
Abstract
A novel coronavirus known as severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) is a potential cause of acute respiratory infection called coronavirus disease 2019 (COVID-19). The binding of SARS-CoV-2 with angiotensin-converting enzyme 2 (ACE2) induces a series of inflammatory cellular events with cytopathic effects leading to cell injury and hyperinflammation. Severe SARS-CoV-2 infection may lead to dysautonomia and sympathetic storm due to dysfunction of the autonomic nervous system (ANS). Therefore, this review aimed to elucidate the critical role of the cholinergic system (CS) in SARS-CoV-2 infection. The CS forms a multi-faceted network performing diverse functions in the body due to its distribution in the neuronal and non-neuronal cells. Acetylcholine (ACh) acts on two main types of receptors which are nicotinic receptors (NRs) and muscarinic receptors (MRs). NRs induce T cell anergy with impairment of antigen-mediated signal transduction. Nicotine through activation of T cell NRs inhibits the expression and release of the pro-inflammatory cytokines. NRs play important anti-inflammatory effects while MRs promote inflammation by inducing the release of pro-inflammatory cytokines. SARS-CoV-2 infection can affect the morphological and functional stability of CS through the disruption of cholinergic receptors. SARS-CoV-2 spike protein is similar to neurotoxins, which can bind to nicotinic acetylcholine receptors (nAChR) in the ANS and brain. Therefore, cholinergic receptors mainly nAChR and related cholinergic agonists may affect the pathogenesis of SARS-CoV-2 infection. Cholinergic dysfunction in COVID-19 is due to dysregulation of nAChR by SARS-CoV-2 promoting the central sympathetic drive with the development of the sympathetic storm. As well, nAChR activators through interaction with diverse signaling pathways can reduce the risk of inflammatory disorders in COVID-19. In addition, nAChR activators may mitigate endothelial dysfunction (ED), oxidative stress (OS), and associated coagulopathy in COVID-19. Similarly, nAChR activators may improve OS, inflammatory changes, and cytokine storm in COVID-19. Therefore, nAChR activators like varenicline in virtue of its anti-inflammatory and anti-oxidant effects with direct anti-SARS-CoV-2 effect could be effective in the management of COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
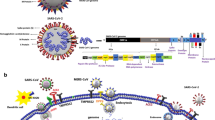
A novel coronavirus known as severe acute respiratory syndrome CoV type 2 (SARS-CoV-2) is the potential cause of acute respiratory infection called coronavirus disease 2019 (COVID-19). SARS-CoV-2 uses specific receptors for entry to human cells; one of the most predominant receptors is an angiotensin-converting enzyme type 2 (ACE2) (Al-Kuraishy et al. 2021b) as shown in Fig. 1. The binding of SARS-CoV-2 with ACE2 leads to a series of inflammatory cellular events with cytopathic effects causing cell injury and hyperinflammation. ACE2 is largely distributed and expressed in diverse cellular systems, including enterocytes, cardiomyocytes, pulmonary alveolar cells, neurons, and testes (Moubarak et al. 2021).

Binding of SARS-CoV-2 to ACE2 receptors
The clinical presentation of COVID-19 is mainly asymptomatic or presented with mild symptoms in 85% of cases. However, 15% of COVID-19 patients presented with moderate to severe form due to the progress of acute lung injury (ALI). As well, 5% of COVID-19 patients may be critical and require supported ventilation due to the development of acute respiratory distress syndrome (ARDS) (Al-Kuraishy et al. 2021a). The clinical presentation of COVID-19 has been reported to be considerably different among types and subtypes of SARS-CoV-2 infection. The relationship between SARS-CoV-2 genetic specificities among variants and clinical presentation is scarce (Al-kuraishy et al. 2022c). One variant with a deletion (∆382) in the open reading frame 8 has been associated with milder infections in Singapore and may have played a role in the very low case fatality rate in this country. Spike mutation D614G which probably occurs in China before its diffusion in Europe is associated with a decreased age of COVID-19 patients possibly due to an increased viral load in younger patients. The N501Y mutation is associated with adaptation to rodents, for instance, mice, and may increase SARS-CoV-2 spike protein binding to ACE2 because of conformational changes, thus increasing its transmissibility. The role of ∆69/∆70 on the spike protein is also potentially involved in the increased transmissibility (Al-Thomali et al. 2022). As well, the Gamma variant of SARS-CoV-2 infection was coincident with an increased COVID-19 incidence in younger patients (Luna-Muschi et al. 2022). The increase in the proportion of COVID-19 cases caused by the Gamma variant in early 2021 was temporally associated with the beginning of the vaccination campaign in Brazil. This context raised the concern that the Gamma variant could evade previous SARS-CoV-2 immune response (Goller et al. 2022). A retrospective study and comparative analyses revealed significant differences between recorded symptoms of BA.2 and BA.5 SARS-CoV-2 variants in infected individuals and found strong correlations of associations between symptoms. In particular, the symptoms chills or sweating, freezing, and runny nose were more frequently reported in BA.2 infections. In contrast, other clinical symptoms appeared more frequently in Omicron infections with BA.5. However, there was no evidence that BA.5 has higher pathogenicity or causes a more severe course of infection than BA.2. (Kopańska et al. 2022). These findings highlighted the difference in the clinical presentation of SARS-CoV-2 and its variants.
SARS-CoV-2 is highly identical to other CoVs like SARS and the Middle East Respiratory Syndrome CoV (MERS-CoV) and shares 80% and 60% genomic similarity correspondingly. In addition, SARS-CoV-2 is highly similar at the genomic level to bat CoV (96% similarity percentage) (Babalghith et al. 2022). Nevertheless, SARS-CoV-2 has 20 times higher binding affinity to ACE2 than other CoVs. ACE2 is a peptidase that metabolizes vasoconstrictor angiotensin II (Ang II) to the vasodilators Ang1-7 and Ang1-9 (Halder and Lal 2021). Consequently, downregulation of ACE2 during SARS-CoV-2 infection induces vasoconstriction and development of endothelial dysfunction (ED), oxidative stress (OS), and inflammatory disorder SARS-CoV-2-induced OS triggers activation of different signaling pathways which counterbalances this type of complication (Moran et al. 2019). It has been shown that severe SARS-CoV-2 infection may lead to dysautonomia and sympathetic storm due to dysfunction of the autonomic nervous system (ANS). In this context, Tizabi et al. (2020) hypothesized that nicotinic CS including nicotinic receptor agonists and partial agonists could be beneficial in COVID-19 management. Thus, nicotine, nicotinic receptor agonists, or positive modulators of these receptors may be of therapeutic potential in a variety of diseases including countering at least some of the harms of COVID-19. A review of the latest research recently conducted by Kopańska et al. (2022) regarding disorders of CS in COVID-19 showed that the presence of the SARS-CoV-2 virus disrupts the activity of the CS, for example, causing the development of myasthenia gravis or a change in acetylcholine (ACh) activity (Kopańska et al. 2022). The SARS-CoV-2 spike protein has a sequence similar to neurotoxins, capable of binding nicotinic acetylcholine receptors (nAChR). Nicotine and caffeine have similar structures to anti-viral drugs, capable of binding ACE 2 epitopes that are recognized by SARS-CoV-2, with the potential to inhibit the formation of the ACE 2/SARS-CoV-2 complex. The blocking is enhanced when nicotine and caffeine are used together with anti-viral drugs (Tizabi et al. 2020). These recent studies confirmed that SARS-CoV-2 infection adversely affects CS. However, the mechanistic role of CS against inflammatory signaling pathways in COVID-19 and how nAChR modulators affect the pathogenic role of SARS-CoV-2 infection need to be elucidated. Therefore, this review aimed to clarify the critical role of CS and its modulators in SARS-CoV-2 infection.
Cholinergic system
CS is one of the main parts of ANS that regulate different body functions including memory, cognitive function, sensation, digestive, cardiovascular, and sexual performance (Moran et al. 2019). ACh is the main neurotransmitter of CS that is synthesized from choline by the action of ACh acyltransferase (ChAT) which is mainly found in the cholinergic neurons. ChAT is also expressed in non-neuronal cells including immune cells and splenic cells serving as a major source of extra-neuronal ACh (Jackisch et al. 2009). ACh is metabolized by ACh esterase (AChE) which is found in two forms; true AChE is found in the neurons and neuromuscular junction (NMJ) while pseudo AChE is present mainly in plasma, and can metabolize other agents like procaine (Moran et al. 2019). AChE inhibitors like neostigmine and physostigmine improve cholinergic neurotransmission in the brain and NMJ (Jackisch et al. 2009).
Certainly, ACh acts on two main types of receptors which are either nicotinic or muscarinic. NRs are ion channels consisting of 4 subunits (α, β, γ, and δ) which are bound in different ratios around the channel central pore (Jackisch et al. 2009). NRs are present either as homomers or heteromers. Human NR α3β4 type which is present in the brain and autonomic ganglions is known as neuronal NR (nNR). However, NRs present in the NMJ are known as muscular NRs (mNRs) (Bekdash 2021).
The MRs are metabotropic receptors, which have seven trans-membrane subunit G-protein-coupled receptors, and respond to both muscarine and ACh. There are five types of MRs. The M1, M3, and M5 receptors mediate activation of phospholipase C, while M2 and M4 act via inhibition of adenylate cyclase with reduction of cAMP (Bekdash 2021). Depending on the physiological distributions of MRs, these receptors provoke many signal transductions in a tissue-specific manner. M1 is present mainly in the brain, M2 in the heart, and M3 in the exocrine glands and intestines. The M3 muscarinic receptor is clinically significant in the bladder, airway, eye, and blood vessels in addition to exocrine glands and intestines. M4 and M5 are distributed non-specifically but mainly in the brain (Jackisch et al. 2009).
Cholinergic system and inflammation
The CS forms a complex network that performs different functions in the body due to its distribution in the neuronal and non-neuronal cells (Yuan et al. 2019). Immune cells have a full machinery system for the synthesis and release of ACh. It has been reported that ACh affects the immune cells in paracrine and autocrine manners (Cox et al. 2020). Different studies revealed that ACh level is elevated in various diseases including periodontal diseases, chronic obstructive airway diseases, ischemic stroke, and atopic dermatitis (Yuan et al. 2019). However, the ACh level is reduced in neurodegenerative-associated inflammatory reactions like multiple sclerosis and vascular dementia. Moreover, ChAT is expressed constitutively in B and T cells, macrophages, and mononuclear lymphocytes. Notably, immunological activation induces transcription of ChAT in the immune cells. ChAT is also expressed in the lung alveolar epithelial cells and lung macrophages. Therefore, ChAT agonists and antagonists used in various neurological disorders may affect the immune cells (Nizri et al. 2006).
