
Abstract
Physical exercise yields beneficial effects on all types of muscle cells, which are essential for the maintenance of cardiovascular homeostasis and good blood circulation. Daily moderate exercise increases systemic antioxidative capacity, which can lead to the prevention of the onset and progression of oxidative stress-related diseases. Therefore, exercise is now widely accepted as one of the best therapeutic strategies for the treatment of ischemic (hypoxic) diseases. Canonical transient receptor potential (TRPC) proteins are non-selective cation channels activated by mechanical stress and/or stimulation of phospholipase C-coupled surface receptors. TRPC channels, especially diacylglycerol-activated TRPC channels (TRPC3 and TRPC6; TRPC3/6), play a key role in the development of cardiovascular remodeling. We have recently found that physical interaction between TRPC3 and NADPH oxidase (Nox) 2 under hypoxic stress promotes Nox2-dependent reactive oxygen species (ROS) production and mediates rodent cardiac plasticity, and inhibition of the TRPC3-Nox2 protein complex results in enhancement of myocardial compliance and flexibility similar to that observed in exercise-treated hearts. In this review, we describe current understanding of the roles of TRPC channels in striated muscle (patho)physiology and propose that targeting TRPC-based protein complexes could be a new strategy to imitate exercise therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Moderate exercise produces pleiotropic effects on health, by stimulating systemic blood circulation and energy metabolism and refreshing the mind. Exercise is also known to reduce cardiovascular risk factors related to many pathological conditions. Although primarily affecting skeletal muscle functions such as increasing the rate of glucose uptake, physical exercise gives rise to several beneficial effects on other remote organs through blood circulation-dependent organ-organ interactions. A critical risk factor for the development of pathological conditions is the increase of oxidative stress caused by overproduction of reactive oxygen species (ROS). ROS are mainly produced as a byproduct of uncoupling of the respiratory chain during mitochondrial dysfunction, or enzymatically, by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) proteins. Physical exercise is well known to induce ROS production, but it also triggers upregulation of antioxidative systems leading to acquisition of resilience against many diseases [65]. We have also revealed that long-term physical exercise reduces the expression of Nox2 in the heart [69]. This review introduces the (patho)physiological roles of Nox and canonical transient receptor potential (TRPC) proteins in skeletal muscles, mainly focusing on the mechanisms common to other muscular organs (i.e., the cardiovascular system), and proposes a novel strategy for imitating exercise therapy.
Roles of Nox proteins in skeletal muscle
There are seven isoforms of Nox proteins. Nox1, Nox2 and Nox4 are reportedly expressed in skeletal muscle [15]. Nox2 and Nox4 are predominantly expressed in the heart. In resting conditions, Nox2 only interacts with the p22phox subunit of the NADPH oxidase, which promotes the expression of Nox2 by preventing its proteasomal degradation. Upon cellular activation, the other cytoplasmic subunits p67phox, p40phox and p47phox, and the small G protein Rac1 are recruited and activate Nox2 protein. Among the cytoplasmic subunits, p47phox is the main regulator of the Nox2 complex formation. To form a complex, phosphorylation of p47phox is necessary. Phosphorylation of p47phox is reported to be mediated by protein kinase C, mitogen-activated protein kinases and p21-activated kinase [13]. The importance of Nox proteins in skeletal muscle is highlighted by their role in contraction-induced ROS production [25]. It is well known that muscle contraction produces ROS and reactive nitrogen species [26, 59]. ROS production plays important roles in skeletal muscle, for example, increasing the activities of antioxidant defense enzymes, force production, glucose uptake and insulin signaling [25, 45]. Application of hydrogen peroxide (H2O2) induces a similar gene expression profile to that of contracting a skeletal muscle, suggesting that muscle contraction signals are mainly conveyed by H2O2 [46]. The regulation and physiological relevance of Nox proteins in skeletal muscle have been reviewed in detail elsewhere [15, 27].
Roles of TRPC channels in skeletal muscle
The trp gene was first identified in 1989 as a causative gene mutant affecting phototransduction in Drosophila [49]. Twenty-eight mammalian TRP homologues have been identified, and these are subdivided into six subfamilies based on their genetic and functional similarities: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin) and TRPA (ankyrin). TRP proteins commonly possess six transmembrane domains and a preserved 25-amino acid sequence called the ‘TRP domain’. There are several reports demonstrating the involvement of TRP channels in exercised skeletal muscles. TRPM8 activation enhances exercise endurance and reduces blood lactic acid and triglycerides by upregulating uncoupling protein 1 (UCP1) and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) in skeletal muscles [36]. TRPV1 activation by dietary capsaicin increases the proportion of oxidative fibers, promotes mitochondrial biogenesis, enhances exercise endurance and prevents high-fat diet-induced metabolic disorders via an increase of PGC1α expression [41]. TRPV1 is reportedly activated by peroxynitrite, a reaction product of nitric oxide and superoxide, and mediates overload-induced skeletal muscle hypertrophy [23, 24]. These TRP channels are likely to function downstream of mechano-signal transduction in skeletal muscle contraction.
The TRPC family proteins, comprising seven mammalian homologues (TRPC1–TRPC7), are believed to be molecular candidates for receptor-activated cation channels (RACCs) [49]. TRPC1 was first suggested as the molecular entity of store-operated Ca2+ entry (SOCE) [38, 78, 95, 96]. TRPC1 contributes to the coordination of elementary Ca2+ signaling events through promoting functional coupling between the endoplasmic reticulum (ER) and the plasma membrane in receptor-induced Ca2+ signaling [50]. Recent findings indicate that TRPC proteins have two important roles: one is to act as a critical component of stretch-activated or store-operated Ca2+-permeable channels, and the other is to act as a signaling platform to amplify receptor-activated Ca2+ signaling via interacting with intracellular signaling molecules [52, 54]. Because of their universal activation mechanism in many cell types, TRPC channels play important roles in basic cellular responses including proliferation, differentiation and death in response to various environmental stimuli. TRPC channels are also linked to physical stimulation such as mechanical stretch, and hypoxia and oxidative stress [62]. TRPC1 and TRPC6 are suggested to be components of the tarantula toxin-sensitive mechanosensitive cation channel [42, 70]. In addition, intracellular lipid mediators such as diacylglycerol and 20-hydroxyeicosatetraenoic acid (20-HETE) mediate activation of TRPC6 induced by oxidative stress [77] and mechanical stretch [22]. Considering the role of TRPC3/6 heterotetramer channels in myocyte hypertrophy, the TRPC6 protein signaling complex, including TRPC1 and TRPC3, may function as a mechanical signal transducer in striated muscle cells (Fig. 1).

