute to the maintenance of normal growth and prevent cancer initiation. In contrast, cancer cells also utilize EVs to overcome this cell competition with non-tumoral epithelial cells during the cancer initiation step. Breast cancer-derived EVs are capable of inducing anchorage-independent growth and survival in mammary epithelial cells [139] (Table 6). Similar to this report, high metastatic hepatocellular carcinoma cell lines also transfer the oncogenic molecules to the non-tumoral immortalized hepatocytes and promote invasive activity of the immortalized hepatocytes [140]. Therefore, cancer-derived EVs contribute to the expansion of cancer cells by transforming non-aberrant normal cells. From these reports, it is conceivable that EVs play an important role in cell competition between cancer cells and non-tumoral epithelial cells. Further investigation will advance our understanding of the progression mechanisms of competitive cellular interaction between cancer cells and epithelial cells.
Conclusion
The precise mechanisms of intercellular communication in the tumor microenvironment remain obscure because there are many important pathways that modulate multiple non-EV factors, such as growth factors, cytokines, and chemokines. However, as described above, the rapid development of EV research elucidated the novel mechanism underlying the intrinsic intercellular communication networks during cancer initiation and progression. EVs possess impressively diverse functions in the intercellular communication networks in the tumor microenvironment. Cancer cells secrete EVs and dictate the phenotypes of surrounding cells to promote cancer progression. Against such “education,” non-tumoral cells utilized EVs to suppress cancer initiation and progression. Understanding the precise mechanisms of EVs in cancer biology may provide a breakthrough in the diagnostic and prognostic tools and therapeutic strategies of cancer.
References
Bhowmick NA, Moses HL (2005) Tumor–stroma interactions. Curr Opin Genet Dev 15(1):97–101. doi:10.1016/j.gde.2004.12.003
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11):1423–1437. doi:10.1038/nm.3394
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Gould SJ, Raposo G (2013) As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles. doi:10.3402/jev.v2i0.20389
Nawaz M, Camussi G, Valadi H, Nazarenko I, Ekstrom K, Wang X, Principe S, Shah N, Ashraf NM, Fatima F, Neder L, Kislinger T (2014) The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat Rev Urol 11(12):688–701. doi:10.1038/nrurol.2014.301
Fujita Y, Yoshioka Y, Ochiya T (2016) Extracellular vesicle transfer of cancer pathogenic components. Cancer Sci 107(4):385–390. doi:10.1111/cas.12896
Yoshioka Y, Konishi Y, Kosaka N, Katsuda T, Kato T, Ochiya T (2013) Comparative marker analysis of extracellular vesicles in different human cancer types. J Extracell Vesicles 2:20424. doi:10.3402/jev.v2i0.20424
Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200(4):373–383. doi:10.1083/jcb.201211138
Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C (2010) Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 123(Pt 10):1603–1611. doi:10.1242/jcs.064386
Akers JC, Gonda D, Kim R, Carter BS, Chen CC (2013) Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol 113(1):1–11. doi:10.1007/s11060-013-1084-8
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516. doi:10.1080/01926230701320337
Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL, Holmgren L (2001) Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc Natl Acad Sci USA 98(11):6407–6411. doi:10.1073/pnas.101129998
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9(6):654–659. doi:10.1038/ncb1596
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nature reviews. Cancer 6(5):392–401. doi:10.1038/nrc1877
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E (2013) Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15(6):637–646. doi:10.1038/ncb2756
Shimoda M, Principe S, Jackson HW, Luga V, Fang H, Molyneux SD, Shao YW, Aiken A, Waterhouse PD, Karamboulas C, Hess FM, Ohtsuka T, Okada Y, Ailles L, Ludwig A, Wrana JL, Kislinger T, Khokha R (2014) Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat Cell Biol 16(9):889–901. doi:10.1038/ncb3021
Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, Goruppi S, Menietti E, Ostano P, Ala U, Provero P, Hoetzenecker W, Neel V, Kilarski WW, Swartz MA, Brisken C, Lefort K, Dotto GP (2015) Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 17(9):1193–1204. doi:10.1038/ncb3228
Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S, Croce O, Sanz-Moreno V, Meneguzzi G, Feral CC, Cristofari G, Gaggioli C (2015) Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun 6:10204. doi:10.1038/ncomms10204
Webber J, Steadman R, Mason MD, Tabi Z, Clayton A (2010) Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res 70(23):9621–9630. doi:10.1158/0008-5472.can-10-1722
Webber JP, Spary LK, Sanders AJ, Chowdhury R, Jiang WG, Steadman R, Wymant J, Jones AT, Kynaston H, Mason MD, Tabi Z, Clayton A (2015) Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 34(3):290–302. doi:10.1038/onc.2013.560
Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, Buchanan M, Hosein AN, Basik M, Wrana JL (2012) Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 151(7):1542–1556. doi:10.1016/j.cell.2012.11.024
Niehrs C (2012) The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol 13(12):767–779. doi:10.1038/nrm3470
Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, Ter Brugge PJ, Jonkers J, Slingerland J, Minn AJ (2014) Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 159(3):499–513. doi:10.1016/j.cell.2014.09.051
Azzam DJ, Zhao D, Sun J, Minn AJ, Ranganathan P, Drews-Elger K, Han X, Picon-Ruiz M, Gilbert CA, Wander SA, Capobianco AJ, El-Ashry D, Slingerland JM (2013) Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in gamma-secretase inhibitor drug responses. EMBO Mol Med 5(10):1502–1522. doi:10.1002/emmm.201302558
Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, Yaswen P, Goga A, Werb Z (2015) Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526(7571):131–135. doi:10.1038/nature15260
Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, Chen X, Yao W (2015) Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci 106(10):1362–1369. doi:10.1111/cas.12747
Josson S, Gururajan M, Sung SY, Hu P, Shao C, Zhau HE, Liu C, Lichterman J, Duan P, Li Q, Rogatko A, Posadas EM, Haga CL, Chung LW (2015) Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 34(21):2690–2699. doi:10.1038/onc.2014.212
Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS, Li Y, Lu ES, Kwan K, Wong KK, Schmandt R, Lu KH, Mok SC (2016) Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun 7:11150. doi:10.1038/ncomms11150
Kohlhapp FJ, Mitra AK, Lengyel E, Peter ME (2015) MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 34(48):5857–5868. doi:10.1038/onc.2015.89
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 324(5930):1029–1033. doi:10.1126/science.1160809
Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, Sotgia F, Lisanti MP (2012) Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1alpha and HIF2alpha in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle (Georgetown, Tex) 11(17):3280–3289. doi:10.4161/cc.21643
Chaudhri VK, Salzler GG, Dick SA, Buckman MS, Sordella R, Karoly ED, Mohney R, Stiles BM, Elemento O, Altorki NK, McGraw TE (2013) Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res: MCR 11(6):579–592. doi:10.1158/1541-7786.mcr-12-0437-t
Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, Alvarez H, Gupta S, Maiti SN, Cooper L, Peehl D, Ram PT, Maitra A, Nagrath D (2016) Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 5:e10250. doi:10.7554/eLife.10250
Warburg O (1956) On the origin of cancer cells. Science (New York, NY) 123(3191):309–314
Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O’Connor ST, Li S, Chin AR, Somlo G, Palomares M, Li Z, Tremblay JR, Tsuyada A, Sun G, Reid MA, Wu X, Swiderski P, Ren X, Shi Y, Kong M, Zhong W, Chen Y, Wang SE (2015) Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 17(2):183–194. doi:10.1038/ncb3094
Jung T, Castellana D, Klingbeil P, Cuesta Hernandez I, Vitacolonna M, Orlicky DJ, Roffler SR, Brodt P, Zoller M (2009) CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia (New York, NY) 11(10):1093–1105
Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G (2011) Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 71(15):5346–5356. doi:10.1158/0008-5472.can-11-0241
Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527(7578):329–335. doi:10.1038/nature15756
Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, Wu Y, Qin J (2015) Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One 10(5):e0125625. doi:10.1371/journal.pone.0125625
de Visser KE, Eichten A, Coussens LM (2006) Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6(1):24–37. doi:10.1038/nrc1782
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23(11):549–555
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16(3):183–194. doi:10.1016/j.ccr.2009.06.017
Jinushi M, Komohara Y (2015) Tumor-associated macrophages as an emerging target against tumors: creating a new path from bench to bedside. Biochim Biophys Acta 1855(2):123–130. doi:10.1016/j.bbcan.2015.01.002
Powell DR, Huttenlocher A (2016) Neutrophils in the tumor microenvironment. Trends Immunol 37(1):41–52. doi:10.1016/j.it.2015.11.008
Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL (2009) Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol (Baltim, Md: 1950) 183(6):3720–3730. doi:10.4049/jimmunol.0900970
Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL (2010) Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One 5(7):e11469. doi:10.1371/journal.pone.0011469
Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, Squarcina P, Accornero P, Lozupone F, Lugini L, Stringaro A, Molinari A, Arancia G, Gentile M, Parmiani G, Fais S (2002) Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med 195(10):1303–1316
Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z (2007) Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res 67(15):7458–7466. doi:10.1158/0008-5472.can-06-3456
Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z (2008) Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol (Baltim, Md: 1950) 180(11):7249–7258
Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E, Le Moulec S, Guigay J, Hirashima M, Guemira F, Adhikary D, Mautner J, Busson P (2009) Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein–Barr virus-infected nasopharyngeal carcinoma cells. Blood 113(9):1957–1966. doi:10.1182/blood-2008-02-142596
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rebe C, Ghiringhelli F (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Investig 120(2):457–471. doi:10.1172/jci40483
de Vrij J, Maas SL, Kwappenberg KM, Schnoor R, Kleijn A, Dekker L, Luider TM, de Witte LD, Litjens M, van Strien ME, Hol EM, Kroonen J, Robe PA, Lamfers ML, Schilham MW, Broekman ML (2015) Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int J Cancer 137(7):1630–1642. doi:10.1002/ijc.29521
Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Urbanowicz B, Branski P, Ratajczak MZ, Zembala M (2006) Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol Immunother: CII 55(7):808–818. doi:10.1007/s00262-005-0075-9
Song X, Ding Y, Liu G, Yang X, Zhao R, Zhang Y, Zhao X, Anderson GJ, Nie G (2016) Cancer cell-derived exosomes induce mitogen-activated protein kinase-dependent monocyte survival by transport of functional receptor tyrosine kinases. J Biol Chem 291(16):8453–8464. doi:10.1074/jbc.M116.716316
Yang M, Chen J, Su F, Yu B, Su F, Lin L, Liu Y, Huang JD, Song E (2011) Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer 10:117. doi:10.1186/1476-4598-10-117
Chow A, Zhou W, Liu L, Fong MY, Champer J, Van Haute D, Chin AR, Ren X, Gugiu BG, Meng Z, Huang W, Ngo V, Kortylewski M, Wang SE (2014) Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-kappaB. Sci Rep 4:5750. doi:10.1038/srep05750
Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, Flament C, Pouzieux S, Faure F, Tursz T, Angevin E, Amigorena S, Zitvogel L (2001) Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 7(3):297–303. doi:10.1038/85438
Taylor DD, Gercel-Taylor C (2005) Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br J Cancer 92(2):305–311. doi:10.1038/sj.bjc.6602316
Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, Li G (2008) Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Therapy: J Am Soc Gene Ther 16(4):782–790. doi:10.1038/mt.2008.1
Harshyne LA, Hooper KM, Andrews EG, Nasca BJ, Kenyon LC, Andrews DW, Hooper DC (2015) Glioblastoma exosomes and IGF-1R/AS-ODN are immunogenic stimuli in a translational research immunotherapy paradigm. Cancer Immunol Immunother: CII 64(3):299–309. doi:10.1007/s00262-014-1622-z