Furthermore, ACh is highly expressed in lymphocytes, macrophages, and dendritic cells. It has been shown that the AChE serum level was increased in irritable bowel syndrome, liver cirrhosis, multiple sclerosis, and Alzheimer’s disease (Snider et al. 2018). AChE inhibitors like rivastigmine, galantamine, and donepezil may affect autoimmunity and inflammation. Nizri et al. (2006) found that rivastigmine can reduce neuroinflammation and immune reactions.
Indeed, choline transporters (ChTs), which are expressed in the macrophages, microvascular cells, and immune cells, have a significant immunomodulatory effect (Leite et al. 2016). Of note, vesicular ACh transporters (VAChTs) are involved in the storage of ACh. Alterations of VAChTs are linked with the release of pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin 1-beta (IL-1β), and interleukin 6 (IL-6) (Leake 2019). The exact functions of VAChTs in the immune cells were not elucidated. The experimental study demonstrated that choline uptake is necessary for macrophage activation and IL-1β-mediated inflammation (Leake 2019; Hernandez et al. 2013).
ACh released from the vagus nerve and via its action on the NRs attenuates the release of the pro-inflammatory cytokine from activated macrophages but does not affect the release of the anti-inflammatory interleukin, IL-10. In endotoxemia, stimulation of the vagus nerve prevents the development of homeostatic disturbance (Hong et al. 2019). However, vagus nerve stimulation fails to attenuate inflammatory reactions in NR knockout mice which develop exaggerated immune response and release of pro-inflammatory cytokine (Hampel et al. 2018). In addition, vagus nerve stimulation inhibits the release of TNF-α which is mediated by endotoxemia. Atropine administration does not reduce the anti-inflammatory effect of vagus nerve stimulation as it is mainly mediated by NRs. Despite these findings, central MRs have anti-inflammatory effects through the vagus nerve (Kabata and Artis 2019). Thus, the spleen represents the connecting point between the CNS and the peripheral immune system. Experimental stimulation of the hypothalamus triggers anti-inflammatory effects in the spleen (Huston et al. 2006).
Cholinergic receptors and immune cells
NRs are involved in the regulation of immune cells; ACh generated at the microenvironment stimulates these receptors with subsequent regulation of the proliferation and activation of T cells. In vitro studies demonstrated that the administration of nicotine blocks the activation of T cells through the inhibition of cytotoxic T lymphocyte-associated protein 4 (CTLAP4) (De Rosa et al. 2009). As well, NRs induce T cell anergy with impairment of antigen-mediated signal transduction. Nicotine through activation of T cell NRs inhibits the expression and release of pro-inflammatory cytokines. Thus, α7nAChR antagonists like methyllycaconitine and bungarotoxin accelerate T cells’ proliferative response. A previous study revealed that nicotine attenuates the experimental autoimmune encephalomyelitis in mice through the polarization of T cells toward anti-inflammatory IL-4-producing T cells. Nicotine-induced activation of α7nAChR on the T cells promotes the proliferation of anti-inflammatory regulatory T cells (Pan et al. 2021).
Notoriously, NRs are essential for the maturation and proliferation of B lymphocytes within the spleen and other lymphoid organs. An experimental study revealed that NR knockout mice experienced noteworthy depletion of circulating B lymphocytes with a reduction of IgG-producing cells (Koval et al. 2018). NRs promote the proliferation and activation of B lymphocytes as well as immune-mediated interaction. Further, NRs inhibit the activation of dendritic cells (DC); thus, nicotine-treated DCs cannot activate T cells and produce pro-inflammatory cytokines. As well, NR agonist induces a robust anti-inflammatory effect in mice with collagen-induced arthritis by inhibiting the expression of CD80 on the DC surface (Kanauchi et al. 2022). However, ACh-treated DCs trigger the release of chemokines which promote the recruitment of Th2 cells at the site of inflammation (Gori et al. 2019). ACh-treated murine DCs increased the lung inflammation through the MR-dependent pathway in mice (Gori et al. 2019). In addition, ACh-treated DCs trigger the expression of anti-inflammatory peroxisome proliferator-activated receptor gamma (PPRA-γ) which favors the Th2 lineage through modulation of the balance of Th1/Th2 (Nouri-Shirazi et al. 2015). Notably, activation of NRs on the immature DCs enhances the expression of costimulatory CD80 and CD86 which accelerate the proliferation of T cells (Nouri-Shirazi et al. 2015).
Moreover, activation of NRs on the macrophages attenuates the release of the pro-inflammatory cytokines. NR knockout mice experience high levels of pro-inflammatory cytokines compared to wild-type mice (Fujii et al. 2007). A previous study conducted by Borovikova et al. (2000) found that ACh attenuates LPS-induced activation of human macrophages, preventing the release of pro-inflammatory cytokines. In addition, NR activation by vagus nerve stimulation hampers systemic inflammation. It was illustrated that nicotine through activation of NRs promotes the expression of IL-1 receptor-associated kinase M (IRAK-M) a negative regulator of TLR4, leading to potent anti-inflammatory effects (Youssef et al. 2021).
On the neutrophils, NRs negatively regulate recruitments and maturations of neutrophils at the site of inflammation. It has been reported that the administration of nicotine inhibits the expression of CD11b molecules on the surface of neutrophils through suppression of actin polymerization with subsequent inhibition of neutrophil recruitments. NR agonists block the interaction between monocytes and endothelial cells through suppression of the expression of adhesion molecules (Wu et al. 2022). Moreover, NR agonists attenuate mast cell activation with the inhibition of the release of pro-inflammatory cytokines and leukotrienes. In addition, NR agonists reduce the interaction and affinity of IgG on the surface of mast cells (Kutukova and Nazarov 2020).
Regarding the effects of MRs, it has been shown that neutrophil chemotactic is mediated by MR activation; thus, tiotropium, a selective M3R antagonist, blocks this interaction. As well, M3R participates in the induction of immunothrombosis due to the generation of NET formation. Blocking of M3R prevents release of pro-inflammatory cytokines (Lo et al. 2018). Profita et al. (2012) observed that sputum from smoker patients with chronic obstructive airway disease had a higher concentration of ACh and TGF-β1, and depletion of TGF-β1 reduces the expression of MRs on the lung epithelial cells. In addition, vagal-induced bronchoconstriction is mediated MR3 activation and expression of TNF-α (Profita et al. 2012). It has been demonstrated that MR agonist carbachol augments the phagocytosis in the peritoneal macrophages. However, activation of M1–M3 induces proliferation of macrophages while activation of M1–M2 induces activation of prostaglandin E2 through stimulation expression of protein kinase C (Profita et al. 2012).
These findings suggest that NRs play important anti-inflammatory effects while MRs promote inflammation by inducing the release of pro-inflammatory cytokines.
Clinical significance
The vagus nerve plays a critical role in the modulation of innate immune response and blood pressure control (Tracey 2009). Vagus nerve activity is reduced in response to hyperinflammation and cytokine storm as in sepsis, systemic lupus erythematosus, and rheumatoid arthritis. Vagus nerve stimulation inhibits the release of pro-inflammatory cytokines in various inflammatory disorders (Yang et al. 2022). Notably, H2-blocker famotidine, through vagus nerve stimulation, inhibits the development of pro-inflammatory cytokines. Because evidence is lacking for a direct anti-viral activity of famotidine, a proposed mechanism of action is blocking the effects of histamine released by mast cells (Mendez-Enriquez et al. 2021). As well, famotidine activates the inflammatory reflex via the brain-integrated vagus nerve mechanism which inhibits inflammation through α7nAChR signal transduction, to prevent cytokine storm. Intracerebroventricular administration of famotidine inhibits the release of pro-inflammatory cytokines independent of mast cell inhibition which could be through vagus nerve stimulation (Mendez-Enriquez et al. 2021). This observation confirmed the vagus nerve-dependent anti-inflammatory effect of famotidine in the setting of cytokine storm which is not replicated by high dosages of other H2R antagonists in clinical use (Seyedabadi et al. 2018).
Additionally, MR agonists like methacholine trigger the activation of inflammation and bronchial hyperresponsiveness through the expression of costimulatory molecules. Of interest, M3R activation is associated with immunopathogenesis of B cell lymphoma and Sjogren syndrome which is mainly mediated due formation of excitatory autoantibodies against the parotid gland (Cox et al. 2019). An elegant study illustrated that use of non-selective MR agonist arecoline in the management of cognitive dysfunction led to a reduction in the size of lymphoid organs and spleen. In contrast, the administration of non-selective MR agonist oxotremorine activates heat shock protein factor in rat hippocampus rats leading to anti-inflammatory and anti-oxidant effects in mice (Cox et al. 2019).
Taken together, ACh, NRs, and MRs have important immunoregulatory effects in the mitigation of different inflammatory disorders.
Cholinergic system and viral infections
ACh in virtue of its anti-inflammatory effects is known to cause vasodilatation a marker of acute inflammation thereby facilitating the recruitment and migration of immune cells to the site of inflammation. MRs expressed by the endothelial cells provoke the generation of nitric oxide (NO) which induces vasodilatation. This effect is abolished by MR antagonists, atropine, or NO inhibitors. Of note, deficiency of T cell CAChT in mice promotes T cell exhaustion during viral infections. Therefore, the expression of CAChT and ACh in the immune cells is necessary to combat viral infections (Sajjanar et al. 2016).
Remarkably, the entry of rabies virus into the host cells is through NRs, and this may induce autoimmune disorders and myasthenia gravis. An experimental study confirmed that rabies virus infection is associated with specific alterations in MRs in the brain stem and hippocampus independent of the viral load. Of interest, a specific peptide that binds NRs can abrogate the binding of the rabies virus to the host cells (Sajjanar et al. 2016). Up to date, rabies virus induces substantial downregulation of nAChR with subsequent release of pro-inflammatory cytokines (Lian et al. 2022). In addition, the persistent infection of neuroblastoma cells by the lymphocytic choriomeningitis virus inhibits the expressions of AChE and AChT with subsequent abnormal anti-viral immune response. Furthermore, M2R, which is an autoreceptor inhibiting the release of ACh in the lung, is highly reduced in viral infection, and associated INF release causing an increased release of ACh. However, dexamethasone augments the expression of M2R with subsequent inhibition release of ACh. Different human and animal model studies observed that M3R on bronchial smooth muscles are downregulated by viral infections due to phosphorylation of these receptors by viral neuraminidases with subsequent reduction in affinity to the ACh (Laguna Merced 2021). An experimental study illustrated that infections by influenza and parainfluenza viruses induce bronchial hyperreactivity by encouraging the release of ACh mainly at day 3, reaches the peak at 2 weeks, and then returns to normal level in dogs through the destruction of M2R (Pawełczyk and Kowalski 2017).
