
Abstract
Avellino corneal dystrophy (ACD) is a common corneal dystrophy that shows allelic homogeneity, R124H mutation in the transforming growth factor beta-induced (TGFBI) gene. There are distinct phenotypes of homozygous Avellino corneal dystrophy, termed types I and II. To investigate if the difference is caused by a modifier mutation, we sequenced the entire coding region of TGFBI of two types of ACDs. The sequences obtained from each type were identical, and we could not find any nucleotide alternations. Instead, we found seven single nucleotide polymorphisms (SNPs) compared with the normal control. Primer extension analysis revealed that all 14 homozygous patients were homozygotes in each SNP, which meant that all the patients shared the same disease haplotype. Subsequent analysis of 45 heterozygous ACD patients showed strong linkage disequilibrium between disease alleles of each SNP and ACD. These results strongly suggest that the allelic homogeneity of TGFBI associated corneal dystrophies (ACD, lattice corneal dystrophy types I and III, granular corneal dystrophy and Reis–Bucklers dystrophy) might not be caused by mutation hot spots but by the founder effects.
Similar content being viewed by others

Introduction
Avellino corneal dystrophy (ACD) is an autosomal dominant corneal dystrophy, of which the clinical aspect is the coexistence of granular deposits and histological amyloidal deposits in the cornea (Folberg et al. 1988; Holland et al. 1992; Rosenwasser et al. 1993). ACD is one of the transforming growth factor beta-induced (TGFBI) associated corneal dystrophies. TGFBI associated corneal dystrophies are quite interesting, and include lattice corneal dystrophy type 1 (CDL1), granular corneal dystrophy (CDGG1), and Reis–Bucklers dystrophy (CDRB). All are caused by mutations in the TGFBI gene and show white deposition accumulating under the sub-epithelium of the cornea. However, the material and shape of the depositions are different in each dystrophy. For example, lattice shaped amyloid deposits are in CDL1, and granular phospholipid deposits are in CDGG1. Moreover, each dystrophy shows significant allelic homogeneity; for example, most of CDL1 is caused by R124C, CDGG1 by R555W, CDRB by R555Q, and ACD by R124H (Munier et al. 1997). The spectrum of these dystrophies is also interesting. In European countries, ACD occupies only 12% of hereditary corneal dystrophies associated with TGFBI (Munier et al. 2002). Sometimes, the families traced their ancestry to the Italian province of Avellino. On the other hand, in Japanese individuals, ACD is the most common hereditary corneal dystrophy, responsible for 72% of corneal dystrophies associated with TGFBI (Mashima et al. 2000). The cause of this unique spectrum is not clear.
ACD is not a lethal disease, and we have reported that the disease is more severe in homozygous patients than in their heterozygous counterparts (Okada et al. 1998). Interestingly, we found that there are two different phenotypes of these homozygous disease types (Watanabe et al. 2001). Type I is a spot-like opacity present in the anterior stroma in which the lesions are confluent. The type II corneal opacity pattern is a reticular opacity in the anterior stroma, with round translucent spaces (Fig. 1). The heterozygous siblings of these two homozygous patients do not show such a clinical variation. The patients with the type I opacity do not share a common birthplace; however, type II patients traced their origin to the Tottori prefecture in western Japan (Watanabe et al. 2001), which suggests the presence of a novel modifier linked to TGFBI in type II patients. In this study, we tried to find such a modifier; however, we could not find any difference between the two types in the coding region of TGFBI. Instead, we noticed the presence of a common haplotype shared by all patients and proved the presence of a founder effect in Japanese ACD patients.

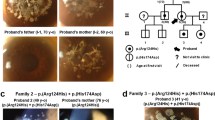
Distinct different opacity pattern between ACD types I and II. a A 29-year-old patient with homozygous ACD type I. Grayish granular opacities present in the anterior stroma cover almost of the entire cornea. Visual acuity is 0.06 with spectacle correction. b A 30-year-old patient with homozygous ACD type II. Compared with type I, more fine grayish deposits cover the entire cornea; however, among the diffuse opacities several round translucent spaces are present. Visual acuity is 0.3 partial with spectacle correction
Materials and methods
Families
We studied 14 homozygous patients in 13 Japanese families and 45 heterozygous patients in 39 Japanese families with ACD through the Department of Ophthalmology at Osaka University of Medicine. Written informed consent was obtained from all patients, and the procedures followed the tenets of the Declaration of Helsinki and Osaka University Ethics Committee. All participants were examined and diagnosed by slit-lamp bio-microscopy by corneal faculty members of the Department of Ophthalmology at Osaka University. We classified the phenotypes of ACD following the previous paper (Watanabe et al. 2001). The homozygous population consisted of ten patients from nine different ACD type I families and four patients of four ACD type II families. All four type II patients traced their origin to the Tottori prefecture in western Japan. The phenotype of homozygous ACD type I is a spot-like confluent opacity in the anterior stroma (Fig. 1a). The phenotype of homozygous type II is reticular opacity in the anterior stroma, with round translucent spaces (Fig. 1b).