Naslund TI, Gehrmann U, Qazi KR, Karlsson MC, Gabrielsson S (2013) Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol (Baltim, Md: 1950) 190(6):2712–2719. doi:10.4049/jimmunol.1203082
Andre F, Chaput N, Schartz NE, Flament C, Aubert N, Bernard J, Lemonnier F, Raposo G, Escudier B, Hsu DH, Tursz T, Amigorena S, Angevin E, Zitvogel L (2004) Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol (Baltim, Md: 1950) 172(4):2126–2136
Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, Valente N, Shreeniwas R, Sutton MA, Delcayre A, Hsu DH, Le Pecq JB, Lyerly HK (2005) A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med 3(1):9. doi:10.1186/1479-5876-3-9
Escudier B, Dorval T, Chaput N, Andre F, Caby MP, Novault S, Flament C, Leboulaire C, Borg C, Amigorena S, Boccaccio C, Bonnerot C, Dhellin O, Movassagh M, Piperno S, Robert C, Serra V, Valente N, Le Pecq JB, Spatz A, Lantz O, Tursz T, Angevin E, Zitvogel L (2005) Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med 3(1):10. doi:10.1186/1479-5876-3-10
Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L (2014) Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol 292(1–2):65–69. doi:10.1016/j.cellimm.2014.09.004
Challagundla KB, Wise PM, Neviani P, Chava H, Murtadha M, Xu T, Kennedy R, Ivan C, Zhang X, Vannini I, Fanini F, Amadori D, Calin GA, Hadjidaniel M, Shimada H, Jong A, Seeger RC, Asgharzadeh S, Goldkorn A, Fabbri M (2015) Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst 107(7):djv135. doi:10.1093/jnci/djv135
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, Tanaka H, Chiba K, Ito S, Watatani Y, Kakiuchi N, Suzuki H, Yoshizato T, Yoshida K, Sanada M, Itonaga H, Imaizumi Y, Totoki Y, Munakata W, Nakamura H, Hama N, Shide K, Kubuki Y, Hidaka T, Kameda T, Masuda K, Minato N, Kashiwase K, Izutsu K, Takaori-Kondo A, Miyazaki Y, Takahashi S, Shibata T, Kawamoto H, Akatsuka Y, Shimoda K, Takeuchi K, Seya T, Miyano S, Ogawa S (2016) Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. doi:10.1038/nature18294
Fabian MR, Sonenberg N, Filipowicz W (2010) Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 79:351–379. doi:10.1146/annurev-biochem-060308-103103
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, Diao L, Wang J, Roybal JD, Patel M, Ungewiss C, Peng D, Antonia S, Mediavilla-Varela M, Robertson G, Jones S, Suraokar M, Welsh JW, Erez B, Wistuba II, Chen L, Peng D, Wang S, Ullrich SE, Heymach JV, Kurie JM, Qin FX (2014) Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 5:5241. doi:10.1038/ncomms6241
Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, Fujiwara T, Tsuta K, Nokihara H, Tamura T, Asamura H, Kawaishi M, Kuwano K, Ochiya T (2015) The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther: J Am Soc Gene Ther 23(4):717–727. doi:10.1038/mt.2015.10
Taylor DD, Gercel-Taylor C, Lyons KS, Stanson J, Whiteside TL (2003) T-cell apoptosis and suppression of T-cell receptor/CD3-zeta by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clinical Cancer Res: Off J Am Assoc Cancer Res 9(14):5113–5119
Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, Zaccheddu A, Colone M, Arancia G, Gentile M, Seregni E, Valenti R, Ballabio G, Belli F, Leo E, Parmiani G, Rivoltini L (2005) Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology 128(7):1796–1804
Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Zembala M (2007) Tumour-derived microvesicles modulate biological activity of human monocytes. Immunol Lett 113(2):76–82. doi:10.1016/j.imlet.2007.07.014
Deng Z, Rong Y, Teng Y, Zhuang X, Samykutty A, Mu J, Zhang L, Cao P, Yan J, Miller D, Zhang HG (2016) Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene. doi:10.1038/onc.2016.229
Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, Ricciardi-Castagnoli P, Raposo G, Amigorena S (1998) Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med 4(5):594–600
Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, Raposo G, Amigorena S (1999) Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol 147(3):599–610
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407(6801):249–257. doi:10.1038/35025220
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2(8):563–572. doi:10.1038/nrc865
Welti J, Loges S, Dimmeler S, Carmeliet P (2013) Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Investig 123(8):3190–3200. doi:10.1172/jci70212
