
Abstract
Pathogenic variants in the PCDH19 gene are associated with epilepsy, intellectual disability (ID) and behavioural disturbances. Only heterozygous females and mosaic males are affected, likely due to a disease mechanism named cellular interference. Until now, only four affected mosaic male patients have been described in literature. Here, we report five additional male patients, of which four are older than the oldest patient reported so far. All reported patients were selected for genetic testing because of developmental delay and/or epilepsy. Custom-targeted next generation sequencing gene panels for epilepsy genes were used. Clinical data were collected from medical records. All patients were mosaic in blood for likely pathogenic variants in the PCDH19 gene. In most, clinical features were very similar to the female phenotype, with normal development before seizure onset, which occurred between 5 and 10 months of age, clustering of seizures and sensitivity to fever. Four out of five patients had mild to severe ID and behavioural problems. We reaffirm the similarity between male and female PCDH19-related phenotypes, now also in a later phase of the disorder (ages 10–14 years).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pathogenic variants in PCDH19 are associated with early onset, clustered epileptic seizures often provoked by fever, intellectual disability (ID) that can be present in variable degrees and behavioural disturbances such as autistic features, attention deficit, hyperactivity and aggression. The clinical features may resemble those of Dravet syndrome (phenotype MIM # 300088) [1,2,3,4,5].
PCDH19 is located at Xq22.1 and codes for protocadherin-19, a transmembrane protein involved in neuronal organization and migration and cell-cell and cell-matrix adhesion [6,7,8,9,10]. It is highly expressed in the central nervous system [1]. PCDH19-related epilepsy shows a remarkable inheritance pattern: generally, females carrying heterozygous pathogenic variants are affected, whereas hemizygous male carriers are asymptomatic or only show psychiatric or behavioural symptoms [1, 2, 11, 12]. However, in 2009, the first affected male mosaic for a PCDH19 pathogenic variant was described [2]. This finding gave rise to a theory of cellular interference as disease mechanism: disease occurs when two different cell populations exist (cells expressing the normal PCDH19 protein and cells expressing a mutant form of the protein), as is true for heterozygous and mosaic pathogenic variants, but not for hemizygous pathogenic variants in males. These non-homogeneous cell populations are likely to disrupt cell-cell interactions, leading to disease [2].
Only four affected mosaic male patients have been described in literature until now, the oldest being 7 years old [2, 13, 14]. Here, we report five additional male patients with mosaic PCDH19 mutations, of ages 2, 10, 13, 13 and 14 years old. We compared the male and female phenotypes.
Methods
Patients and molecular analysis
All five patients were selected for diagnostic genetic testing because of developmental delay and/or epilepsy. Genetic testing on DNA from lymphocytes was performed using a custom targeted next generation sequencing (NGS) gene panel for epilepsy genes (see online resource 1 for details). Mosaicism of PCDH19 variants was determined based on the simultaneous presence of a variant allele and the reference allele, as PCDH19 is located on the X-chromosome and all patients were male. The percentage of mosaicism was based on the percentage of reads that showed the alternate allele. PCDH19 mosaic male patients from different centres were collected through personal communication between authors, meaning no structured cohort was tested. Detailed clinical data were collected from medical records. The parents of all patients gave informed consent for the publication of clinical data.
Literature search
A literature study was carried out in the PubMed database to identify previously described patients with PCDH19 pathogenic variants.
Results
Molecular analysis
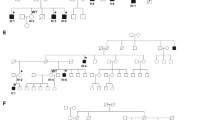
All five patients carried a PCDH19 variant in various degrees of mosaicism (patient A: c.1864G > C, p.Gly622Arg, 60%; patient B: c.840C > G, p.Tyr280*, 22%; patient C: c.462C > G, p.Tyr154*, 65%; patient D: c.1682C > G, p.Pro561Arg, 78%; patient E: c.799G > T p.Glu267*, 20%, RefSeq NM_001184880). According to American College of Medical Genetics and Genomics (ACMG) criteria, the variant of patient A was classified as likely pathogenic, and the variants of patient B-D were classified as pathogenic. The variants of patient B, C and E lead to a premature stopcodon; the variants of patient A and D were both predicted probably damaging and damaging by PolyPhen and SIFT, respectively. None of the variants was present in control databases (Database of Single Nucleotide Polymorphisms (dbSNP), NHLBI Exome Sequencing Project (ESP) and the the Exome Aggregation Consortium (ExAC) database). The Pro561Arg variant has been previously described in two affected female siblings with ID, microcephaly and seizures and was paternally transmitted [15]; the other variants are novel. All variants were confirmed de novo, except for the variant in patient D, whose parents were not tested. No other variants that could explain the phenotypes were found.