Genomic DNA preparation
Twenty milliliters of peripheral blood was drawn from each patient, and genomic DNA was extracted from the leukocytes using a DNA extraction kit (Stratagene, La Jolla, CA, USA). Samples were quantified by spectrophotometry and diluted to 50 ng/μl for polymerase chain reaction (PCR) amplification and other examinations.
Mutation analysis and polymorphisms
We performed PCR for samples and followed Munier’s paper for all exons (Munier et al. 1997). Some primers were designed to amplify all exons, including exon–intron boundaries of the TGFBI gene. Each PCR was carried out in a 15 μl reaction mixture containing 20 ng of genomic DNA, 20 pmol of each primer, 1.0–2.5 mM MgCl2, 1× reaction buffer (Takara, Japan), 200 mM of each dNTP, and 1.0 unit of Ex Taq polymerase (Takara). Samples were amplified in 35 cycles of 30 s at 94°C for denaturing, 30 s at optimum annealing temperature, and 30 s at 72°C for extension in a GeneAmp 9600 thermocycler (Applied Biosystems, Foster City, CA, USA). Amplified fragments were purified using the QIAquick PCR purification Kit (Qiagen) and directly sequenced using ABI PRISM big-dye terminator cycle and ABI3100, 310 or 377 sequencer (Applied Biosystems).
We genotyped homozygous and heterozygous ACD families in the region of the single nucleotide polymorphisms (SNPs) we found by the primer extension method. Each genomic segment containing SNPs was amplified by PCR and purified with 1 unit of ExoI and 0.5 unit of SAP. After purification, the primer extension reactions were performed using the Snap Shot kit (Applied Biosystems) according to the manufacturer’s recommended methods and analyzed with a 3100 genetic analyzer and Genescan software (Applied Biosystems). The following primers for SNP analysis were used;
418G->A: ACTCAGCTGTACACGGACC; 698C->G: TGTGAACTGTGCCCGGCT; 1028A->G: ATCGTTGCGGGGCTGTCTGT; 1463C->T: ATGCAGCTGTTCTCAATGCA; IVS12 + 23G->A: TAAAGACCAACTTAAGTACAC; IVS14 + 44T->C: TGCAGCCAGTCCTTTTCT; 2636T->G: AAGAAATGGTATGTAGAGCTTAGAT.
Results
To screen the sequence difference between two types of homozygous ACDs, we initially studied two patients with type I, two patients with type II, and one healthy control by direct sequencing of the complete coding region of the TGFBI gene. We anticipated some genetic difference between the two types, which causes the clinical difference; however, PCR amplification and direct sequencing of all exons revealed that there was no difference in the sequence between the two types of homozygous patients. The results were completely identical in the sequences from the four affected patients. We could not find even a silent alternation in the coding region. On the other hand, there were seven differences of sequence, including R124H between both types of ACD and the healthy control (Fig. 2). All four patients of both types of ACD were homozygous in all these alterations, which meant that all the patients shared the same haplotype. Table 1 and Fig. 3 summarize the seven nucleotide alterations that were observed in homozygous ACD patients and the healthy control. One of them, IVS14 + 44T->C, has not been reported previously. One is the R124H, which has been reported as being responsible for ACD. The other three are in the coding region of TGFBI; however, they did not change the amino acid sequence during translation. One is in the 3′ UTR of TGFBI, and the other is a SNP in intron 12. These SNPs spanned only 18 kb on chromosome 5 (Fig. 3). This homogeneity brought us to question if the other ACD homozygous patients in Japan possess the same haplotype. To investigate this possibility, we performed primer extension analysis, which revealed all are homozygous for the disease alleles in all seven SNPs. All of the disease chromosomes share the same disease haplotype of these intra-genetic markers. All 14 homozygous ACD patients share the same disease chromosome (Fig. 4a).