Notably, herpes virus infection can induce reactive immunoreactive antibodies which block NRs at NMJ leading to the development of myasthenia gravis. Capo-Velez et al. (2018) disclosed that the expression of α7nAChR in the immune cells and neurons could be a possible link between the pathogenesis of HIV-1 infection and associated cognitive dysfunction in AIDS patients (Pawełczyk and Kowalski 2017). It has been shown that molecular mimicry between gp120 and viral coat protein with α7nAChR may trigger the generation of neurotoxin-affecting NRs in the NMJ with the development of myasthenia. A clinical study involved an AIDS patient treated with pyridostigmine which is an effective agent in the management of under-reactive bladder and myasthenia gravis illustrated that this drug inhibits T cell over-activity with induction of the release of pro-inflammatory IL-10 and increases circulating CD4 level (Valdés-Ferrer et al. 2017). Furthermore, HIV-induced neuroinflammation can affect the expression of anti-inflammatory α7nAChR on the microglia cells. Besides, anti-retroviral drugs like indinavir act as positive allosteric modular of this receptor but it acts as an inhibitor for them at higher concentrations (Ekins et al. 2017). These observations suggest that α7nAChR is highly disturbed in HIV infection and associated with the development of neuroinflammation in AIDS patients.
These observations suggest that CS is highly distorted during various viral infections with subsequent propagation of pro-inflammatory response. Taken together, downregulation of NRs and over-expression of MRs together with dysregulation of M2R in various viral infections lead to hyperinflammation and associated tissue injury.
Cholinergic dysfunction in COVID-19
SARS-CoV-2 infection can affect the morphological and functional stability of the cholinergic system through the disruption of cholinergic receptors. SARS-CoV-2 spike protein is similar to the neurotoxins that can bind nAChR in the ANS, NMJ, and brain (Kopańska et al. 2022). Interestingly, SARS-CoV-2 spike protein epitopes have higher similarity with neurotoxins affecting nAChR (Lagoumintzis et al. 2021). Different in silico studies demonstrated that SARS-CoV-2 spike protein interacts with nAChR through cryptic epitopes similar to that of snake toxins (Batiha et al. 2021).
Of interest, nicotine can bind to ACE2, thus preventing the interaction between SARS-CoV-2 and ACE2 (Al-Kuraishy et al. 2022b). Therefore, cholinergic receptors mainly nAChR and related cholinergic agonists may affect the pathogenesis of SARS-CoV-2 infection (Al-Kuraishy et al. 2021c). It has been observed that the SARS-CoV-2 spike protein has a higher affinity to bind nAChR and produce an antagonist effect. Notably, SARS-CoV-2 spike protein and other proteins have an identical sequence to the nicotinic receptor antagonists indicating that SARS-CoV-2 blocks the functional activity and stability of nAChR (Kloc et al. 2020). Tizabi et al (2020) proposed that downregulation of nAChR during SARS-CoV-2 infection promotes the release of pro-inflammatory cytokines. This effect is due to the suppression of anti-inflammatory nAChR on the immune cells. In turn, the pro-inflammatory cytokines inhibit the expression and functional capacity of nAChR (Tizabi et al. 2020). This finding suggests that nAChR are inhibited either directly by SARS-CoV-2 or indirectly by exaggerated immune response and high level of pro-inflammatory cytokines.
Therefore, the interaction between SARS-CoV-2 spike protein and nAChR triggers hyperinflammation and the development of cytokine storm in COVID-19. In addition, dysregulation of brain nAChR in SARS-CoV-2 infection exaggerates neuroinflammation in COVID-19 (Farsalinos et al. 2020). Remarkably, a positive allosteric modulator of nAChR ivermectin can modulate SARS-CoV-2 infection-induced dysautonomia and neuroinflammation (Pąchalska et al. 2021).
Moreover, inhibition of platelet nAChR by SARS-CoV-2 promotes platelet hyperreactivity and thrombosis a hallmark of COVID-19. Of note, ACh acts as an endogenous inhibitor of platelet aggregation; thus, augmentation of ACh and activation of platelet nAChR could be a promising option in the mitigation of SARS-CoV-2-induced thrombosis (Farsalinos et al. 2020).
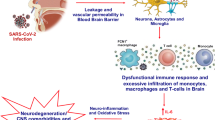
Cholinergic dysfunction in COVID-19 is due to dysregulation of nAChR by SARS-CoV-2 which promotes the central sympathetic drive with the development of sympathetic storm (Alexandris et al. 2021). In turn, sympathetic storm triggers oxidative stress and hyperinflammation by increasing the generation of ROS and release of pro-inflammatory cytokines (Al-Kuraishy et al. 2021c). Therefore, the use of cholinergic agonists or β-adrenoceptor antagonists might be beneficial in severely affected COVID-19 patients. Of note, β-blockers are effective in the mitigation of sympathetic and cytokine storms. Uncontrolled sympathetic activation in COVID-19 is due to the inhibition of counterbalance CS. Therefore, activation of CS and inhibition of the sympathetic nervous system (SNS) could be an effective therapeutic strategy in the mitigation of COVID-19 complications (Al-Kuraishy et al. 2021d).
Furthermore, a prospective study involving 37 COVID-19 patients compared to 14 healthy controls revealed that the expression of the nAChR gene was reduced in COVID-19 due to exaggerated pro-inflammatory cytokines (Courties et al. 2021). Of note, IL-6 and TNF-α, which are the major pro-inflammatory cytokines, inhibit the expression of anti-inflammatory effect of nAChR (Courties et al. 2021). Consequently, reduction of nAChR promotes the release of pro-inflammatory cytokines with increasing risk for development of cytokine storm (Mehranfard and Speth 2022).
Higher expression of pro-inflammatory cytokines inhibits expression of the nAChR gene (Courties et al. 2021). As well, AChE activity was dysregulated in severely affected COVID-19 patients causing an additional impact on the cholinergic activity. Bahloul et al. (2022) observed that low level of AChE activity was associated with high mortality so it can be used as a prognostic biomarker of COVID-19 patient severity. A retrospective study, which involved 137 COVID-19 patients, illustrated that AChE activity was reduced in critically affected COVID-19 patients compared to the mild one (Bahloul et al. 2022). AChE is reduced in sepsis due to the inflammatory process and acute phase reaction. AChE level at the time of admission is regarded as an independent predictor for mortality in COVID-19 patients (Nakajima et al. 2021).
Taken together, as the virus replicates, the virions may interact with the nAChRs blocking the action of the cholinergic anti-inflammatory pathway. If the initial immune response is not enough to combat the viral invasion at an early stage, the extensive and prolonged replication of the virus will ultimately disturb the cholinergic anti-inflammatory pathway seriously compromising its ability to control and regulate the immune response (Farsalinos et al. 2020). The uncontrolled action of pro-inflammatory cytokines will result in the development of cytokine storm, with ALI leading to ARDS, coagulation disturbances, and multi-organ failure. Based on this hypothesis, COVID-19 appears to eventually become a disease of the nicotinic CS. Nicotine could maintain or restore the function of the cholinergic anti-inflammatory system and thus control the release and activity of pro-inflammatory cytokines. This could prevent or suppress the cytokine storm (Mazloom 2020).
Overall, the changes in the CS in viral infection are not consistent with those in COVID-19 which might due to distinct binding activity of SARS-CoV-2 to nAChR. These verdicts proposed that deregulation of nAChR expression and AChE activity in severe SARS-CoV-2 lead to propagation of immunoinflammatory disorders associated with COVID-19 severity.
nAChR and inflammatory signaling pathways in COVID-19
SARS-CoV-2 infection and irregular immune response are linked with the commencement of various types of inflammatory signaling pathways leading to hyperinflammation. NLRP3 inflammasome is extremely activated during exaggerated immune response leading to multiple complications in COVID-19 (Mostafa-Hedeab et al. 2022). Targeting of NLRP3 inflammasome pathway by selective inhibitors may reduce COVID-19-induced complications (Al-Kuraishy et al. 2022e). Activation of anti-inflammatory nAChR inhibits various inflammatory signaling pathways like NLRP3 inflammasome via inhibition of β-arrestin 1in mice. Deng and coworkers observed that activation of nAChR reduces the development of pulmonary hypertension induced by monocrotaline in rats (Deng et al. 2019). Consequently, nAChR activators could play important role in the reduction of SARS-CoV-2 infection-induced inflammation through suppression of NLRP3 inflammasome in COVID-19 (Ke et al. 2017).
As well, activation of NLRP3 inflammasome is associated with more expression of NF-κB which increases inflammatory disorders via the release of pro-inflammatory cytokines in COVID-19 (Al-Kuraishy et al. 2022g). Worth mentioning, nAChR activators attenuate the expression of NF-κB leading to potent anti-inflammatory effects. nAChR activators prevent the development of ALI in mice by suppressing the NF-κB pathway (Patel et al. 2017).
Notably, immune response during acute cell injury persuades expression of toll-like receptor 4 (TLR4) and high-mobility box protein 1 (HMBP1). As well, nAChR activators reduce the expression of pro-inflammatory cytokines including MCP-1, TNF-α, IL-6, and IL-1β by inhibiting the recruitment of RNA polymerase II and macrophage activation. Thus, nAChR activators attenuate the development of inflammatory changes through modulation of the expression of the pro-inflammatory cytokines (Mizrachi et al. 2021).
In COVID-19, the expressions of TLR4 and HMBP1 are exaggerated leading to induction of the release of pro-inflammatory cytokine and immunothrombosis, respectively. HMBP1 induction in COVID-19 provokes the recruitment of neutrophils with the formation of neutrophil extracellular traps (NETs) which cause more inflammation and thrombosis termed immunothrombosis. Interestingly, nAChR activators suppress inflammatory reactions and inhibit the expression of MCP-1, TNF-α, IL-6, and IL-1β as well as the expression of adhesion molecules (Abdallah et al. 2007).