Kim CW, Lee HM, Lee TH, Kang C, Kleinman HK, Gho YS (2002) Extracellular membrane vesicles from tumor cells promote angiogenesis via sphingomyelin. Cancer Res 62(21):6312–6317
Millimaggi D, Mari M, D’Ascenzo S, Carosa E, Jannini EA, Zucker S, Carta G, Pavan A, Dolo V (2007) Tumor vesicle-associated CD147 modulates the angiogenic capability of endothelial cells. Neoplasia (New York, NY) 9(4):349–357
Chan YK, Zhang H, Liu P, Tsao SW, Lung ML, Mak NK, Ngok-Shun Wong R, Ying-Kit Yue P (2015) Proteomic analysis of exosomes from nasopharyngeal carcinoma cell identifies intercellular transfer of angiogenic proteins. Int J Cancer 137(8):1830–1841. doi:10.1002/ijc.29562
Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, Ochiya T (2013) Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem 288(15):10849–10859. doi:10.1074/jbc.M112.446831
Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T (2010) Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem 285(23):17442–17452. doi:10.1074/jbc.M110.107821
Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F, Ochiya T (2012) Competitive interactions of cancer cells and normal cells via secretory microRNAs. J Biol Chem 287(2):1397–1405. doi:10.1074/jbc.M111.288662
Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH (2014) Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 124(25):3748–3757. doi:10.1182/blood-2014-05-576116
Tadokoro H, Umezu T, Ohyashiki K, Hirano T, Ohyashiki JH (2013) Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J Biol Chem 288(48):34343–34351. doi:10.1074/jbc.M113.480822
Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127(4):679–695. doi:10.1016/j.cell.2006.11.001
Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W, Lim SK, Sze SK (2010) Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol Cell Proteomics: MCP 9(6):1085–1099. doi:10.1074/mcp.M900381-MCP200
King HW, Michael MZ, Gleadle JM (2012) Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 12:421. doi:10.1186/1471-2407-12-421
Hannafon BN, Carpenter KJ, Berry WL, Janknecht R, Dooley WC, Ding WQ (2015) Exosome-mediated microRNA signaling from breast cancer cells is altered by the anti-angiogenesis agent docosahexaenoic acid (DHA). Mol Cancer 14:133. doi:10.1186/s12943-015-0400-7
Arshad F, Wang L, Sy C, Avraham S, Avraham HK (2010) Blood-brain barrier integrity and breast cancer metastasis to the brain. Pathol Res Int 2011:920509. doi:10.4061/2011/920509
Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis 16(1):1–13. doi:10.1016/j.nbd.2003.12.016
Wrobel JK, Toborek M (2016) Blood-brain barrier remodeling during brain metastasis formation. Mol Med (Camb, Mass). doi:10.2119/molmed.2015.00207
Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K, Lotvall J, Nakagama H, Ochiya T (2015) Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun 6:6716. doi:10.1038/ncomms7716
Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, Yu Y, Chow A, O’Connor ST, Chin AR, Yen Y, Wang Y, Marcusson EG, Chu P, Wu J, Wu X, Li AX, Li Z, Gao H, Ren X, Boldin MP, Lin PC, Wang SE (2014) Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 25(4):501–515. doi:10.1016/j.ccr.2014.03.007
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, Xiang J, Zhang T, Theilen TM, Garcia-Santos G, Williams C, Ararso Y, Huang Y, Rodrigues G, Shen TL, Labori KJ, Lothe IM, Kure EH, Hernandez J, Doussot A, Ebbesen SH, Grandgenett PM, Hollingsworth MA, Jain M, Mallya K, Batra SK, Jarnagin WR, Schwartz RE, Matei I, Peinado H, Stanger BZ, Bromberg J, Lyden D (2015) Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17(6):816–826. doi:10.1038/ncb3169
Gesierich S, Berezovskiy I, Ryschich E, Zoller M (2006) Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res 66(14):7083–7094. doi:10.1158/0008-5472.can-06-0391
Nazarenko I, Rana S, Baumann A, McAlear J, Hellwig A, Trendelenburg M, Lochnit G, Preissner KT, Zoller M (2010) Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res 70(4):1668–1678. doi:10.1158/0008-5472.can-09-2470
Ekstrom EJ, Bergenfelz C, von Bulow V, Serifler F, Carlemalm E, Jonsson G, Andersson T, Leandersson K (2014) WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol Cancer 13:88. doi:10.1186/1476-4598-13-88
Conigliaro A, Costa V, Lo Dico A, Saieva L, Buccheri S, Dieli F, Manno M, Raccosta S, Mancone C, Tripodi M, De Leo G, Alessandro R (2015) CD90 + liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol Cancer 14:155. doi:10.1186/s12943-015-0426-x