Typical sequence analysis of TGFBI in ACD patients. Typical results of sequence analysis. IVS12 + 23 and IVS14 + 44 are shown. Upper panels show disease allele sequence and bottom panels show the normal allele. Mutation positions are underlined

Genomic structure of TGFBI and positions of each SNP marker. Each exon is showed in a box. Solid boxes indicate coding region of TGFBI. Note all SNP markers are placed within 18 kb from the R124H mutation

Typical results of the primer extension analysis. Results of 1028A->G polymorphism are shown. a A genotype of homozygous disease alleles from a homozygous ACD patient. b A genotype of heterozygous disease alleles from an ACD heterozygote. c A genotype of homozygous normal alleles from a healthy control
The SNP analyses of homozygotes indicate the possibility that all were disease chromosomes, which, not only in homozygotes but also in heterozygotes, carry the same haplotype. To investigate this, we genotyped 45 heterozygous patients from the 39 Japanese heterozygous ACD families and 66 healthy Japanese controls, using primer extension (Fig. 4b, c). Table 2 summarizes the results except those for R124H (all patients possess the heterozygous R124H mutation). In these samples, we could construct the disease haplotype only in five heterozygote families. However, at least the haplotype is completely the same as that of the homozygote families. Even in the case where the haplotype could not be constructed, all the patients possessed the diseased allele observed in homozygote families in all seven SNPs. Moreover, the allele frequencies were quite different from those in the healthy control. Chi-square analysis revealed that each of the seven disease alleles showed strong linkage disequilibrium with the ACD phenotype (Table 2). These results strongly suggest that Japanese ACD patients share a founder disease chromosome.
Discussion
ACD does not show complete dominance, and the homozygotes of this mutation display a more severe phenotype than the heterozygotes do (Okada et al. 1998). Interestingly, there are two distinct phenotypes of Japanese homozygous patients with ACD, which we have reported (Watanabe et al. 2001): ACD types I and II. In our studies, all four type II families traced their origin to the Tottori prefecture in western Japan, so we anticipated a relatively recent modifier mutation aside from R124H, which gives the type II phenotype, and the mutation was shared and reserved in all type II families. However, we could not find any nucleotide differences in the coding region of the TGFBI gene between the two types. We have not studied the mutations in all the introns and the promoter region, which may affect the expression levels of TGFBI, so we could not conclude whether there is no genetic difference between the two types in the TGFBI region. However, there is no amino acid alternation in the TGFBI coding region between the two types of ACDs.
Allelic homogeneity is a prominent feature of TGFBI associated corneal dystrophies, including ACD. The occurrence of the identical TGFBI mutations, R124H, has been reported in both Japanese and European patients with ACD. This identical mutation, i.e., allelic homogeneity, does not necessarily implicate a common ancestry (founder effect). An alternative explanation would be the presence of a mutation hot spot. In Europe, some patients with ACD traced their origins to the Italian province of Avellino, which favors a common founder. In spite of that fact, it has been reported that the allelic homogeneity observed in European patients with TGFBI corneal dystrophies, including ACD, did not reflect a founder effect but was caused by CpG mutation hot spots, when genetic markers near TGFBI were used (Korvatska et al. 1998). However, those results were obtained by microsatellite markers, which were located more than 300 kb from the R124H mutation, and for ACD the authors examined only four families. So we think the cause of homogeneity is still under question, even in those families.
In this paper we reported seven intragenic nucleotide alternations, including R124H. These markers are very close to R124H; one of them is placed within 1 kb from R124H, which enabled us to detect a very narrow conserved region. All 14 Japanese homozygous ACD patients, ten patients from nine ACD type I families and four patients of four ACD type II families, were homozygous for disease alleles in each of the seven SNPs. Subsequent genotyping of 45 heterozygous patients from 39 Japanese families with ACD revealed that, in each SNP, all patients possessed disease alleles, which were observed in homozygous Japanese ACD patients. In these studies, all constructed disease haplotypes are identical. Moreover, each disease allele shows quite a significant linkage disequilibrium related to heterozygous ACD. These results strongly favor common ancestry rather than the CpG mutation hot spot for this base substitution causing R124H. ACD is a common corneal dystrophy; more than 30 mutation reports have been published. However, we could not find any de novo mutations of R124H. It supports our results indicating the common ancestry. Our observations also suggest that a single founder R124H mutation event had occurred in the chromosome carrying the disease haplotype In six SNP alternations, except for R124H, in ancient age.