In addition, the mechanistic target of the rapamycin (mTOR) pathway promotes viral replication and growth in COVID-19. Notably, mTOR is a serine/threonine kinase that controls cell growth by enhancing mTOR1 and mTOR2. Previous research identified that metformin which is an agent widely used to treat type 2 diabetes due to AMPK activation was effective in treating influenza virus infection through inhibition of the mTOR inhibitor pathway (Karam et al. 2021). It has been suggested that the mTOR pathway is essential for the replication of SARS-CoV-2, and the use of mTOR inhibitors and modulators like sapanisertib and metformin, respectively, may reduce COVID-19 severity. Witayateeraporn et al. (2020) showed that nAChR blocks migration and proliferation of non-small-cell lung cancer via inhibition of the mTOR pathway. Besides, nAChR activators and allosteric modulators mitigate inflammatory changes in inflammatory bowel disease by inhibiting autophagy and expression of mTOR (Witayateeraporn et al. 2020).
In this state, nAChR activators may decrease the severity of SARS-CoV-2 infection and associated complications via modulation of the mTOR pathway. Noticeably, advanced glycation end products (AGEs) encourage the release of pro-inflammatory cytokines and induce the propagation of OS and inflammatory disorders. Receptors for AGEs (RAGE) expressed in pulmonary epithelial alveolar cells are intricate in the pathogenesis of SARS-CoV-2 infection and associated lung inflammation, ALI, and ARDS (Perrone et al. 2020). Both diabetes and the aging process accelerate the production of AGEs, which via interaction with RAGE on the macrophages, triggers lung inflammation in COVID-19. It has been shown that nAChR activators decrease AGE-induced inflammatory changes in myasthenia gravis. Preclinical evidence showed that RAGE and its legends are exaggerated in experimental autoimmune myasthenia gravis which is associated with nAChR dysfunction in the NMJ (Lim et al. 2021). AGEs are produced due to the glycation of DNA, proteins, and lipids that participates a critical role in the pathogenesis of metabolic and inflammatory disorders. Besides, nAChR is reduced in different metabolic disorders including diabetes mellitus. This may explain the susceptibility of patients with metabolic disorders to the risk of SARS-CoV-2 infection and COVID-19 severity. Therefore, activation of nAChR may attenuate AGE-mediated hyperinflammation in COVID-19 patients (Xie et al. 2020).
Similarly, soluble RAGE (sRAGE) plasma level is correlated with COVID-19 severity. A prospective cohort study included 164 COVID-19 patients compared to 23 non-COVID-19 pneumonia illustrating that high sRAGE plasma level was linked with the need for oxygen therapy and 30-day mortality (Lim et al. 2021). Hence, sRAGE is regarded as a potential biomarker that predicts COVID-19 severity and mortality. Furthermore, signal transducer and activator of transcription 3 (STAT3) are exaggerated in SARS-CoV-2 infection leading to hyperinflammation, thrombosis, and lung fibrosis (Al-Kuraishy et al. 2022f). In addition, STAT3 impairs the anti-viral immune response and the development of lymphopenia. In this, targeting STAT3 in COVID-19 may mitigate hyperinflammation and related fatal complications. Likewise, ADAM-metalloproteinase domain 17 (ADAM17) activates TNF-α and shedding of ACE2, with the facilitation of SARS-CoV-2 entry (Palau et al. 2020). It has been recognized that nAChR activators reduce COVID-19 severity through inhibition of the ADAM17 pathway. Thus, nAChR activators play a crucial role in the attenuation of inflammatory disorders in COVID-19 through the inhibition of STAT3 and ADAM17 pathways (Palau et al. 2020).
This greater activation is accompanied by greater activity in the vagus efferent nerve that can increase the release of acetylcholine through direct or indirect pathways and mediated by T lymphocytes (Al-kuraishy et al. 2022d). Thus, the components of this pathway act in a way to reduce the production and release of pro-inflammatory cytokines, such as TNF-α and IL-6, of the immune system. In this perspective, when the severity of the cytokine storm in the pathological scenario of COVID-19 and its involvement with MS is understood, the activation of the anti-inflammatory reflex mechanism can modulate the exaggeration of the effects of pro-inflammatory cytokines and reduce the symptom level (Al-Kuraishy et al. 2022a).
These verdicts pointed out that nAChR activators are intricate with diverse signaling pathways to reduce the risk of inflammatory disorders in COVID-19.
nAChR and endothelial dysfunction in COVID-19
SARS-CoV-2 infection primarily affects the vascular endothelium leading to ED and the development of coagulopathy. SARS-CoV-2 infects the endothelial cells directly due to abundant expression of ACE2 causing cellular injury and apoptosis with subsequent reduction of endothelial cells’ capacity to release anti-thrombotic factors (Gavriilaki et al. 2020). In addition, injury of pulmonary vascular endothelial cells, by direct cytopathic effects of SARS-CoV-2 or due to OS and hyperinflammation, leads to pulmonary microthrombosis, a marked feature of COVID-19 (Bonaventura et al. 2021). Notably, circulating endothelial cells and soluble intercellular adhesion molecule 1 levels are augmented in severely affected COVID-19 patients. ED is regarded as a risk factor for the development of microvascular dysfunction and immunothrombosis due to NET formation and platelet activation. It has been reported that nicotine improves endothelial function via the enhancement of the nAChR on the endothelial progenitor cells. As well, nAChR plays an essential role in the prevention of endothelial injury and rupture of atherosclerotic plaque. Besides, activation of non-neuronal endothelial nAChR improves inflammatory changes, OS, migration, and survival of endothelial cells (Vieira-Alves et al. 2020).
Moreover, the vascular endothelium is an active paracrine, endocrine, and autocrine organ that is indispensable for the regulation of vascular tone and the maintenance of vascular homeostasis. ED is a principal determinant of microvascular dysfunction by shifting the vascular equilibrium towards more vasoconstriction with subsequent organ ischemia, inflammation with associated tissue edema, and a pro-coagulant state (Varga et al. 2020). SARS-CoV-2 infection facilitates the induction of endothelins in several organs as a direct consequence of viral involvement and of the host inflammatory response. In addition, induction of apoptosis and pyroptosis might have an important role in endothelial cell injury in patients with COVID-19. COVID-19-endotheliitis might explain the systemic impaired microcirculatory function in different vascular beds and their clinical squeal in patients with COVID-19 (Simões et al. 2021; Elekhnawy et al. 2022). Vascular endothelins and consequent ED can be a principal determinant of microvascular failure, which would favor impaired perfusion, tissue hypoxia, and subsequent organ failure. In severe COVID-19, emerging data indicate a crucial role of widespread endothelins as a key player in provoking scattered microvascular disruption and eventually dysfunction of different organ systems (Hattori et al. 2022; Attallah et al. 2021). Calabretta et al. (2021) suggest that SARS-CoV-2 directly targets endothelial cells, promoting the release of pro-inflammatory and pro-thrombotic molecules. ED appears to be a crucial initiating step in the pathogenesis of the disease and its ensuing morbidity and mortality. Endothelins with the hyperproduction of cytokines lead to cytokine-releasing syndrome, hypercoagulability, and thrombotic microangiopathy which are hallmarks shared by COVID-19 and endothelial injury syndromes (Calabretta et al. 2021). Most importantly, endothelial-protective agents represent a promising and rational therapeutic strategy for COVID-19. As a unifying concept, heparanase inhibition, with the modulation of related pathways and other effects on endothelial stress responses, may thus be crucial in mediating anti-viral and anti-inflammatory activity (Calabretta et al. 2021; Elekhnawy and Negm 2022).
Furthermore, nAChR activators attenuate the development of immunothrombosis and coagulopathy through mitigation of hyperinflammation and OS which is involved in the propagation of ED and associated coagulopathy (Yilmaz et al. 2010; Al-kuraishy et al. 2022c). These verdicts suggested that nAChR activators may mitigate ED, OS, and associated coagulopathy in COVID-19.
nAChR and renin angiotensin system in COVID-19
SARS-CoV-2 infection provokes the downregulation of ACE2 which is involved in the metabolism of angiotensin II (AngII) to angiotensin 1–7 (Ang1–7). This interaction leads to the over-expression of pro-inflammatory AngII and reduction of anti-inflammatory Ang1-7 with subsequent development of ALI/ARDS. Dysregulation of the renin-angiotensin system (RAS) is connected with the development of OS and inflammation by increasing the expression of NADPH and pro-inflammatory cytokines, respectively (Alomair et al. 2022). Advanced circulating AngII level inhibits endogenous anti-oxidant capacity and may inhibit the expression of nAChR. It has been shown that nicotine affects RAS homeostasis through upregulation of the expression of ACE, AngII, and AT2R with downregulation of the expression of ACE2 and Ang1-7 (Batiha et al. 2022). However, an experimental study showed that activation of nAChR reduced AngII-induced vascular smooth cell senescence by promoting the expression of nicotinamide adenine dinucleotide (NAD) (Li et al. 2016). Therefore, dysregulation of RAS in COVID-19 could be a possible reason behind the reduction of nAChR activity. Thus, angiotensin receptor blockers (AT1R) may be beneficial in preventing OS and inflammatory reactions via upregulation of the anAChR pathway (Mogi et al. 2012).
nAChR and cytokine storm in COVID-19
Cytokine storm is developed in severe cases of COVID-19 due to exaggerated immune response and release of a huge amount of pro-inflammatory cytokines in parallel with the reduction of anti-inflammatory cytokine (Zanza et al. 2022). Cytokine storm is also developed due to cholinergic dysfunction and sympathetic over-activity in severe COVID-19. Direct inhibition of nAChR by SARS-CoV-2 induces hyperinflammation and immunopathogenesis (Tillman et al. 2022). In addition, pro-inflammatory cytokines which cross the blood–brain barrier (BBB) can inhibit the autonomic center and central anti-inflammatory nAChR with the development of neuroinflammation (Jiang et al. 2021). In turn, neuroinflammation and dysregulated central nAChR trigger sympathetic discharge with the development of a sympathetic storm (Yue et al. 2015). Notably, dysregulated nAChR on the immune cells by SARS-CoV-2 provokes the release of pro-inflammatory cytokines with the development of cytokine storm. These observations proposed that dysregulation of central and peripheral nAChR during SARS-CoV-2 infection could be the potential mechanism for the development of cytokine storm in COVID-19 (Yue et al. 2015; Khudhair et al. 2022).