Canonical transient receptor potential (TRPC) channels function as mechanosignal transducers to Nox proteins during skeletal muscle contraction. Nox-mediated reactive oxygen species (ROS) production plays critical roles in skeletal muscle homeostasis
TRPC1
Vandebrouck et al. first demonstrated that TRPC1/2/3/4 and TRPC6 were detected both at the transcript and protein levels in skeletal muscle cells, with TRPC2 and TRPC3 being found in intracellular compartments, and TRPC1/4 and TRPC6 at the plasma membrane [75]. The abnormal Ca2+ influx observed in adult skeletal muscle fibers from dystrophic (mdx) mice was partially mediated by TRPC channels [75]. Later, the same group demonstrated that TRPC1 is associated with the PSD95-discs large-zonula occludens protein (PDZ) domain-possessing scaffold proteins α1-syntrophin and dystrophin and suggested that the mechanosensitive activation of TRPC1 is supported by these interactions (Fig. 1) [74]. Stiber et al. demonstrated that Homer1 determines the localization and activation timing by mechanical stretch of TRPC1 channels. Therefore, the absence of Homer1 induces spontaneous TRPC1 activation and Ca2+ overload which results in myopathy [71]. Another group demonstrated that protein levels of TRPC1 and Caveolin-3 (Cav3) were increased in skeletal muscle from mdx mice and that TRPC1 was activated by ROS in an Src kinase-dependent manner (Fig. 2) [18].

Canonical transient receptor potential (TRPC) channel function in striated muscle cells. TRPC1 channel activity is regulated via interaction with the dystrophin-associated protein complex (DAPC). TRPC1 also functions as a Ca2+ leak channel in the sarcoplasmic reticulum. TRPC3 channels are localized in T-tubules
TRPC1 mediates SOCE in the C2C12 myoblast cell line. siRNA-mediated knockdown of TRPC1 suppressed myotube formation of C2C12 cells. Interestingly, TRPC1 mRNA expression transiently increased immediately after the onset of differentiation (1 day) and returned to the basal level 4–6 days after the start of differentiation. Increased TRPC1 activity was correlated with the activity of calpain [40]. TRPC1 proteins were also transiently upregulated 24 h after the induction of differentiation and returned to the basal level at 72 h. Formigli et al. also demonstrated that TRPC1 is not only activated by store depletion, but also mechanical stretch, in C2C12 cells. Mechanical stretch facilitates myoblast differentiation in a sphingosine 1-phosphate (S1P)-dependent manner [12]. S1P application to C2C12 cells markedly increased TRPC1 expression, concomitant with an increase in stretch-activated channel expression [17]. S1P-mediated activation of TRPC1 induces m-calpain activity and subsequent expression of connexin43 [47]. TRPC1 overexpression in C2C12 cells increased the rate and amplitude of SOCE. Interestingly, in those cells levels of stromal interaction molecule 1 (STIM1) and sarcoendoplasmic reticulum calcium ATPase (SERCA) expression were lower, which resulted in insufficient Ca2+ clearance after the depolarization-induced Ca2+ increase. Furthermore, Ca2+ dyshomeostasis induced by TRPC1 overexpression attenuated the nuclear factor of activated T cells (NFAT) signaling pathway and myotube formation [57]. In human myoblasts, TRPC1 downregulation caused by siRNA expression or overexpression of a dominant negative mutant clearly suppressed SOCE, myogenic driver MEF2 expression and fusion of myoblasts into myotubes [3]. TRPC1 activation is regulated by STIM1L, a long isoform of STIM1 [2]. TRPC1 forms a heterotetramer with TRPC3 via interaction at the ankyrin repeat of TRPC3. The short protein comprising the N-terminal 37 amino acids of TRPC3 can inhibit TRPC1-TRPC3 heteromultimerization, which reduces resting cytosolic Ca2+ in murine skeletal myotubes [82].
TRPC1 is highly expressed in skeletal muscle stem cell satellite cells. Fibroblast growth factor 2 (FGF2) treatment increased the intracellular Ca2+ concentration and nuclear accumulation of NFATc3 and NFATc2 in these cells. The broad TRPC blocker SKF-96365 inhibits these responses [39]. Therefore, TRPC1 plays a critical role in the regeneration process following muscle injury, by contributing to satellite cell activation.
A TRPC1 knockout (TRPC1−/−) mouse showed decreased endurance for physical activity. Histological analysis showed a reduced cross-sectional area of skeletal muscle fibers and myofibrillar protein content. Isolated muscle fibers from TRPC1+/+ mice showed instances of small, spontaneous activity which are absent in those from TRPC1−/− mice. In primary muscle fibers, TRPC1 does not participate in store-operated or stretch-activated calcium influx. However, there is a marked reduction of force production in both the soleus and extensor digitorum longus (EDL) muscles of TRPC1−/− mice. Furthermore, muscle fatigue is accelerated in the soleus and EDL muscles from TRPC1−/− mice compared with those from TRPC1+/+ mice [88]. TRPC1-YFP transgenic mice also exhibited no significant differences in the electrical properties of skeletal muscle fibers. However, calcium clearance after repetitive contractile stimuli was delayed in TRPC1−/− mice, and responses to cyclopiazonic acid were enhanced, suggesting that TRPC1 functions in the intracellular Ca2+ store membrane as a calcium leak channel (Fig. 2) [7]. In mdx mice, the diaphragm muscle had higher expression of TRPC1 compared with the sternomastoid and limb muscles. The levels of TRPC1 expression in mdx mice correlate well with the degree of pathological changes observed in skeletal muscles, i.e., the diaphragm shows the most severe pathological phenotype [43]. In a model of cardiotoxin-induced muscle injury, TRPC1−/− mice showed significant hypotrophy and increased proportions of centrally nucleated muscle fibers. It is suggested that TRPC1−/− myoblasts cannot correctly differentiate into myotubes because myogenic factors are downregulated. These phenotypes of TRPC1-depleted skeletal muscle were attributed to the suppression of the phosphatidylinositol-3-kinase-mammalian target of rapamycin (PI3K-mTOR) pathway. The TRPC1-mediated Ca2+ increase is critical for the activation of PI3K [89]. TRPC1−/− muscle is resistant to repeated eccentric contraction. This phenotype is similar to that observed in muscle treated with streptomycin, a stretch-activated channel inhibitor. Although force reduction caused by repeated eccentric contraction was not affected by the absence of TRPC1, the loss of sarcolemmal proteins and reduced resting stiffness were suppressed by both TRPC1 knockout and streptomycin treatment, suggesting that TRPC1 contributes to stretch-activated Ca2+ entry in skeletal muscle [90].