The presence of the founder effect well explains why patients with ACD are observed more frequently in Japan. One of the other TGFBI associated corneal dystrophies, lattice corneal dystrophy type IIIA (LCDIIIA), is also observed mainly in Japan. Using intragenetic markers, we have demonstrated that allelic homogeneity (P501T) of Japanese LCDIIIA may result from a single founder mutation (Tsujikawa et al. 2002). Because the disease alleles in these markers were not found in ACD patients, there is no relationship between the founders of ACD and LCDIIIA. However, the founder effects observed in ACD and LCDIIIA are interesting genetic phenomena in corneal dystrophies in Japanese patients. It will be interesting to investigate haplotype of other TGFBI associated dystrophies, i.e., CDL1 in Japan and ACD in other countries, using the intragenic markers we reported. We have also demonstrated the other founder effect in gelatinous drop-like corneal dystrophy, which is not associated with TGFBI (Tsujikawa et al. 1999, 2000). These dystrophies also are more frequently observed in Japan than in Western countries. These founder effects may be caused by the fact that corneal dystrophies do not influence the survival rate of patients. These three founder effects may give great influence in the spectrum of Japanese corneal dystrophies and are an important aspect in Japanese corneal inherited dystrophies, and they facilitate the genetic diagnoses in these diseases.
References
Folberg R, Alfonso E, Croxatto JO, Driezen NG, Panjwani N, Laibson PR, Boruchoff SA, Baum J, Malbran ES, Fernandez-Meijide R, et al (1988) Clinically atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits. A study of these families. Ophthalmology 95:46–51
Holland EJ, Daya SM, Stone EM, Folberg R, Dobler AA, Cameron JD, Doughman DJ (1992) Avellino corneal dystrophy. Clinical manifestations and natural history. Ophthalmology 99:1564–1568
Korvatska E, Munier FL, Djemai A, Wang MX, Frueh B, Chiou AG, Uffer S, Ballestrazzi E, Braunstein RE, Forster RK, Culbertson WW, Boman H, Zografos L, Schorderet DF (1998) Mutation hot spots in 5q31-linked corneal dystrophies. Am J Hum Genet 62:320–324
Mashima Y, Yamamoto S, Inoue Y, Yamada M, Konishi M, Watanabe H, Maeda N, Shimomura Y, Kinoshita S (2000) Association of autosomal dominantly inherited corneal dystrophies with BIGH3 gene mutations in Japan. Am J Ophthalmol 130:516–517
Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G, Schorderet DF (1997) Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet 15:247–251
Munier FL, Frueh BE, Othenin-Girard P, Uffer S, Cousin P, Wang MX, Heon E, Black GC, Blasi MA, Balestrazzi E, Lorenz B, Escoto R, Barraquer R, Hoeltzenbein M, Gloor B, Fossarello M, Singh AD, Arsenijevic Y, Zografos L, Schorderet DF (2002) BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci 43:949–954
Okada M, Yamamoto S, Inoue Y, Watanabe H, Maeda N, Shimomura Y, Ishii Y, Tano Y (1998) Severe corneal dystrophy phenotype caused by homozygous R124H keratoepithelin mutations. Invest Ophthalmol Vis Sci 39:1947–1953
Rosenwasser GO, Sucheski BM, Rosa N, Pastena B, Sebastiani A, Sassani JW, Perry HD (1993) Phenotypic variation in combined granular-lattice (Avellino) corneal dystrophy. Arch Ophthalmol 111:1546–1552
Tsujikawa K, Tsujikawa M, Yamamoto S, Fujikado T, Tano Y (2002) Allelic homogeneity due to a founder mutation in Japanese patients with lattice corneal dystrophy type IIIA. Am J Med Genet 113:20–22
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, Nakamura Y (1999) Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet 21:420–423
Tsujikawa M, Tsujikawa K, Maeda N, Watanabe H, Inoue Y, Mashima Y, Shimomura Y, Tano Y (2000) Rapid detection of M1S1 mutations by the protein truncation test. Invest Ophthalmol Vis Sci 41:2466–2468
Watanabe H, Hashida Y, Tsujikawa K, Tsujikawa M, Maeda N, Inoue Y, Yamamoto S, Tano Y (2001) Two patterns of opacity in corneal dystrophy caused by the homozygous BIG-H3 R124H mutation. Am J Ophthalmol 132:211–216
Acknowledgments
We would like to thank Drs. Sharon Schechter, Seema Lyer, Tomoyuki Inoue, and Andria Schibler for many helpful comments. This study was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (to M.T.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsujikawa, K., Tsujikawa, M., Watanabe, H. et al. Allelic homogeneity in Avellino corneal dystrophy due to a founder effect. J Hum Genet 52, 92–97 (2007). https://doi.org/10.1007/s10038-006-0083-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0083-4