On the other hand, nAChR activators can inhibit the expression of pro-inflammatory cytokines and the development of cytokine storm in COVID-19. Above all, nAChR has an important role in the regulation of immune response and the promulgation of inflammation. It decreases the expression of pro-inflammatory cytokines including MCP-1, TNF-α, IL-6, and IL-1β by inhibiting the recruitment of RNA polymerase II and macrophage activation. Diverse experimental studies confirmed that nAChR activators inhibit the expression and release of pro-inflammatory cytokines (Jiang et al. 2021). Likewise, nAChR activators attenuate the development of airway inflammatory disorders, ED, and lung inflammation. The nAChR activators inhibit the activation of different inflammatory signaling pathways including NLRP3 inflammasome, TLR4, HMBP1, NF-κB, and STAT3 that are involved in the development of cytokine storm. Amazingly, nAChR activators block the OS pathway which activates inflammatory signaling pathways like NF-κB and NLRP3 inflammasome (Liu et al. 2015). Similarly, nAChR activators inhibit abnormal and exaggerated immune through the inhibition of INF activation, thereby preventing the excessive release of pro-inflammatory cytokines. Thus, nAChR activators could be effective in the attenuation of SARS-CoV-2 infection-induced cytokine storm (Ren et al. 2018). Therefore, nAChR activators may alleviate oxidative stress and inflammatory changes as well as prevent the development of cytokine storm in COVID-19.
Role of nAChR agonists/antagonists in COVID-19
Varenicline
Different nAChR agonists had been repurposed in treating SARS-CoV-2 infection. Varenicline is a selective nAChR agonist; it affords a full agonist effect on the α7 subunit and a partial agonist effect on the other subunit of nAChR. Varenicline is an FDA drug for the treatment of smoking cessation and was recently hypothesized as a potential drug for the treatment of SARS-CoV-2 infection by inhibiting binding and proliferation processes (Nau et al. 2021). In silico study demonstrated that varenicline binds SARS-CoV-2 spike protein with higher affinity (Ramírez-Salinas et al. 2020). Topical administration of varenicline via the trans-nasal route may attenuate the transmission of SARS-CoV-2 at the site of infection through modulation of nAChR on the olfactory bulb. In vitro study confirmed that varenicline had an anti-SARS-CoV-2 effect without a negative effect on cell viability (Nau et al. 2021). Therefore, varenicline can inhibit transmission and pathogenesis of SARS-CoV-2 infection via inhibition of the interaction between SARS-CoV-2 and ACE2. Notoriously, varenicline can inhibit the proliferation of different variants of SARS-CoV-2 infection (Ramírez-Salinas et al. 2020). It has been revealed that varenicline attenuates inflammatory changes in mice with an experimental stroke model. Ho et al. demonstrated that varenicline could attenuate ischemic-reperfusion injury and associated inflammation in mice following testicular torsion via inhibition of the release of pro-inflammatory cytokines. In addition, varenicline reduces LPS-induced hyperinflammation by inhibiting macrophage activity. A prospective study that involved 100 smokers randomized into varenicline and nicotine replacement therapy for 3 months showed that varenicline replacement therapy was more effective in reduction of OS and ED (Ikonomidis et al. 2017). The activation of nAChRs by either endogenous or exogenous agonists is induced by opening the ion channel in the receptor, allowing the flow of cations, and results in a variety of biological responses. nAChR antagonists, such as α-neurotoxins, compete with typical agonists for binding, and their binding is restricted to nAChR α-subunits. Nicotine and other nicotinic cholinergic agonists are FDA-approved drugs for several pathologies including for smoking cessation and may reverse this binding, by competing for binding with the SARS-CoV-2 spike glycoprotein and promoting the activity of the cholinergic anti-inflammatory pathway (Seyedaghamiri et al. 2022). These verdicts suggest that varenicline in virtue of its anti-inflammatory and anti-oxidant effects with direct anti-SARS-CoV-2 effect could be effective in the management of COVID-19.
Memantine
Memantine is an FDA-proofed drug for the treatment of Alzheimer’s disease acts by blocking N-methyl-D-aspartate (NMDA) receptors (Zhu et al. 2020). It acts by different mechanisms; it blocks α7nAChR which may explain the worsening of cognitive function of patients with Alzheimer’s disease during the early phase of treatment. Later on, the α7nAChRs are rapidly upregulated, and this may explain the positive effect in the enhancement of the cognitive function of patients with Alzheimer’s disease. In vitro experiments illustrated that memantine inhibits the SARS-CoV-2 E protein which is necessary for pathogenesis and viral proliferation (Matsunaga et al. 2018). As well, NMDA antagonists like memantine and amantadine could be used as prophylactic agents and reduce neuropsychiatric complications in COVID-19 patients (Marinescu et al. 2020). Memantine also reduced HCoV‐OC43 replication in the CNS, indicating that it could be utilized as an anti-viral agent and improve neurological diseases. Besides coronavirus OC43, memantine has also been investigated regarding other neurotrophic viruses such as rabies and Japanese encephalitis virus (JEV), and while of limited effectiveness in a rabies challenge experiment, mildly extending survival time in mice, in JEV, survival time was significantly increased, inflammatory cell infiltrates, and intravascular cuffing were significantly reduced and mouse brain JEV content was less (Sun et al. 2019). There were several patients with multiple sclerosis (n = 10), Parkinson’s disease (n = 5), and cognitive impairment (n = 7), who were treated with adamantanes, 15 with amantadine, and seven with memantine; all the patients had SARS‐CoV‐2 documented by real‐time polymerase chain reaction nasopharyngeal swabs, undergoing 2-week quarantine since documented exposure, and none developed clinical disease (Rejdak and Grieb 2020). Hasanaic and Serdarevic (2020) illustrated that memantine could be effective against SARS-CoV-2 infection through the downregulation of ACE2 and reduction of glutamate excitotoxicity. ACE2 is protective rather than harmful as expected earlier in the COVID-19 pandemic. Recombinant ACE2 has protective effects against ALI/ARDS in COVID-19 through its anti-inflammatory effects (Wang et al. 2022). Children and female sex are less susceptible to SARS-CoV-2 infection due to higher expression of ACE2. As well, low expression of ACE2 in the old age group makes them more vulnerable to SARS-CoV-2 infection. Particularly, drugs that increase the expression of ACE2 were more effective in the management of COVID-19. Thus, the protective effect of memantine against SARS-CoV-2 infection could be independent of nAChR/ACE2 axis (Wang et al. 2022).
Further, memantine has a potent anti-inflammatory effect through the inhibition of the release of the pro-inflammatory cytokine and the development of cytokine storm. Likewise, memantine reduces the expression of NF-κB which can prevent microglial activation and associated neuroinflammation in COVID-19 (Jiménez-Jiménez et al. 2020). Despite these findings, clinical evidence for use of memantine in COVID-19 patients is limited. A questionnaire-based study that involved 10 patients with multiple sclerosis and 5 patients with Parkinson’s disease showed that those patients remain asymptomatic despite a positive PCR test for SARS-CoV-2 (Rejdak and Grieb 2020). So, memantine could be effective against the development of symptomatic COVID-19. As well, memantine can provide protection for olfactory neurocircuits, decrease the aggressiveness of NMDA receptor hyperactivity, alveolar protection, and regulate cytokine mechanisms (Marinescu et al. 2020).
Therefore, memantine can decrease COVID-19 severity through its anti-inflammatory effect and modulation of NMDA activity independent of nAChR blocking effect and ACE2 expression as shown in Fig. 2.

Effect of nicotine agonist on the pathogenesis of SARS-CoV-2 infection
Taken together, both memantine and varenicline seem to be effective in the prevention and treatment of SARS-CoV-2 infection through modulation of the anti-inflammatory CS. In this state, clinical trials for use of memantine and varenicline in COVID-19 patients are recommended.
Vagus nerve stimulation in COVID-19
It has been reported that non-invasive vagus nerve stimulation was approved by FDA for the treatment of cluster headache and migraine. Vagus nerve stimulation is effective in SARS-CoV-2 infection via suppression of the release of the pro-inflammatory cytokines. Remarkably, vagus nerve stimulation attenuates the development of cytokine storm and the need for invasive oxygen therapy and mechanical ventilation in severely affected COVID-19 patients (Mwamburi et al. 2017). Stimulation of the vagus nerve triggers bronchial parasympathetic reflex with induction of the anti-inflammatory cholinergic pathway in a nAChR-dependent manner (Mwamburi et al. 2017). Of interest, vagus nerve stimulation inhibits the expression and the release of IL-6, a pro-inflammatory cytokine involved in tissue injury and the development of cytokine storm (Koopman et al. 2016). Indeed, vagus nerve stimulation provokes the release of ACh which through activation of nAChR inhibits the expression of NF-κB with subsequent release of pro-inflammatory cytokines and propagation of cytokine storm (Mehta et al. 2020). It was observed that vagus nerve stimulation blocks fibrinolysis and coagulation activation in rats with endotoxemia. Thrombotic events are hallmarks associated with COVID-19 severity (Levy et al. 2021).
Moreover, famotidine activates the vagus nerve leading to a significant anti-inflammatory effect in COVID-19. Yang et al. (2022) illustrated that famotidine prevents the development of cytokine storm in COVID-19 via activation of the vagus nerve. Taken together, vagus nerve stimulation plays a critical role in preventing cytokine storm and coagulation disorders in COVID-19 (Yang et al. 2022).