The mechanical unloading seen in long-term bed rest patients and astronauts evokes muscle loss via oxidative stress. Ca2+ influx is critical for myoblast proliferation and controls exit from the G2/M phase of the cell cycle. Simulated microgravity, an in vitro model of mechanical unloading in space, reduced the expression of TRPC1 [6]. Hind limb unloading induces soleus muscle atrophy and reduction of tetanic force. During unloading, TRPC1 protein expression was reduced [84, 91] and recovered 14 days after reloading. The recovery of TRPC1 expression was preceded by and dependent on NFAT pathway activation. siRNA-mediated TRPC1 downregulation in vivo attenuated skeletal muscle regrowth of the soleus muscle, manifested by reduced cross-sectional area and type I myosin heavy chain expression [84]. These results suggest that proper mechanical signaling is important for skeletal muscle homeostasis, and TRPC1 plays a critical role in this.
Consistent with the accumulated data from the mdx mouse model, human myoblasts isolated from Duchenne muscular dystrophy (DMD) patients showed a significant increase in SOCE but no increase in levels of TRPC1, Stim1 or Orai1. However, pharmacological inhibition of phospholipase C or protein kinase C, which are components of a signaling complex with TRPC1, restores SOCE to the normal level [19].
Omega-3 fatty acid administration slows DMD progression, partly due to a reduction in TRPC1 expression [44].
Step up/down exercise involves concentric contraction in the right vastus lateralis (VL) muscle and eccentric contraction in the left VL muscle. Satellite cells in the left VL muscle only are activated, as indicated by an increase of expression of hepatocyte growth factor and MyoD, a myogenic transcription factor. As stated above, TRPC1 likely plays an important role in satellite cell activation. Consistent with this, TRPC1 expression was significantly increased in satellite cells of the left VL muscle, suggesting that eccentric but not concentric exercise activates satellite cells in a TRPC1-dependent manner [21].
TRPC3
TRPC3 expression is relatively high in skeletal muscle tissue [32]. TRPC3 mRNA expression was increased after 3–4 days of differentiation in the C2C12 myoblast cell line [10, 40]. In the model of hind limb unloading, TRPC3 expression was lower in the early phase after the reloading process [91], suggesting that TRPC3 is downregulated during the regeneration process, possibly because undifferentiated myoblasts have lower levels of TRPC3 expression. TRPC3 channel expression in skeletal muscle is increased after neuromuscular activity by NFAT-dependent transcriptional upregulation. TRPC3 expression is higher in muscles enriched in slow oxidative fibers than those enriched in fast glycolytic fibers. Voluntary free-wheel running increased TRPC3 expression either 1 or 3 weeks after treatment in both the soleus and EDL muscles. Furthermore, electrical neurostimulation at 10 Hz increased levels of TRPC3 transcripts in the tibialis anterior (TA) muscle [66]. TRPC3 expression was significantly increased in TRPV4−/− mouse skeletal muscle, in which the proportion of oxidative fibers was also increased [33]. These results suggest the importance of TRPC3 channels, especially in oxidative slow muscle fibers. TRPC3 interacts with ryanodine receptor type 1 (RyR1) in skeletal muscle (Fig. 2) [35, 80]. Loss of TRPC3 reduces the expression of RyR1, and vice versa [35], suggesting that TRPC3 plays a critical role in the modulation of RyR1. Indirect positive regulation of RyR by TRPC3 via Nox2-mediated ROS production has also been demonstrated in cardiomyocytes [29, 63, 64]. This TRPC3-Nox2-RyR coupling might also play important roles in skeletal muscle. TRPC3 also interacts with glucose transporter 4 (Glut-4) in T-tubules, and silencing of TRPC3 by siRNA reduced insulin-mediated glucose uptake by skeletal muscle. In accordance with these data, obese mice showed less oleoyl-acyl-sn-glycerol (OAG)-induced TRPC3 current [34]. TRPC3 also interacts with mitsugumin 29 (MG29), which is involved in the fatigue and aging processes of skeletal muscle. TRPC3-binding-deficient MG29 expression reduced the excitation-induced Ca2+ response in skeletal myotubes, indicating that MG29 plays a critical role in the regulation of TRPC3 channel function in skeletal muscle (Fig. 2) [83]. It has also been demonstrated that MG53 can interact with TRPC3 in skeletal muscle [1]. Myoblasts from muscular dysgenic mouse skeletal muscle failed to differentiate into myotubes when TRPC3 was knocked down [81].
TRPC3-overexpressing transgenic mice show a pathological phenotype similar to muscular dystrophy, suggesting that excess Ca2+ influx mediated by TRPC channels is sufficient to cause the disease. Using a TRPC6 dominant negative mutant, suppression of TRPC channels ameliorated the dystrophic myofibers of delta-sarcoglycan-null (Scgd−/−) mice [48].
TRPC4
TRPC4 is also expressed in skeletal muscle cells, and its expression is increased in mdx mice. TRPC4 can form a heterotetramer with TRPC1. Similar to TRPC1, TRPC4 can interact with alpha-syntrophin and is part of the dystrophin-associated protein complex (DAPC) [67]. In human myoblasts, TRPC4 downregulation by siRNA or overexpression of a dominant negative mutant clearly suppressed SOCE, expression of the myogenic driver MEF2 and fusion of myoblasts into myotubes [2]. In these contexts, TRPC4 couples with TRPC1 and is regulated by STIM1L [3].
TRPC6
TRPC6 expression is increased in mdx mouse skeletal muscle. Immunostaining revealed that TRPC6 is localized to the sarcoplasmic membrane [31]. Inhibition or deletion of TRPC6 has been reported to blunt the chronic mechanical stress-induced muscular contraction in mouse myocytes with Duchenne muscular dystrophy [68]. TRPC6 expression was significantly increased in TRPV4−/− mouse skeletal muscle, in which the numbers of oxidative fibers were increased more than glycolytic fibers [33].