Clinical characteristics of the newly reported and previously described male patients with de novo PCDH19 pathogenic variants are summarized and compared to those of previously published females in Table 1. See online resource for extensive clinical descriptions (online resource 2). Overall, all patients had normal development before seizure onset, occurring between 5 and 10 months of age, except for patient E. This patient had a delay in speech development, like his father, before onset of seizures and seizure onset occurred later, at 31 months. First seizure types were generalized clonic or clusters of focal or complex partial seizures. Later seizures were mainly complex partial seizures and primary or secondary generalized clonic and tonic-clonic seizures. In all five patients, seizures tended to cluster and could be provoked by fever. Four out of five patients had mild to severe ID, autism and additional behavioural problems.
Discussion
We here report five male patients with mosaic PCDH19 likely pathogenic variants, which raises the total number of described male patients to nine [2, 13, 14]. Four of the currently described male patients are the oldest reported so far (ages 10, 13 twice and 14 years old), which gives the opportunity to investigate whether PCDH19-related phenotypes evolve the same way in male and female patients. Our current findings confirm previously reported observations of similar clinical features in male and female patients, also for older children [2, 13, 14]. Focal seizures with affective symptoms (fearful screaming) are very common in female patients and become more prevalent with an increasing age [26]. This distinctive seizure type is also reported in three male patients [14, current study]. These similarities suggest that there are no differences in clinical consequences between phenotypes caused by postzygotic PCDH19 pathogenic variants in mosaic males, and phenotypes caused by heterozygous PCDH19 pathogenic variants in females. This lends further support to the hypothesis that cellular interference is the main disease mechanism, as proposed by Depienne et al. [2] The increasing number of affected mosaic male patients undermines the theory of a compensating effect by the nonparalogous PCDH11Y gene in male patients, as proposed by Dibbens et al. [1]. It is highly unlikely that this gene would only compensate for a complete absence of PCDH19 in hemizygous affected males, but not for a partial loss of PCDH19 in mosaic males.
In female patients, seizure frequency often diminishes around puberty [3, 5, 11, 28,29,30], possibly due to hormonal changes [30]. Our 13-year old patient has been seizure free since the age of 10 years old; our 10-year-old patient only has one cluster of seizures per year. However, our other 13- and 14-year-old patients still have ongoing seizures. Although four of our male patients are the oldest reported until now, none of them has reached adolescence yet, and the numbers are still small, making it hard to draw definite conclusions. Nevertheless, since some of the few reported male patients already show a reduction in seizure frequency with increasing age, it seems unlikely that a declining seizure frequency is exclusively occurring in females and is related to female specific hormones.
We hypothesised that males with a mutated allele percentage around 50% in the brain, who would have an inherent high level of cellular interference, may show a more severe clinical picture than males with a lower or higher percentage of mosaicism. High or low percentages of mosaicism would resemble skewed X-inactivation in female patients, which has also been suggested to lead to a milder phenotype [5, 15], although no clear correlation has been shown [25, 29]. Indeed, in our cohort, the three patients with the lowest and highest percentages of mosaicism in blood are the least severely affected (patients B, D and E), and two previously described patients, one with borderline ID and one with normal intelligence showed percentages of mosaicism of 10 and 90%, respectively (patients H and I). However, patient G shows an equal number of mutated and wild-type alleles in blood and is also mildly affected, and patient F shows no mosaicism in blood cells at all, only in fibroblasts, but has moderate to severe ID and ongoing seizures. It is thus not possible to predict the phenotype based on the percentage of mosaicism in blood, most likely because it does not necessarily equal the percentage of mosaicism in the brain.
The number of identified male patients mosaic for PCDH19 pathogenic variants increases [2, 13, 14], probably due to our improving abilities to detect mosaicism in general by using NGS techniques. It is now clear that PCDH19 pathogenic variants can cause epilepsy both in males and females and that mosaicism for PCDH19 pathogenic variants in males might be more common than previously thought. Because our five described patients were gathered through personal communication between authors from different diagnostic centres, no structured cohort with clearly defined inclusion criteria was tested, which makes it difficult to estimate the frequency of mosaic pathogenic PCDH19 variants in male patients. Since the PCDH19 and Dravet syndrome phenotypes show many similarities, testing a cohort of SCN1A-negative, male Dravet syndrome patients for mosaic PCDH19 pathogenic variants could give more insight in the true incidence. In this cohort and in males with clinical features characteristic of a PCDH19-related disorder, NGS techniques with high coverage should be used to look for PCDH19 pathogenic variants, as traditional Sanger Sequencing is not sensitive enough to reliably detect mosaicism. This overview helps create more knowledge about the disease course in male patients, which is extremely relevant for counselling those affected and their families. Reporting on more (older) patients in the future is essential for establishing a good understanding of prognosis in male patients.