Conclusions
The binding of SARS-CoV-2 with ACE2 leads to a series of inflammatory cellular events with cytopathic effects causing cell injury and hyperinflammation. Severe SARS-CoV-2 infection may lead to dysautonomia and sympathetic storm due to dysfunction of ANS. The CS forms a complex network doing different functions in the body due to its distribution in the neuronal and non-neuronal cells. ACh acts on two main types of receptors which are NRs and MRs. NRs induce T cell anergy with impairment of antigen-mediated signal transduction. Nicotine through activation of T cell NRs inhibits the expression and the release of pro-inflammatory cytokines. NRs play important anti-inflammatory effects while MRs promote inflammation by inducing the release of pro-inflammatory cytokines. SARS-CoV-2 infection can affect the morphological and functional stability of the cholinergic system through the disruption of cholinergic receptors. SARS-CoV-2 spike protein is similar to the neurotoxins that can bind nAChR in the ANS, NMJ, and brain. Interestingly, SARS-CoV-2 spike protein epitopes have higher similarity with neurotoxins affecting nAChR. Therefore, cholinergic receptors mainly nAChR and related cholinergic agonists may affect the pathogenesis of SARS-CoV-2 infection. Cholinergic dysfunction in COVID-19 due to dysregulation of nAChR by SARS-CoV-2 promotes central sympathetic drive with the development of the sympathetic storm. In turn, sympathetic storm triggers oxidative stress and hyperinflammation by increasing the generation of ROS and release of pro-inflammatory cytokines. As well, nAChR activators are intricate with diverse signaling pathways to reduce the risk of inflammatory disorders in COVID-19. In addition, nAChR activators may mitigate ED, OS, and associated coagulopathy in COVID-19. Similarly, nAChR activators may alleviate OS and inflammatory changes as well as prevent the development of cytokine storm in COVID-19. Thus, nAChR activator varenicline in virtue of its anti-inflammatory and anti-oxidant effects with direct anti-SARS-CoV-2 effect could be effective in the management of COVID-19. Remarkably, vagus nerve stimulation attenuates the development of cytokine storm and the need for invasive oxygen therapy and mechanical ventilation in severely affected COVID-19 patients. Indeed, vagus nerve stimulation blocks fibrinolysis and coagulation activation which are hallmarks associated with COVID-19 severity. Taken together, the nAChR activator and vagus nerve stimulation could be a possible therapeutic strategy for COVID-19.
Data availability
All data are available in the manuscript.
References
Abdallah BM, Boissy P, Tan Q, Dahlgaard J, Traustadottir GA, Kupisiewicz K, Laborda J, Delaisse J-M, Kassem M (2007) dlk1/FA1 regulates the function of human bone marrow mesenchymal stem cells by modulating gene expression of pro-inflammatory cytokines and immune response-related factors. J Biol Chem 282:7339–7351
Al-Kuraishy HM, Al-Fakhrany OM, Elekhnawy E, Al-Gareeb AI, Alorabi M, De Waard M, Albogami SM, Batiha GE-S (2022a) Traditional herbs against COVID-19: back to old weapons to combat the new pandemic. Eur J Med Res 27:1–11
Al-Kuraishy HM, Al-Gareeb AI, Al-Maiahy TJ, Alexiou A, Mukerjee N, Batiha GE-S (2022b) An insight into the placental growth factor (PlGf)/angii axis in COVID-19: a detrimental intersection. Biotechnol Gen Eng Rev, 1–20
Al-kuraishy HM, Al-Gareeb AI, Alkhuriji AF, Al-Megrin WAI, Elekhnawy E, Negm WA, De Waard M, Batiha GE-S (2022a) Investigation of the impact of rosuvastatin and telmisartan in doxorubicin-induced acute cardiotoxicity. Biomed Pharmacother 154:113673
Al-Kuraishy HM, Al-Gareeb AI, Almulaiky YQ, Cruz-Martins N, Batiha GE-S (2021a) Role of leukotriene pathway and montelukast in pulmonary and extrapulmonary manifestations of COVID-19: the enigmatic entity. Eur J Pharmacol 904:174196
Al-Kuraishy HM, Al-Gareeb AI, Alzahrani KJ, Alexiou A, Batiha GE-S (2021b) Niclosamide for COVID-19: bridging the gap. Mol Biol Rep 48:8195–8202
Al-kuraishy HM, Al-Gareeb AI, Elekhnawy E, Batiha GE-S (2022b) Dipyridamole and adenosinergic pathway in COVID-19: a juice or holy grail. Egypt J Med Human Gen 23:1–6
Al-Kuraishy HM, Al-Gareeb AI, Mostafa-Hedeab G, Dubey R, Prabhakar PK, Batiha G (2022e) COVID-19 and diabetes: will novel drugs for diabetes help in COVID-19? Curr Mol Pharmacol
Al-Kuraishy HM, Al-Gareeb AI, Mostafa-Hedeab G, Kasozi KI, Zirintunda G, Aslam A, Allahyani M, Welburn SC, Batiha GE-S (2021c) Effects of β-blockers on the sympathetic and cytokines storms in COVID-19. Front Immunol, 12
Al-Kuraishy HM, Al-Gareeb AI, Negm WA, Alexiou A, Batiha GE-S (2022f) Ursolic acid and SARS-CoV-2 infection: a new horizon and perspective. Inflammopharmacology, 1–9
Al-Kuraishy HM, Al-Gareeb AI, Qusti S, Alshammari EM, Gyebi GA, Batiha GE-S (2021d) COVID-19-induced dysautonomia: a menace of sympathetic storm. ASN neuro, 13 17590914211057635
Al-Kuraishy HM, Al-Gareeb AI, Welson NN, Batiha GE-S (2022g) Trimetazidine and COVID-19-induced acute cardiac injury: a missed key. Intl J Clin Pharm, 1–2
Al-Thomali AW, Al-Kuraishy HM, Al-Gareeb AI, Al-buhadiliy AK, De Waard M, Sabatier J-M, Khan Khalil AA, Saad HM, Batiha GE-S (2022) Role of neuropilin 1 in COVID-19 patients with acute ischemic stroke. Biomedicines 10:2032
Alexandris N, Lagoumintzis G, Chasapis CT, Leonidas DD, Papadopoulos GE, Tzartos SJ, Tsatsakis A, Eliopoulos E, Poulas K, Farsalinos K (2021) Nicotinic cholinergic system and COVID-19: in silico evaluation of nicotinic acetylcholine receptor agonists as potential therapeutic interventions. Toxicol Rep 8:73–83
Alomair BM, Al-Kuraishy HM, Al-Buhadily AK, Al-Gareeb AI, De Waard M, Elekhnawy E, Batiha GE-S (2022) Is sitagliptin effective for SARS-CoV-2 infection: false or true prophecy? Inflammopharmacology, 1–5
Attallah NG, El-Kadem AH, Negm WA, Elekhnawy E, El-Masry TA, Elmongy EI, Altwaijry N, Alanazi AS, Al-Hamoud GA, Ragab AE (2021) Promising antiviral activity of Agrimonia pilosa phytochemicals against severe acute respiratory syndrome coronavirus 2 supported with in vivo mice study. Pharmaceuticals 14:1313
Babalghith AO, Al-Kuraishy HM, Al-Gareeb AI, De Waard M, Sabatier J-M, Saad HM, Batiha GE-S (2022) The potential role of growth differentiation factor 15 in COVID-19: a corollary subjective effect or not? Diagnostics 12:2051
Bahloul M, Kharrat S, Makni S, Baccouche N, Ammar R, Eleuch A, Berrajah L, Chtourou A, Turki O, Chelly H (2022) Prognostic value of serum cholinesterase activity in severe SARS-CoV-2-infected patients requiring intensive care unit admission. Am J Trop Med Hyg, tpmd210934-tpmd210934
Batiha GE-S, Al-Gareeb AI, Elekhnawy E, Al-Kuraishy HM (2022) Potential role of lipoxin in the management of COVID-19: a narrative review. Inflammopharmacology, 1–9
Batiha GE-S, Gari A, Elshony N, Shaheen HM, Abubakar MB, Adeyemi SB, Al-Kuraishy HM (2021) Hypertension and its management in COVID-19 patients: the assorted view. Intl J Cardiol Cardiovasc Risk Prev 11:200121
Bekdash RA (2021) The cholinergic system, the adrenergic system and the neuropathology of Alzheimer’s disease. Int J Mol Sci 22:1273
Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, Dentali F, Montecucco F, Massberg S, Levi M (2021) Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol 21:319–329
Calabretta E, Moraleda JM, Iacobelli M, Jara R, Vlodavsky I, O’Gorman P, Pagliuca A, Mo C, Baron RM, Aghemo A (2021) COVID-19-induced endotheliitis: emerging evidence and possible therapeutic strategies. Br J Haematol 193:43–51
Courties A, Boussier J, Hadjadj J, Yatim N, Barnabei L, Péré H, Veyer D, Kernéis S, Carlier N, Pène F (2021) Regulation of the acetylcholine/α7nAChR anti-inflammatory pathway in COVID-19 patients. Sci Rep 11:1–8
Cox M, Bassi C, Saunders M, Nechanitzky R, Morgado-Palacin I, Zheng C, Mak T (2020) Beyond neurotransmission: acetylcholine in immunity and inflammation. J Intern Med 287:120–133
Cox MA, Duncan GS, Lin GH, Steinberg BE, Yu LX, Brenner D, Buckler LN, Elia AJ, Wakeham AC, Nieman B (2019) Choline acetyltransferase-expressing T cells are required to control chronic viral infection. Science 363:639–644
De Rosa MJ, Dionisio L, Agriello E, Bouzat C, del Carmen Esandi M (2009) Alpha 7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci 85:444–449
Deng Y, Guo S-L, Wei B, Gao X-C, Zhou Y-C, Li J-Q (2019) Activation of nicotinic acetylcholine α7 receptor attenuates progression of monocrotaline-induced pulmonary hypertension in rats by downregulating the NLRP3 inflammasome. Front Pharmacol 10:128
Ekins S, Mathews P, Saito EK, Diaz N, Naylor D, Chung J, McMurtray AM (2017) α7-Nicotinic acetylcholine receptor inhibition by indinavir: implications for cognitive dysfunction in treated HIV disease. AIDS 31:1083–1089
Elekhnawy E, Negm WA (2022) The potential application of probiotics for the prevention and treatment of COVID-19. Egypt J Med Human Gen 23:1–9
Elekhnawy E, Negm WA, El-Sherbeni SA, Zayed A (2022) Assessment of drugs administered in the Middle East as part of the COVID-19 management protocols. Inflammopharmacology, 1–20
Farsalinos K, Niaura R, Le Houezec J, Barbouni A, Tsatsakis A, Kouretas D, Vantarakis A, Poulas K (2020) Nicotine and SARS-CoV-2: COVID-19 may be a disease of the nicotinic cholinergic system. Toxicol Rep 7:658
Fujii YX, Fujigaya H, Moriwaki Y, Misawa H, Kasahara T, Grando SA, Kawashima K (2007) Enhanced serum antigen-specific IgG1 and proinflammatory cytokine production in nicotinic acetylcholine receptor α7 subunit gene knockout mice. J Neuroimmunol 189:69–74
Gavriilaki E, Anyfanti P, Gavriilaki M, Lazaridis A, Douma S, Gkaliagkousi E (2020) Endothelial dysfunction in COVID-19: lessons learned from coronaviruses. Curr Hypertens Rep 22:1–12
Goller KV, Moritz J, Ziemann J, Kohler C, Becker K, Hübner N-O, C.-G. S. Group (2022) Differences in clinical presentations of omicron infections with the lineages BA. 2 and BA. 5 in Mecklenburg-Western Pomerania, Germany, between April and July 2022. Viruses, 14, 2033.