Other TRPC channels
Compared with the aforementioned TRPC channels, the roles of TRPC2, TRPC5 and TRPC7 in striated muscles have been less well studied. The expression of TRPC2 is highly restricted, being present only in sperm and the vomeronasal sensory system [87]. Furthermore, TRPC2 is a pseudogene in the human genome. These facts imply that TRPC2 does not contribute significantly to striated muscle physiology. Although its specific function in striated muscles has not been demonstrated even with knockout mice, an involvement of TRPC5 in SOCE in cardiomyocytes has been implied. Recently, we demonstrated that extracellular ATP-induced Ca2+ influx mediated by TRPC5 induces nitric oxide synthase (NOS) activation and protects cardiomyocytes from hypertrophic responses [72]. TRPC7 was originally cloned from a cDNA library of mouse heart [56]. However, its function in cardiac and skeletal muscle remains elusive. The pathological significance of the closely related homologues TRPC3 and TRPC6 in striated muscles has been established, as mentioned above. Therefore, TRPC7 might play an important role in striated muscles, although confirmation of this will require a thorough analysis of knockout mice.
Cardioprotective effect of exercise
Physical activity affects not only skeletal muscle cells but also other remote organs. Several factors secreted from skeletal muscle after exercise have been identified, and these are termed myokines [60]. However, not all effects of exercise have been reproduced by the administration of myokines, suggesting that the beneficial effect of exercise is not solely attributable to these limited factors but is a systematic change of whole tissues [28]. The heart is an example of an organ that is very sensitive to the effects of exercise [28]. Patients suffering from heart failure are recommended to engage in supervised physical activity to prevent disease progression and assist cardiac rehabilitation [5]. Therefore, a systematic understanding of the beneficial effects of exercise will be fundamental for developing more effective drugs against cardiac diseases.
Physical exercise as a therapeutic intervention for DOX-induced cardiotoxicity
Doxorubicin (DOX) is a highly effective anticancer agent used to treat a variety of hematologic and solid malignancies [8, 79, 85, 92]. However, its dose-dependent cardiotoxicity limits its clinical use. The cardiotoxic effects of DOX range from asymptomatic increases in left ventricular (LV) wall stress to reductions in ejection fraction, arrhythmias and highly symptomatic congestive heart failure, which are all associated with high mortality [8, 14]. DOX initially causes the heart to shrink, which leads to induction of myocardial apoptosis and interstitial fibrosis at later stages of LV dilated cardiomyopathy [11, 94]. Numerous animal studies suggest that physical exercise training is the best intervention for preventing DOX-induced cardiac toxicity. In sedentary mice, DOX treatment resulted in a statistically significant decrease in heart function compared with control animals, which was mitigated by moderate aerobic exercise during DOX treatment. However, these protective effects of exercise were not observed when exercise was started after completion of DOX treatment. DOX caused not only a decrease in heart function but also cardiac atrophy and loss of body weight that were prevented by exercise, whereas non-trained mice exhibited no changes in these measurements. DOX delivery to the hearts of trained mice was reduced by consistent moderate aerobic exercise before DOX treatment [76]. Resistance training preserved cardiac function and attenuated the α- to β-myosin heavy chain shift that occurs with DOX treatment. No significant differences in lipid peroxidation were observed between sedentary and resistance-trained animals treated with DOX. [61]. Ashraf et al. demonstrated that after endurance training for 3 weeks, the DOX-induced increase in oxidative stress in the heart was significantly attenuated, possibly due to the upregulation of antioxidant systems such as superoxide dismutases (SOD) [4].
TRPC3-Nox2 coupling in DOX-induced cardiac toxicity
Nox2 proteins are known to play a critical role in DOX-induced cardiotoxicity. Indeed, Nox2 knockout (Nox2−/−) mice exhibit preserved contractile function after DOX treatment. In Nox2−/− mice, DOX-induced structural changes of the heart including atrophy, interstitial fibrosis and apoptosis of cardiomyocytes were all diminished [93]. DOX treatment increased Nox2 expression in the heart [37, 69, 93]. Reduction of Nox2 expression by treatment with paeoniflorin ameliorated DOX-induced cardiomyocyte apoptosis [37]. We recently identified that TRPC3 is a critical regulator of Nox2 activity in the mechanically stressed heart [29]. TRPC3 functions in the signaling platform by providing Ca2+ to the downstream signaling molecules such as protein kinase C, which also plays a key role in Nox2 activation [51, 54]. TRPC3 knockout (TRPC3−/−) mice exhibit preserved cardiac function and heart mass compared with WT mice [69]. Furthermore, the DOX-induced increase of Nox2 protein abundance was significantly suppressed in the hearts of TRPC3−/− mice. Although important for Nox2-mediated ROS production by providing Ca2+ for the enzymatic activation, TRPC3 may increase Nox2 protein abundance by preventing Nox2 from proteasome-mediated degradation through physical interaction (Fig. 3) [29]. Pharmacological inhibition or disruption of the TRPC3-Nox2 interaction ameliorates DOX-induced cardiac dysfunction [30, 69]. Therefore, suppression of TRPC3 function and/or expression will be promising therapeutic approaches against DOX-induced cardiac toxicity. Because TRPC3 expression is upregulated in various types of stressed hearts [9, 29, 53], the reduction of TRPC3 expression will lead to the reduction of ROS production by Nox2. Physical exercise reduced basal expression levels of TRPC3 and Nox2 in the mouse heart [69].