Zhuang G, Wu X, Jiang Z, Kasman I, Yao J, Guan Y, Oeh J, Modrusan Z, Bais C, Sampath D, Ferrara N (2012) Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J 31(17):3513–3523. doi:10.1038/emboj.2012.183
Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, Krause U, Blake J, Schwager C, Eckstein V, Ansorge W, Ho AD (2005) Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp Hematol 33(11):1402–1416. doi:10.1016/j.exphem.2005.07.003
Bergfeld SA, DeClerck YA (2010) Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev 29(2):249–261. doi:10.1007/s10555-010-9222-7
Keating A (2012) Mesenchymal stromal cells: new directions. Cell Stem Cell 10(6):709–716. doi:10.1016/j.stem.2012.05.015
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449(7162):557–563. doi:10.1038/nature06188
Tsai KS, Yang SH, Lei YP, Tsai CC, Chen HW, Hsu CY, Chen LL, Wang HW, Miller SA, Chiou SH, Hung MC, Hung SC (2011) Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology 141(3):1046–1056. doi:10.1053/j.gastro.2011.05.045
Chowdhury R, Webber JP, Gurney M, Mason MD, Tabi Z, Clayton A (2015) Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 6(2):715–731. doi:10.18632/oncotarget.2711
Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, Kim YJ, Adam J, Lichter P, Solary E, Berchem G, Moussay E (2015) Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 126(9):1106–1117. doi:10.1182/blood-2014-12-618025
Kurtz JM, Spitalier JM, Amalric R (1983) Late breast recurrence after lumpectomy and irradiation. Int J Radiat Oncol Biol Phys 9(8):1191–1194
(EBCTCG) EBCTCG (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet (London, England) 365(9472):1687–1717. doi:10.1016/s0140-6736(05)66544-0
Sgroi DC, Sestak I, Cuzick J, Zhang Y, Schnabel CA, Schroeder B, Erlander MG, Dunbier A, Sidhu K, Lopez-Knowles E, Goss PE, Dowsett M (2013) Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 14(11):1067–1076. doi:10.1016/s1470-2045(13)70387-5
Ono M, Kosaka N, Tominaga N, Yoshioka Y, Takeshita F, Takahashi RU, Yoshida M, Tsuda H, Tamura K, Ochiya T (2014) Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signal 7(332):ra63. doi:10.1126/scisignal.2005231
Rombouts K, Carloni V, Mello T, Omenetti S, Galastri S, Madiai S, Galli A, Pinzani M (2013) Myristoylated alanine-rich protein kinase C substrate (MARCKS) expression modulates the metastatic phenotype in human and murine colon carcinoma in vitro and in vivo. Cancer Lett 333(2):244–252. doi:10.1016/j.canlet.2013.01.040
Lee JK, Park SR, Jung BK, Jeon YK, Lee YS, Kim MK, Kim YG, Jang JY, Kim CW (2013) Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One 8(12):e84256. doi:10.1371/journal.pone.0084256
Du T, Ju G, Wu S, Cheng Z, Cheng J, Zou X, Zhang G, Miao S, Liu G, Zhu Y (2014) Microvesicles derived from human Wharton’s jelly mesenchymal stem cells promote human renal cancer cell growth and aggressiveness through induction of hepatocyte growth factor. PLoS One 9(5):e96836. doi:10.1371/journal.pone.0096836
Zhu W, Huang L, Li Y, Zhang X, Gu J, Yan Y, Xu X, Wang M, Qian H, Xu W (2012) Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett 315(1):28–37. doi:10.1016/j.canlet.2011.10.002
Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, Azab F, Flores LM, Campigotto F, Weller E, Anderson KC, Scadden DT, Ghobrial IM (2013) BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Investig 123(4):1542–1555. doi:10.1172/jci66517
Gu J, Qian H, Shen L, Zhang X, Zhu W, Huang L, Yan Y, Mao F, Zhao C, Shi Y, Xu W (2012) Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-beta/Smad pathway. PLoS One 7(12):e52465. doi:10.1371/journal.pone.0052465
De Veirman K, Wang J, Xu S, Leleu X, Himpe E, Maes K, De Bruyne E, Van Valckenborgh E, Vanderkerken K, Menu E, Van Riet I (2016) Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett 377(1):17–24. doi:10.1016/j.canlet.2016.04.024
Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18(6):883–891. doi:10.1038/nm.2753
Vallabhaneni KC, Penfornis P, Dhule S, Guillonneau F, Adams KV, Mo YY, Xu R, Liu Y, Watabe K, Vemuri MC, Pochampally R (2015) Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget 6(7):4953–4967. doi:10.18632/oncotarget.3211
Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E, Lahoutte T, De Wever O, Vanderkerken K, Menu E (2014) Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 124(4):555–566. doi:10.1182/blood-2014-03-562439
Katakowski M, Buller B, Zheng X, Lu Y, Rogers T, Osobamiro O, Shu W, Jiang F, Chopp M (2013) Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett 335(1):201–204. doi:10.1016/j.canlet.2013.02.019
Brandes AA, Franceschi E, Tosoni A, Hegi ME, Stupp R (2008) Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res: Off J Am Assoc Cancer Res 14(4):957–960. doi:10.1158/1078-0432.ccr-07-1810
Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J (2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 10(5):619–624. doi:10.1038/ncb1725
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10(12):1470–1476. doi:10.1038/ncb1800
Biernat W, Huang H, Yokoo H, Kleihues P, Ohgaki H (2004) Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol (Zurich, Switz) 14(2):131–136
Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, Paucar A, Yang H, Ohashi M, Zhu S, Wykosky J, Reed R, Nelson SF, Cloughesy TF, James CD, Rao PN, Kornblum HI, Heath JR, Cavenee WK, Furnari FB, Mischel PS (2014) Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science (New York, NY) 343(6166):72–76. doi:10.1126/science.1241328
Le MT, Hamar P, Guo C, Basar E, Perdigao-Henriques R, Balaj L, Lieberman J (2014) miR-200-containing extracellular vesicles promote breast cancer cell metastasis. J Clin Investig 124(12):5109–5128. doi:10.1172/jci75695
Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, Wang X, Wang Y, Xu ZY, Gao L, Yang Q, Xu B, Li YM, Fang ZY, Xu ZP, Bao Y, Wu DS, Miao X, Sun HY, Sun YH, Wang HY, Wang LH (2016) Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 29(5):653–668. doi:10.1016/j.ccell.2016.03.004
Fedele C, Singh A, Zerlanko BJ, Iozzo RV, Languino LR (2015) The alphavbeta6 integrin is transferred intercellularly via exosomes. J Biol Chem 290(8):4545–4551. doi:10.1074/jbc.C114.617662
Kogure T, Lin WL, Yan IK, Braconi C, Patel T (2011) Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology (Baltimore, Md) 54(4):1237–1248. doi:10.1002/hep.24504
Singh R, Pochampally R, Watabe K, Lu Z, Mo YY (2014) Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol Cancer 13:256. doi:10.1186/1476-4598-13-256
Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL, Ji MH, Hu Q, Luo Z, Wu JZ, Tang JH (2014) Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS One 9(4):e95240. doi:10.1371/journal.pone.0095240
Felicetti F, De Feo A, Coscia C, Puglisi R, Pedini F, Pasquini L, Bellenghi M, Errico MC, Pagani E, Care A (2016) Exosome-mediated transfer of miR-222 is sufficient to increase tumor malignancy in melanoma. J Transl Med 14(1):56. doi:10.1186/s12967-016-0811-2
Johnston LA (2009) Competitive interactions between cells: death, growth, and geography. Science (New York, NY) 324(5935):1679–1682. doi:10.1126/science.1163862
Wagstaff L, Kolahgar G, Piddini E (2013) Competitive cell interactions in cancer: a cellular tug of war. Trends Cell Biol 23(4):160–167. doi:10.1016/j.tcb.2012.11.002
Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, Holowka DA, Cerione RA (2011) Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci USA 108(12):4852–4857. doi:10.1073/pnas.1017667108
He M, Qin H, Poon TC, Sze SC, Ding X, Co NN, Ngai SM, Chan TF, Wong N (2015) Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 36(9):1008–1018. doi:10.1093/carcin/bgv081
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number 15K21646 and JSPS Fellows 15K10473 and in part by a Grant-in-Aid for the Japan Science and Technology Agency (JST) through the Center of Open Innovation Network for Smart Health (COINS) initiated by the Council for Science; a Grant-in-Aid for Basic Science and Platform Technology Program for Innovative Biological Medicine; and a Grant-in-Aid for the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct); and Grant-in-Aid for the Project for Cancer Research And Therapeutic Evolution (P-CREATE) from the Japan Agency for Medical Research and development, AMED.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Naito, Y., Yoshioka, Y., Yamamoto, Y. et al. How cancer cells dictate their microenvironment: present roles of extracellular vesicles. Cell. Mol. Life Sci. 74, 697–713 (2017). https://doi.org/10.1007/s00018-016-2346-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2346-3