Gori S, Alcain J, Vanzulli S, Moreno Ayala MA, Candolfi M, Jancic C, Geffner J, Vermeulen M, Salamone G (2019) Acetylcholine-treated murine dendritic cells promote inflammatory lung injury. PLoS One 14:e0212911
Halder N, Lal G (2021) Cholinergic system and its therapeutic importance in inflammation and autoimmunity. Front Immunol 12:660342
Hampel H, Mesulam M-M, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, Khachaturian AS, Vergallo A, Cavedo E, Snyder PJ (2018) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141:1917–1933
Hattori Y, Hattori K, Machida T, Matsuda N (2022) Vascular endotheliitis associated with infections: its pathogenetic role and therapeutic implication. Biochem Pharmacol, 114909
Hernandez CP, Morrow K, Velasco C, Wyczechowska DD, Naura AS, Rodriguez PC (2013) Effects of cigarette smoke extract on primary activated T cells. Cell Immunol 282:38–43
Hong GS, Zillekens A, Schneiker B, Pantelis D, de Jonge WJ, Schaefer N, Kalff JC, Wehner S (2019) Non-invasive transcutaneous auricular vagus nerve stimulation prevents postoperative ileus and endotoxemia in mice. Neurogastroenterol Motil 31:e13501
Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B (2006) Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 203:1623–1628
Ikonomidis I, Marinou M, Vlastos D, Kourea K, Andreadou I, Liarakos N, Triantafyllidi H, Pavlidis G, Tsougos E, Parissis J (2017) Effects of varenicline and nicotine replacement therapy on arterial elasticity, endothelial glycocalyx and oxidative stress during a 3-month smoking cessation program. Atherosclerosis 262:123–130
Jackisch R, Förster S, Kammerer M, Rothmaier AK, Ehret A, Zentner J, Feuerstein TJ (2009) Inhibitory potency of choline esterase inhibitors on acetylcholine release and choline esterase activity in fresh specimens of human and rat neocortex. J Alzheimers Dis 16:635–647
Jiang Y, Ma H, Wang X, Wang Z, Yang Y, Li L, Feng T (2021) Protective effect of the α7 nicotinic receptor agonist PNU-282987 on dopaminergic neurons against 6-hydroxydopamine, regulating anti-neuroinflammatory and the immune balance pathways in rat. Front Aging Neurosci 12:606927
Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agúndez JA (2020) Anti-inflammatory effects of amantadine and memantine: possible therapeutics for the treatment of COVID-19? J Person Med 10:217
Kabata H, Artis D (2019) Neuro-immune crosstalk and allergic inflammation. J Clin Investig 129:1475–1482
Kanauchi Y, Yamamoto T, Yoshida M, Zhang Y, Lee J, Hayashi S, Kadowaki M (2022) Cholinergic anti-inflammatory pathway ameliorates murine experimental Th2-type colitis by suppressing the migration of plasmacytoid dendritic cells. Sci Rep 12:1–18
Karam BS, Morris RS, Bramante CT, Puskarich M, Zolfaghari EJ, Lotfi-Emran S, Ingraham NE, Charles A, Odde DJ, Tignanelli CJ (2021) mTOR inhibition in COVID-19: a commentary and review of efficacy in RNA viruses. J Med Virol 93:1843–1846
Ke P, Shao BZ, Xu ZQ, Chen XW, Wei W, Liu C (2017) Activating α7 nicotinic acetylcholine receptor inhibits NLRP 3 inflammasome through regulation of β-arrestin-1. CNS Neurosci Ther 23:875–884
Khudhair DH, Al-Gareeb AI, Al-Kuraishy HM, El-Kadem AH, Elekhnawy E, Negm WA, Saber S, Cavalu S, Tirla A, Alotaibi SS (2022) Combination of vitamin C and curcumin safeguards against methotrexate-induced acute liver injury in mice by synergistic antioxidant effects. Front Med, 9
Kloc M, Ghobrial RM, Kubiak JZ (2020) How nicotine can inhibit cytokine storm in the lungs and prevent or lessen the severity of COVID-19 infection? Immunol Lett 224:28–29
Koopman FA, Chavan SS, Miljko S, Grazio S, Sokolovic S, Schuurman PR, Mehta AD, Levine YA, Faltys M, Zitnik R (2016) Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Natl Acad Sci 113:8284–8289
Kopańska M, Batoryna M, Bartman P, Szczygielski J, Banaś-Ząbczyk A (2022) Disorders of the cholinergic system in COVID-19 era—a review of the latest research. Int J Mol Sci 23:672
Koval L, Kalashnyk O, Lykhmus O, Skok M (2018) α7 nicotinic acetylcholine receptors are involved in suppression of the antibody immune response. J Neuroimmunol 318:8–14
Kutukova N, Nazarov P (2020) Modulation of functional activity of mast cells by cholinergic agents. Russ J Immunol 12:335–341
Lagoumintzis G, Chasapis CT, Alexandris N, Kouretas D, Tzartos S, Eliopoulos E, Farsalinos K, Poulas K (2021) Nicotinic cholinergic system and COVID-19: in silico identification of interactions between α7 nicotinic acetylcholine receptor and the cryptic epitopes of SARS-Co-V and SARS-CoV-2 spike glycoproteins. Food Chem Toxicol 149:112009
Laguna Merced A (2021) Vasorelaxant role of antigen-experienced CD8+ T cells during acute viral infection
Leake I (2019) Choline uptake is vital for IL-1β-driven inflammation. Nat Rev Rheumatol 15:320–320
Leite HR, de Oliveira-Lima OC, de Melo Pereira L, de Moura Oliveira VE, Prado VF, Prado MAM, Pereira GS, Massensini AR (2016) Vesicular acetylcholine transporter knock down-mice are more susceptible to inflammation, c-Fos expression and sickness behavior induced by lipopolysaccharide. Brain Behav Immun 57:282–292
Levy JH, Iba T, Olson LB, Corey KM, Ghadimi K, Connors JM (2021) COVID-19: thrombosis, thromboinflammation, and anticoagulation considerations. Int J Lab Hematol 43:29–35
Lian M, Hueffer K, Weltzin M (2022Aug) Interactions between the rabies virus and nicotinic acetylcholine receptors: a potential role in rabies virus induced behavior modifications. Heliyon 28:e10434
Li D-J, Huang F, Ni M, Fu H, Zhang L-S, Shen F-M (2016) α7 nicotinic acetylcholine receptor relieves angiotensin II-induced senescence in vascular smooth muscle cells by raising nicotinamide adenine dinucleotide-dependent SIRT1 activity. Arterioscler Thromb Vasc Biol 36:1566–1576
Lim A, Radujkovic A, Weigand MA, Merle U (2021) Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COVID-19 disease severity and indicator of the need for mechanical ventilation, ARDS and mortality. Ann Intensive Care 11:1–13
Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor DH, Ji J, Fan W, Huang Z, Hu J (2015) Activation of α7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: implications for Parkinson’s disease. Neuropharmacology 91:87–96
Lo D, Kennedy JL, Kurten RC, Panettieri RA, Koziol-White CJ (2018) Modulation of airway hyperresponsiveness by rhinovirus exposure. Respir Res 19:1–11
Luna-Muschi A, Borges IC, de Faria E, Barboza AS, Maia FL, Leme MD, Guedes AR, Mendes-Correa MC, Kallas EG, Segurado AC (2022) Clinical features of COVID-19 by SARS-CoV-2 Gamma variant: a prospective cohort study of vaccinated and unvaccinated healthcare workers. J Infect 84:248–288
Marinescu I, Marinescu D, Mogoantă L, Efrem IC, Stovicek PO (2020) SARS-CoV-2 infection in patients with serious mental illness and possible benefits of prophylaxis with Memantine and Amantadine. Rom J Morphol Embryol 61:1007
Matsunaga S, Kishi T, Nomura I, Sakuma K, Okuya M, Ikuta T, Iwata N (2018) The efficacy and safety of memantine for the treatment of Alzheimer’s disease. Expert Opin Drug Saf 17:1053–1061
Mazloom R (2020) Feasibility of therapeutic effects of the cholinergic anti-inflammatory pathway on COVID-19 symptoms. J Neuroimmune Pharmacol 15:165–166
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. The Lancet 395:1033–1034
Mendez-Enriquez E, Alvarado-Vazquez PA, Abma W, Simonson OE, Rodin S, Feyerabend TB, Rodewald HR, Malinovschi A, Janson C, Adner M (2021) Mast cell-derived serotonin enhances methacholine-induced airway hyperresponsiveness in house dust mite-induced experimental asthma. Allergy 76:2057–2069
Mehranfard D, Speth RC. Cholinergic anti-inflammatory pathway and COVID-19. BioImpacts: BI. 2022;12(2):171
Mizrachi T, Marsha O, Brusin K, Ben-David Y, Thakur GA, Vaknin-Dembinsky A, Treinin M, Brenner T (2021) Suppression of neuroinflammation by an allosteric agonist and positive allosteric modulator of the α7 nicotinic acetylcholine receptor GAT107. J Neuroinflammation 18:1–14
Mogi M, Iwanami J, Horiuchi M (2012) Roles of brain angiotensin II in cognitive function and dementia. International Journal of Hypertension, 2012
Moran SP, Maksymetz J, Conn PJ (2019) Targeting muscarinic acetylcholine receptors for the treatment of psychiatric and neurological disorders. Trends Pharmacol Sci 40:1006–1020
Mostafa-Hedeab G, Al-Kuraishy HM, Al-Gareeb AI, Welson NN, Batiha GE-S, Conte-Junior CA (2022) Selinexor and COVID-19: the neglected warden. Front Pharmacol, 13
Moubarak M, Kasozi KI, Hetta HF, Shaheen HM, Rauf A, Al-Kuraishy HM, Qusti S, Alshammari EM, Ayikobua ET, Ssempijja F (2021) The rise of SARS-CoV-2 variants and the role of convalescent plasma therapy for management of infections. Life 11:734