TRPC3-Nox2 coupling in cardiomyocytes. TRPC3 co-localizes and interacts with Nox2 in T-tubules in cardiomyocytes. Reactive oxygen species (ROS) oxidize ryanodine receptors (RyRs), which sensitizes them to Ca2+ during diastole
Perspective
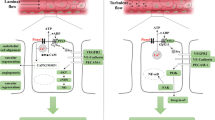
The beneficial effects of physical activity for the prevention and improvement of several diseases are well accepted. However, the molecular details underlying those effects are still largely obscure. The most accepted concept is that exercise initially induces moderate ROS production that may be metabolized by promoting the endogenous antioxidant system [65, 73]. Because of an increase in endogenous antioxidants, the body becomes more resistant to the oxidative stresses which cause several pathological conditions. The Ca2+ influx induced by physical exercise plays an important role in mediating enhancement of muscular contractility and energy metabolism. As a signal mediator, Ca2+ also plays a critical role in the activation of Nox proteins. Increased activity of TRPC channels has been reported in skeletal muscle after physical activity [21, 66]. In skeletal muscle, acute exercise increases Nox2 protein activity. The increase of Nox2 activity and ROS is critical for the upregulation of manganese superoxide dismutase (MnSOD), glutathione peroxidase (GPx), citrate synthase (CS), mitochondrial transcription factor A (tfam) and interleukin-6 (IL-6) [20]. IL-6 is one of the myokines released by skeletal muscle during exercise, and its release is decreased by treatment with antioxidant [16, 86]. This evidence suggests that TRPC and Nox coupling is likely to be enhanced by physical exercise and contributes to the upregulation of adaptive responses against oxidative stresses in skeletal muscle. Furthermore, the increased activity of the antioxidative system in skeletal muscle is transduced to the whole body via secreted factors such as myokines to modify metabolic homeostasis (Fig. 4). In contrast, physical activity reduces Nox2 expression levels in heart, suggesting downregulation of the endogenous TRPC3-Nox2 protein complex (Fig. 4) [69]. Thus, the mechanical stress-induced upregulation of TRPC3 and Nox2 proteins is actually an important compensative mechanism to enhance Ca2+-dependent muscular contractility, and moderate exercise negatively regulates the formation of the TRPC3-Nox2 stable protein complex. It is clear that exercise-induced upregulation of TRPC3 and Nox2 is sufficient to upregulate endogenous antioxidant systems in skeletal muscles. However, it is unclear whether the formation of the TRPC3-Nox2 complex in skeletal muscles has the ability to enhance antioxidant systems. Recently, we have obtained the interesting finding that the upregulation of TRPC6 can suppress TRPC3-Nox2 functional coupling in hyperglycemic cardiomyocytes [55]. Although it has been widely accepted that TRPC6 forms a heterotetramer with TRPC3 and works cooperatively [58], the expression balance of TRPC channels might be flexibly changed and function to maintain homeostatic TRPC channel activity in a cellular context-dependent manner. Future studies focusing on the formation of the TRPC3-Nox2 complex in skeletal muscles will resolve the pathological significance of TRPC3-Nox2 protein-protein interaction in muscular organs, and we suggest that perturbation of the TRPC3-Nox2 complex may be an innovative strategy to imitate exercise-induced beneficial effects on cardiovascular systems.

Physiological significance of canonical transient receptor potential (TRPC) channels in exercised human body. Exercise may increase the abundance of TRPCs and Nox proteins in skeletal muscle, while it may downregulate TRPC3 and Nox2 in the heart. Exercise-induced upregulation of TRPCs is concomitant with the upregulation of antioxidants, which may lead to a reduction of disease risk in remote organs, such as the cardiac pathological remodeling mediated by the TRPC3-Nox2 complex formation
References
Ahn MK, Lee KJ, Cai C, Huang M, Cho CH, Ma J, Lee EH (2016) Mitsugumin 53 regulates extracellular Ca(2+) entry and intracellular Ca(2+) release via Orai1 and RyR1 in skeletal muscle. Sci Rep 6:36909. https://doi.org/10.1038/srep36909
Antigny F, Koenig S, Bernheim L, Frieden M (2013) During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca(2)(+) entry. J Cell Sci 126:2525–2533. https://doi.org/10.1242/jcs.122911
Antigny F, Sabourin J, Sauc S, Bernheim L, Koenig S, Frieden M (2017) TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca(2+) release in human myotubes. Biochim Biophys Acta 1864:806–813. https://doi.org/10.1016/j.bbamcr.2017.02.003
Ashraf J, Roshan VD (2012) Is short-term exercise a therapeutic tool for improvement of cardioprotection against DOX-induced cardiotoxicity? An experimental controlled protocol in rats. Asian Pac J Cancer Prev 13:4025–4030
Balady GJ, Ades PA, Bittner VA, Franklin BA, Gordon NF, Thomas RJ, Tomaselli GF, Yancy CW, American Heart Association Science A, Coordinating C (2011) Referral, enrollment, and delivery of cardiac rehabilitation/secondary prevention programs at clinical centers and beyond: a presidential advisory from the American Heart Association. Circulation 124:2951–2960. https://doi.org/10.1161/CIR.0b013e31823b21e2
Benavides Damm T, Richard S, Tanner S, Wyss F, Egli M, Franco-Obregon A (2013) Calcium-dependent deceleration of the cell cycle in muscle cells by simulated microgravity. FASEB J 27:2045–2054. https://doi.org/10.1096/fj.12-218693
Berbey C, Weiss N, Legrand C, Allard B (2009) Transient receptor potential canonical type 1 (TRPC1) operates as a sarcoplasmic reticulum calcium leak channel in skeletal muscle. J Biol Chem 284:36387–36394. https://doi.org/10.1074/jbc.M109.073221
Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmstrom A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB, Wu JC (2016) Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med 22:547–556. https://doi.org/10.1038/nm.4087
Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA (2006) Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem 281:33487–33496. https://doi.org/10.1074/jbc.M605536200
Cheung KK, Yeung SS, Au SW, Lam LS, Dai ZQ, Li YH, Yeung EW (2011) Expression and association of TRPC1 with TRPC3 during skeletal myogenesis. Muscle Nerve 44:358–365. https://doi.org/10.1002/mus.22060
Cove-Smith L, Woodhouse N, Hargreaves A, Kirk J, Smith S, Price SA, Galvin M, Betts CJ, Brocklehurst S, Backen A, Radford J, Linton K, Roberts RA, Schmitt M, Dive C, Tugwood JD, Hockings PD, Mellor HR (2014) An integrated characterization of serological, pathological, and functional events in doxorubicin-induced cardiotoxicity. Toxicol Sci 140:3–15. https://doi.org/10.1093/toxsci/kfu057
Donati C, Cencetti F, Bruni P (2013) Sphingosine 1-phosphate axis: a new leader actor in skeletal muscle biology. Front Physiol 4:338. https://doi.org/10.3389/fphys.2013.00338
El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F (2009) p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med 41:217–225. https://doi.org/10.3858/emm.2009.41.4.058
Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK (2000) Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 342:1077–1084. https://doi.org/10.1056/NEJM200004133421502
Ferreira LF, Laitano O (2016) Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med 98:18–28. https://doi.org/10.1016/j.freeradbiomed.2016.05.011
Fischer CP, Hiscock NJ, Penkowa M, Basu S, Vessby B, Kallner A, Sjoberg LB, Pedersen BK (2004) Supplementation with vitamins C and E inhibits the release of interleukin-6 from contracting human skeletal muscle. J Physiol 558:633–645. https://doi.org/10.1113/jphysiol.2004.066779
Formigli L, Sassoli C, Squecco R, Bini F, Martinesi M, Chellini F, Luciani G, Sbrana F, Zecchi-Orlandini S, Francini F, Meacci E (2009) Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J Cell Sci 122:1322–1333. https://doi.org/10.1242/jcs.035402
Gervasio OL, Whitehead NP, Yeung EW, Phillips WD, Allen DG (2008) TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J Cell Sci 121:2246–2255. https://doi.org/10.1242/jcs.032003
Harisseh R, Chatelier A, Magaud C, Deliot N, Constantin B (2013) Involvement of TRPV2 and SOCE in calcium influx disorder in DMD primary human myotubes with a specific contribution of alpha1-syntrophin and PLC/PKC in SOCE regulation. Am J Physiol Cell Physiol 304:C881–C894. https://doi.org/10.1152/ajpcell.00182.2012
Henriquez-Olguin C, Diaz-Vegas A, Utreras-Mendoza Y, Campos C, Arias-Calderon M, Llanos P, Contreras-Ferrat A, Espinosa A, Altamirano F, Jaimovich E, Valladares DM (2016) NOX2 inhibition impairs early muscle gene expression induced by a single exercise bout. Front Physiol 7:282. https://doi.org/10.3389/fphys.2016.00282
Imaoka Y, Kawai M, Mori F, Miyata H (2015) Effect of eccentric contraction on satellite cell activation in human vastus lateralis muscle. J Physiol Sci 65:461–469. https://doi.org/10.1007/s12576-015-0385-4
Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y (2009) Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res 104:1399–1409. https://doi.org/10.1161/CIRCRESAHA.108.193227
Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y, Takeda S (2013) Capsaicin mimics mechanical load-induced intracellular signaling events: involvement of TRPV1-mediated calcium signaling in induction of skeletal muscle hypertrophy. Channels (Austin) 7:221–224. https://doi.org/10.4161/chan.24583
Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y, Takeda S (2013) Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat Med 19:101–106. https://doi.org/10.1038/nm.3019
Jackson MJ (2009) Redox regulation of adaptive responses in skeletal muscle to contractile activity. Free Radic Biol Med 47:1267–1275. https://doi.org/10.1016/j.freeradbiomed.2009.09.005
Jackson MJ, McArdle A (2011) Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J Physiol 589:2139–2145. https://doi.org/10.1113/jphysiol.2011.206623
Ji LL (2015) Redox signaling in skeletal muscle: role of aging and exercise. Adv Physiol Educ 39:352–359. https://doi.org/10.1152/advan.00106.2014
Joyner MJ, Green DJ (2009) Exercise protects the cardiovascular system: effects beyond traditional risk factors. J Physiol 587:5551–5558. https://doi.org/10.1113/jphysiol.2009.179432
Kitajima N, Numaga-Tomita T, Watanabe M, Kuroda T, Nishimura A, Miyano K, Yasuda S, Kuwahara K, Sato Y, Ide T, Birnbaumer L, Sumimoto H, Mori Y, Nishida M (2016) TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci Rep 6:37001. https://doi.org/10.1038/srep37001
Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y (2009) Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A 106:5400–5405. https://doi.org/10.1073/pnas.0808793106
Kruger J, Kunert-Keil C, Bisping F, Brinkmeier H (2008) Transient receptor potential cation channels in normal and dystrophic mdx muscle. Neuromuscul Disord 18:501–513. https://doi.org/10.1016/j.nmd.2008.04.003
Kunert-Keil C, Bisping F, Kruger J, Brinkmeier H (2006) Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics 7:159. https://doi.org/10.1186/1471-2164-7-159
Kusudo T, Wang Z, Mizuno A, Suzuki M (1985) Yamashita H (2012) TRPV4 deficiency increases skeletal muscle metabolic capacity and resistance against diet-induced obesity. J Appl Physiol 112:1223–1232. https://doi.org/10.1152/japplphysiol.01070.2011
Lanner JT, Bruton JD, Assefaw-Redda Y, Andronache Z, Zhang SJ, Severa D, Zhang ZB, Melzer W, Zhang SL, Katz A, Westerblad H (2009) Knockdown of TRPC3 with siRNA coupled to carbon nanotubes results in decreased insulin-mediated glucose uptake in adult skeletal muscle cells. FASEB J 23:1728–1738. https://doi.org/10.1096/fj.08-116814
Lee EH, Cherednichenko G, Pessah IN, Allen PD (2006) Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J Biol Chem 281:10042–10048. https://doi.org/10.1074/jbc.M600981200
Li C, Li J, Xiong X, Liu Y, Lv Y, Qin S, Liu D, Wei R, Ruan X, Zhang J, Xu L, Wang X, Chen J, Zhang Y, Zheng L (2018) TRPM8 activation improves energy expenditure in skeletal muscle and exercise endurance in mice. Gene 641:111–116. https://doi.org/10.1016/j.gene.2017.10.045
Li JZ, Yu SY, Wu JH, Shao QR, Dong XM (2012) Paeoniflorin protects myocardial cell from doxorubicin-induced apoptosis through inhibition of NADPH oxidase. Can J Physiol Pharmacol 90:1569–1575. https://doi.org/10.1139/y2012-140
Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS (2007) Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(−/−) mice. Proc Natl Acad Sci U S A 104:17542–17547. https://doi.org/10.1073/pnas.0701254104
Liu Y, Schneider MF (2014) FGF2 activates TRPC and Ca(2+) signaling leading to satellite cell activation. Front Physiol 5:38. https://doi.org/10.3389/fphys.2014.00038
Louis M, Zanou N, Van Schoor M, Gailly P (2008) TRPC1 regulates skeletal myoblast migration and differentiation. J Cell Sci 121:3951–3959. https://doi.org/10.1242/jcs.037218