Mwamburi M, Liebler EJ, Tenaglia AT (2017) Cost-effectiveness of gammaCore (non-invasive vagus nerve stimulation) for acute treatment of episodic cluster headache. Am J Manag Care 23:S300–S306
Nakajima K, Abe T, Saji R, Ogawa F, Taniguchi H, Yamaguchi K, Sakai K, Nakagawa T, Matsumura R, Oi Y (2021) Serum cholinesterase associated with COVID-19 pneumonia severity and mortality. J Infect 82:282–327
Nau J, Luthra P, Lanzer K, Szaba F, Cookenham T, Carlson E (2021) Varenicline prevents SARS-CoV-2 infection in vitro and in rhesus macaques. bioRxiv
Nizri E, Hamra-Amitay Y, Sicsic C, Lavon I, Brenner T (2006) Anti-inflammatory properties of cholinergic up-regulation: a new role for acetylcholinesterase inhibitors. Neuropharmacology 50:540–547
Nouri-Shirazi M, Kahlden C, Nishino P, Guinet E (2015) Nicotine exposure alters the mRNA expression of notch ligands in dendritic cells and their response to Th1-/Th2-promoting stimuli. Scand J Immunol 81:110–120
Pąchalska M, Góral-Półrola J, Chojnowska-Ćwiąkała I (2021) Effect of individually-tailored TDCS and symbolic art therapy for chronic associative prosopagnosia after infection by SARS-COV-2, neuroCOVID-19 and ischemic stroke. Acta Neuropsychologica, 19
Palau V, Riera M, Soler MJ (2020) ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol Dial Transplant 35:1071–1072
Pan S, Wu Y-J, Zhang S-S, Cheng X-P, Olatunji OJ, Yin Q, Zuo J (2021) The effect of α7nAChR signaling on T cells and macrophages and their clinical implication in the treatment of rheumatic diseases. Neurochem Res, 1–14
Patel H, McIntire J, Ryan S, Dunah A, Loring R (2017) Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-κB pathway and activation of the Nrf2 pathway. J Neuroinflammation 14:1–15
Pawełczyk M, Kowalski ML (2017) The role of human parainfluenza virus infections in the immunopathology of the respiratory tract. Curr Allergy Asthma Rep 17:1–10
Perrone A, Giovino A, Benny J, Martinelli F (2020) Advanced glycation end products (AGEs): biochemistry, signaling, analytical methods, and epigenetic effects. Oxidative Med Cell Longevity, 2020
Profita M, Bonanno A, Montalbano AM, Albano GD, Riccobono L, Siena L, Ferraro M, Casarosa P, Pieper MP, Gjomarkaj M (2012) β2 long-acting and anticholinergic drugs control TGF-β1-mediated neutrophilic inflammation in COPD. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1822, 1079–1089
Ramírez-Salinas GL, Martínez-Archundia M, Correa-Basurto J, García-Machorro J (2020) Repositioning of ligands that target the spike glycoprotein as potential drugs for SARS-CoV-2 in an in silico study. Molecules 25:5615
Rejdak K, Grieb P (2020) Adamantanes might be protective from COVID-19 in patients with neurological diseases: multiple sclerosis, parkinsonism and cognitive impairment. Multiple Sclerosis and Related Disorders 42:102163
Ren C, Li X-H, Wang S-B, Wang L-X, Dong N, Wu Y, Yao Y-M (2018) Activation of central alpha 7 nicotinic acetylcholine receptor reverses suppressed immune function of T lymphocytes and protects against sepsis lethality. Int J Biol Sci 14:748
Sajjanar B, Saxena S, Bisht D, Singh AK, Reddy GM, Singh R, Singh R, Kumar S (2016) Effect of nicotinic acetylcholine receptor alpha 1 (nAChRα1) peptides on rabies virus infection in neuronal cells. Neuropeptides 57:59–64
Seyedabadi M, Rahimian R, Ghia J-E (2018) The role of alpha7 nicotinic acetylcholine receptors in inflammatory bowel disease: involvement of different cellular pathways. Expert Opin Ther Targets 22:161–176
Seyedaghamiri F, Hosseini L, Kazmi S, Mahmoudi J, Shanehbandi D, Ebrahimi-Kalan A, Rahbarghazi R, Sadigh-Eteghad S, Farhoudi M (2022) Varenicline improves cognitive impairment in a mouse model of mPFC ischemia: the possible roles of inflammation, apoptosis, and synaptic factors. Brain Res Bull 181:36–45
Simões JLB, de Araújo JB, Bagatini MD (2021) Anti-inflammatory therapy by cholinergic and purinergic modulation in multiple sclerosis associated with SARS-CoV-2 infection. Mol Neurobiol 58:5090–5111
Snider SA, Margison KD, Ghorbani P, LeBlond ND, O’Dwyer C, Nunes JR, Nguyen T, Xu H, Bennett SA, Fullerton MD (2018) Choline transport links macrophage phospholipid metabolism and inflammation. J Biol Chem 293:11600–11611
Sun L, Zhou M, Liu C, Tang Y, Xiao K, Dai J, Gao Z, Siew L, Cao G, Wu X (2019) Memantine can relieve the neuronal impairment caused by neurotropic virus infection. J Med Virol 91:935–940
Tillman TS, Xu Y, Tang P (2022) Impact of SARS-CoV-2 spike protein on α7 nicotinic acetylcholine receptor in cells. Biophys J 121:243a–244a
Tizabi Y, Getachew B, Copeland RL, Aschner M (2020) Nicotine and the nicotinic cholinergic system in COVID-19. FEBS J 287:3656–3663
Tracey KJ (2009) Reflex control of immunity. Nat Rev Immunol 9:418–428
Valdés-Ferrer SI, Crispín JC, Belaunzarán-Zamudio PF, Rodríguez-Osorio CA, Cacho-Díaz B, Alcocer-Varela J, Cantú-Brito C, Sierra-Madero J (2017) Add-on Pyridostigmine enhances CD4+ T-cell recovery in HIV-1-infected immunological non-responders: a proof-of-concept study. Front Immunol 8:1301
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H (2020) Endothelial cell infection and endotheliitis in COVID-19. The Lancet 395:1417–1418
Vieira-Alves I, Coimbra-Campos L, Sancho M, Da Silva RF, Cortes SF, Lemos VS (2020) Role of the α7 nicotinic acetylcholine receptor in the pathophysiology of atherosclerosis. Front Physiol, 1719
Wang Z, Xiang L, Lin F, Cai Z, Ruan H, Wang J, Liang J, Wang F, Lu M, Cui W (2022) Inhaled ACE2-engineered microfluidic microsphere for intratracheal neutralization of COVID-19 and calming of the cytokine storm. Matter 5:336–362
Witayateeraporn W, Arunrungvichian K, Pothongsrisit S, Doungchawee J, Vajragupta O, Pongrakhananon V (2020) α7-Nicotinic acetylcholine receptor antagonist QND7 suppresses non-small cell lung cancer cell proliferation and migration via inhibition of Akt/mTOR signaling. Biochem Biophys Res Commun 521:977–983
Wu C-H, Inoue T, Nakamura Y, Uni R, Hasegawa S, Maekawa H, Sugahara M, Wada Y, Tanaka T, Nangaku M (2022) Activation of α7 nicotinic acetylcholine receptors attenuates monocyte–endothelial adhesion through FUT7 inhibition. Biochem Biophys Res Commun 590:89–96
Xie H, Yepuri N, Meng Q, Dhawan R, Leech CA, Chepurny OG, Holz GG, Cooney RN (2020) Therapeutic potential of α7 nicotinic acetylcholine receptor agonists to combat obesity, diabetes, and inflammation. Rev Endocr Metab Disord 21:431–447
Yang H, George SJ, Thompson DA, Silverman HA, Tsaava T, Tynan A, Pavlov VA, Chang EH, Andersson U, Brines M (2022) Famotidine activates the vagus nerve inflammatory reflex to attenuate cytokine storm. Mol Med 28:1–13
Yilmaz Z, Ozarda Y, Cansev M, Eralp O, Kocaturk M, Ulus IH (2010) Choline or CDP-choline attenuates coagulation abnormalities and prevents the development of acute disseminated intravascular coagulation in dogs during endotoxemia. Blood Coag Fibrinol 21:339–348
Youssef ME, Moustafa Y, Abdelrazek H (2021) Molecular mechanisms of α7-nAchR-mediated anti-inflammatory effects. Indian J Physiol Pharmacol 64:158–173
Yuan M, Han B, Xia Y, Liu Y, Wang C, Zhang C (2019) Augmentation of peripheral lymphocyte-derived cholinergic activity in patients with acute ischemic stroke. BMC Neurol 19:1–9
Yue Y, Liu R, Cheng W, Hu Y, Li J, Pan X, Peng J, Zhang P (2015) GTS-21 attenuates lipopolysaccharide-induced inflammatory cytokine production in vitro by modulating the Akt and NF-κB signaling pathway through the α7 nicotinic acetylcholine receptor. Int Immunopharmacol 29:504–512
Zanza C, Romenskaya T, Manetti AC, Franceschi F, La Russa R, Bertozzi G, Maiese A, Savioli G, Volonnino G, Longhitano Y (2022) Cytokine storm in COVID-19: immunopathogenesis and therapy. Medicina 58:144
Zhu C-C, Fu S-Y, Chen Y-X, Li L, Mao R-L, Wang J-Z, Liu R, Liu Y, Wang X-C (2020) Advances in drug therapy for Alzheimer’s disease. Curr Med Sci 40:999–1008
Funding
French Agence Nationale de la Recherche and the Région Pays de la Loire provided financial support on COVID-19 research (ANR Flash COVID-19 call — name: CoV2-E-TARGET — grant number: 2020 07132).
Author information
Authors and Affiliations
Contributions
H. M. A., A. I. A., E. E., and G. E. B. contributed to the study conception and design. Material preparation, data collection and analysis, and writing the first draft were performed by H. M. A., A. I. A., E. E., and G. E. B. E. H. N., S. M. A., M. A., and M. D. W. revised the manuscript. E. H. N., H. M. A., A. I. A., E. E., S. M. A., M. A., G. B. E., and M. D. W. read and approved the final manuscript. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nadwa, E.H., Al-Kuraishy, H.M., Al-Gareeb, A.I. et al. Cholinergic dysfunction in COVID-19: frantic search and hoping for the best. Naunyn-Schmiedeberg's Arch Pharmacol 396, 453–468 (2023). https://doi.org/10.1007/s00210-022-02346-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-022-02346-9