Luo Z, Ma L, Zhao Z, He H, Yang D, Feng X, Ma S, Chen X, Zhu T, Cao T, Liu D, Nilius B, Huang Y, Yan Z, Zhu Z (2012) TRPV1 activation improves exercise endurance and energy metabolism through PGC-1alpha upregulation in mice. Cell Res 22:551–564. https://doi.org/10.1038/cr.2011.205
Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP (2005) TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol 7:179–185. https://doi.org/10.1038/ncb1218
Matsumura CY, Taniguti AP, Pertille A, Santo Neto H, Marques MJ (2011) Stretch-activated calcium channel protein TRPC1 is correlated with the different degrees of the dystrophic phenotype in mdx mice. Am J Physiol Cell Physiol 301:C1344–C1350. https://doi.org/10.1152/ajpcell.00056.2011
Mauricio AF, de Carvalho SC, Santo Neto H, Marques MJ (2017) Effects of dietary omega-3 on dystrophic cardiac and diaphragm muscles as evaluated by (1)H magnetic resonance spectroscopy: metabolic profile and calcium-related proteins. Clin Nutr ESPEN 20:60–67. https://doi.org/10.1016/j.clnesp.2017.03.005
McArdle A, Pattwell D, Vasilaki A, Griffiths RD, Jackson MJ (2001) Contractile activity-induced oxidative stress: cellular origin and adaptive responses. Am J Physiol Cell Physiol 280:C621–C627. https://doi.org/10.1152/ajpcell.2001.280.3.C621
McArdle F, Spiers S, Aldemir H, Vasilaki A, Beaver A, Iwanejko L, McArdle A, Jackson MJ (2004) Preconditioning of skeletal muscle against contraction-induced damage: the role of adaptations to oxidants in mice. J Physiol 561:233–244. https://doi.org/10.1113/jphysiol.2004.069914
Meacci E, Bini F, Sassoli C, Martinesi M, Squecco R, Chellini F, Zecchi-Orlandini S, Francini F, Formigli L (2010) Functional interaction between TRPC1 channel and connexin-43 protein: a novel pathway underlying S1P action on skeletal myogenesis. Cell Mol Life Sci 67:4269–4285. https://doi.org/10.1007/s00018-010-0442-3
Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD (2009) Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci U S A 106:19023–19028. https://doi.org/10.1073/pnas.0906591106
Montell C, Rubin GM (1989) Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2:1313–1323
Mori Y, Wakamori M, Miyakawa T, Hermosura M, Hara Y, Nishida M, Hirose K, Mizushima A, Kurosaki M, Mori E, Gotoh K, Okada T, Fleig A, Penner R, Iino M, Kurosaki T (2002) Transient receptor potential 1 regulates capacitative Ca(2+) entry and Ca(2+) release from endoplasmic reticulum in B lymphocytes. J Exp Med 195:673–681
Nishida M, Sugimoto K, Hara Y, Mori E, Morii T, Kurosaki T, Mori Y (2003) Amplification of receptor signalling by Ca2+ entry-mediated translocation and activation of PLCgamma2 in B lymphocytes. EMBO J 22:4677–4688. https://doi.org/10.1093/emboj/cdg457
Nishida M, Hara Y, Yoshida T, Inoue R, Mori Y (2006) TRP channels: molecular diversity and physiological function. Microcirculation 13:535–550. https://doi.org/10.1080/10739680600885111
Numaga-Tomita T, Oda S, Shimauchi T, Nishimura A, Mangmool S, Nishida M (2017) TRPC3 channels in cardiac fibrosis. Front Cardiovasc Med 4:56. https://doi.org/10.3389/fcvm.2017.00056
Numaga T, Nishida M, Kiyonaka S, Kato K, Katano M, Mori E, Kurosaki T, Inoue R, Hikida M, Putney JW, Jr., Mori Y (2010) Ca2+ influx and protein scaffolding via TRPC3 sustain PKCbeta and ERK activation in B cells. J Cell Sci 123:927–938 DOI https://doi.org/10.1242/jcs.061051
Oda S, Numaga-Tomita T, Kitajima N, Toyama T, Harada E, Shimauchi T, Nishimura A, Ishikawa T, Kumagai Y, Birnbaumer L, Nishida M (2017) TRPC6 counteracts TRPC3-Nox2 protein complex leading to attenuation of hyperglycemia-induced heart failure in mice. Sci Rep 7:7511. https://doi.org/10.1038/s41598-017-07903-4
Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y (1999) Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca(2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. In: J biol Chem 274:27359–70 DOI
Olah T, Fodor J, Ruzsnavszky O, Vincze J, Berbey C, Allard B, Csernoch L (2011) Overexpression of transient receptor potential canonical type 1 (TRPC1) alters both store operated calcium entry and depolarization-evoked calcium signals in C2C12 cells. Cell Calcium 49:415–425. https://doi.org/10.1016/j.ceca.2011.03.012
Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H (2006) TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J 25:5305–5316. https://doi.org/10.1038/sj.emboj.7601417
Palomero J, Pye D, Kabayo T, Spiller DG, Jackson MJ (2008) In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid Redox Signal 10:1463–1474. https://doi.org/10.1089/ars.2007.2009
Pedersen BK, Febbraio MA (2012) Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8:457–465. https://doi.org/10.1038/nrendo.2012.49
Pfannenstiel K, Hayward R (2018) Effects of resistance exercise training on doxorubicin-induced cardiotoxicity. J Cardiovasc Pharmacol 71:332–339. https://doi.org/10.1097/FJC.0000000000000574
Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C, Groschner K (2006) TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem 281:13588–13595 DOI https://doi.org/10.1074/jbc.M512205200
Prosser BL, Ward CW, Lederer WJ (2011) X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333:1440–1445. https://doi.org/10.1126/science.1202768
Prosser BL, Khairallah RJ, Ziman AP, Ward CW, Lederer WJ (2013) X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca(2)(+)]i. J Mol Cell Cardiol 58:172–181. https://doi.org/10.1016/j.yjmcc.2012.11.011
Radak Z, Chung HY, Goto S (2008) Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic Biol Med 44:153–159. https://doi.org/10.1016/j.freeradbiomed.2007.01.029
Rosenberg P, Hawkins A, Stiber J, Shelton JM, Hutcheson K, Bassel-Duby R, Shin DM, Yan Z, Williams RS (2004) TRPC3 channels confer cellular memory of recent neuromuscular activity. Proc Natl Acad Sci U S A 101:9387–9392. https://doi.org/10.1073/pnas.0308179101
Sabourin J, Cognard C, Constantin B (2009) Regulation by scaffolding proteins of canonical transient receptor potential channels in striated muscle. J Muscle Res Cell Motil 30:289–297. https://doi.org/10.1007/s10974-010-9206-9
Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ, Marino JP, Jr., Birnbaumer L, Schnackenberg CG, Kass DA (2014) Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A 111:1551–1556 DOI https://doi.org/10.1073/pnas.1308963111
Shimauchi T, Numaga-Tomita T, Ito T, Nishimura A, Matsukane R, Oda S, Hoka S, Ide T, Koitabashi N, Uchida K, Sumimoto H, Mori Y, Nishida M (2017) TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy. JCI Insight 2https://doi.org/10.1172/jci.insight.93358, 2
Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL (2006) A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci U S A 103:16586–16591. https://doi.org/10.1073/pnas.0606894103
Stiber JA, Zhang ZS, Burch J, Eu JP, Zhang S, Truskey GA, Seth M, Yamaguchi N, Meissner G, Shah R, Worley PF, Williams RS, Rosenberg PB (2008) Mice lacking Homer 1 exhibit a skeletal myopathy characterized by abnormal transient receptor potential channel activity. Mol Cell Biol 28:2637–2647. https://doi.org/10.1128/MCB.01601-07
Sunggip C, Shimoda K, Oda S, Tanaka T, Nishiyama K, Mangmool S, Nishimura A, Numaga-Tomita T, Nishida M (2018) TRPC5-eNOS Axis negatively regulates ATP-induced cardiomyocyte hypertrophy. Front Pharmacol 9:523. https://doi.org/10.3389/fphar.2018.00523
Thirupathi A, Pinho RA (2018) Effects of reactive oxygen species and interplay of antioxidants during physical exercise in skeletal muscles. J Physiol Biochem 74:359–367. https://doi.org/10.1007/s13105-018-0633-1
Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A, Raymond G, Cognard C, Bourmeyster N, Constantin B (2007) Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J 21:608–617. https://doi.org/10.1096/fj.06-6683com
Vandebrouck C, Martin D, Colson-Van Schoor M, Debaix H, Gailly P (2002) Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol 158:1089–1096. https://doi.org/10.1083/jcb.200203091
Wang F, Iskra B, Kleinerman E, Alvarez-Florez C, Andrews T, Shaw A, Chandra J, Schadler K, Aune GJ (2018) Aerobic exercise during early murine doxorubicin exposure mitigates cardiac toxicity. J Pediatr Hematol Oncol 40:208–215. https://doi.org/10.1097/MPH.0000000000001112
Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA, Dietrich A (2012) Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun 3:649 DOI https://doi.org/10.1038/ncomms1660, 3, 649
Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C (1995) TRPC1, a human homolog of a Drosophila store-operated channel. Proc Natl Acad Sci U S A 92:9652–9656
Witteles RM, Bosch X (2015) Myocardial protection during cardiotoxic chemotherapy. Circulation 132:1835–1845. https://doi.org/10.1161/CIRCULATIONAHA.114.010486
Woo JS, Kim DH, Allen PD, Lee EH (2008) TRPC3-interacting triadic proteins in skeletal muscle. Biochem J 411:399–405. https://doi.org/10.1042/bj20071504
Woo JS, Cho CH, Kim DH, Lee EH (2010) TRPC3 cation channel plays an important role in proliferation and differentiation of skeletal muscle myoblasts. Exp Mol Med 42:614–627. https://doi.org/10.3858/emm.2010.42.9.061
Woo JS, Lee KJ, Huang M, Cho CH, Lee EH (2014) Heteromeric TRPC3 with TRPC1 formed via its ankyrin repeats regulates the resting cytosolic Ca2+ levels in skeletal muscle. Biochem Biophys Res Commun 446:454–459. https://doi.org/10.1016/j.bbrc.2014.02.127
Woo JS, Hwang JH, Huang M, Ahn MK, Cho CH, Ma J, Lee EH (2015) Interaction between mitsugumin 29 and TRPC3 participates in regulating ca(2+) transients in skeletal muscle. Biochem Biophys Res Commun 464:133–139. https://doi.org/10.1016/j.bbrc.2015.06.096
Xia L, Cheung KK, Yeung SS, Yeung EW (2016) The involvement of transient receptor potential canonical type 1 in skeletal muscle regrowth after unloading-induced atrophy. J Physiol 594:3111–3126. https://doi.org/10.1113/JP271705
Yeh ET, Tong AT, Lenihan DJ, Yusuf SW, Swafford J, Champion C, Durand JB, Gibbs H, Zafarmand AA, Ewer MS (2004) Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation 109:3122–3131. https://doi.org/10.1161/01.CIR.0000133187.74800.B9
Yfanti C, Fischer CP, Nielsen S, Akerstrom T, Nielsen AR, Veskoukis AS, Kouretas D, Lykkesfeldt J, Pilegaard H (1985) Pedersen BK (2012) role of vitamin C and E supplementation on IL-6 in response to training. J Appl Physiol 112:990–1000. https://doi.org/10.1152/japplphysiol.01027.2010
Yildirim E, Birnbaumer L (2007) TRPC2: molecular biology and functional importance. Handb Exp Pharmacol:53–75. https://doi.org/10.1007/978-3-540-34891-7_3
Zanou N, Shapovalov G, Louis M, Tajeddine N, Gallo C, Van Schoor M, Anguish I, Cao ML, Schakman O, Dietrich A, Lebacq J, Ruegg U, Roulet E, Birnbaumer L, Gailly P (2010) Role of TRPC1 channel in skeletal muscle function. Am J Physiol Cell Physiol 298:C149–C162. https://doi.org/10.1152/ajpcell.00241.2009
Zanou N, Schakman O, Louis P, Ruegg UT, Dietrich A, Birnbaumer L, Gailly P (2012) Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J Biol Chem 287:14524–14534. https://doi.org/10.1074/jbc.M112.341784
Zhang BT, Whitehead NP, Gervasio OL, Reardon TF, Vale M, Fatkin D, Dietrich A, Yeung EW (1985) Allen DG (2012) pathways of Ca(2)(+) entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J Appl Physiol 112:2077–2086. https://doi.org/10.1152/japplphysiol.00770.2011
Zhang BT, Yeung SS, Cheung KK, Chai ZY, Yeung EW (2014) Adaptive responses of TRPC1 and TRPC3 during skeletal muscle atrophy and regrowth. Muscle Nerve 49:691–699. https://doi.org/10.1002/mus.23952
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET (2012) Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18:1639–1642. https://doi.org/10.1038/nm.2919
Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, McDermott BJ, Grieve DJ (2010) Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Res 70:9287–9297. https://doi.org/10.1158/0008-5472.CAN-10-2664
Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W, Field LJ (2009) Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119:99–106. https://doi.org/10.1161/CIRCULATIONAHA.108.799700
Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L (1996) trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 85:661–671
Zitt C, Zobel A, Obukhov AG, Harteneck C, Kalkbrenner F, Luckhoff A, Schultz G (1996) Cloning and functional expression of a human Ca2+−permeable cation channel activated by calcium store depletion. Neuron 16:1189–1196
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (16H05092 to M.N.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). We thank Melony Black, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
This article is part of the special issue on Exercise Physiology: future opportunities and challenges in Pflügers Archiv – European Journal of Physiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Numaga-Tomita, T., Oda, S., Nishiyama, K. et al. TRPC channels in exercise-mimetic therapy. Pflugers Arch - Eur J Physiol 471, 507–517 (2019). https://doi.org/10.1007/s00424-018-2211-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2211-3