References
Dibbens LM, Tarpey PS, Hynes K et al (2008) X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 40:776–781. doi:10.1038/ng.149.X-linked
Depienne C, Bouteiller D, Keren B et al (2009) Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 5:e1000381. doi:10.1371/journal.pgen.1000381
Specchio N, Marini C, Terracciano A et al (2011) Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 52:1251–1257. doi:10.1111/j.1528-1167.2011.03063.x
Camacho A, Simón R, Sanz R et al (2012) Cognitive and behavioral profile in females with epilepsy with PDCH19 mutation: two novel mutations and review of the literature. Epilepsy Behav 24:134–137. doi:10.1016/j.yebeh.2012.02.023
Depienne C, Leguern E (2012) PCDH19-related infantile epileptic encephalopathy: an unusual X-linked inheritance disorder. Hum Mutat 33:627–634. doi:10.1002/humu.22029
Kim SY, Chung HS, Sun W, Kim H (2007) Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 147:996–1021. doi:10.1016/j.neuroscience.2007.03.052
Emond MR, Biswas S, Blevins CJ, Jontes JD (2011) A complex of protocadherin-19 and n-cadherin mediates a novel mechanism of cell adhesion. J Cell Biol 195:1115–1121. doi:10.1083/jcb.201108115
Compagnucci C, Petrini S, Higuraschi N et al (2015) Characterizing PCDH19 in human induced pluripotent stem cells (iPSCs) and iPSC-derived developing neurons: emerging role of a protein involved in controlling polarity during neurogenesis. Oncotarget 6:26804–26813. doi:10.18632/oncotarget.5757
Cooper SR, Emond MR, Duy PQ et al (2015) Protocadherins control the modular assembly of neuronal columns in the zebrafish optic tectum. J Cell Biol 211:807–814. doi:10.1083/jcb.201507108
Pederick DT, Homan CC, Jaehne EJ et al (2016) Pcdh19 loss-of-function increases neuronal migration in vitro but is dispensable for brain development in mice. Sci Rep 6:26765. doi:10.1038/srep26765
Scheffer IE, Turner SJ, Dibbens LM et al (2008) Epilepsy and mental retardation limited to females: an under-recognized disorder. Brain 131:918–927. doi:10.1093/brain/awm338
Van Harssel JJT, Weckhuysen S, Van Kempen MJA et al (2013) Clinical and genetic aspects of PCDH19-related epilepsy syndromes and the possible role of PCDH19 mutations in males with autism spectrum disorders. Neurogenetics 14:23–34. doi:10.1007/s10048-013-0353-1
Thiffault I, Farrow E, Smith L et al (2016) PCDH19-related epileptic encephalopathy in a male mosaic for a truncating variant. Am J Med Genet Part A 170:1585–1589. doi:10.1002/ajmg.a.37617
Terracciano A, Trivisano M, Cusmai R et al (2016) PCDH19-related epilepsy in two mosaic male patients. Epilepsia 57:e51–e55. doi:10.1111/epi.13295
Depienne C, Trouillard O, Bouteiller D et al (2011) Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females. Hum Mutat 32:1959–1975. doi:10.1002/humu.21373
Cappelletti S, Specchio N, Moavero R et al (2015) Cognitive development in females with PCDH19 gene-related epilepsy. Epilepsy Behav 42:36–40. doi:10.1016/j.yebeh.2014.10.019
Dibbens LM, Kneen R, Bayly MA et al (2011) Recurrence risk of epilepsy and mental retardation in females due to parental mosaicism of PCDH19 mutations. Neurology 76:1514–1519. doi:10.1212/WNL.0b013e318217e7b6
Dimova PS, Kirov A, Todorova A et al (2012) A novel PCDH19 mutation inherited from an unaffected mother. Pediatr Neurol 46:397–400. doi:10.1016/j.pediatrneurol.2012.03.004
Gagliardi M, Annesi G, Sesta M et al (2015) PCDH19 mutations in female patients from southern Italy. Seizure 24:118–120. doi:10.1016/j.seizure.2014.08.010
Higurashi N, Shi X, Yasumoto S et al (2012) PCDH19 mutation in Japanese females with epilepsy. Epilepsy Res 99:28–37. doi:10.1016/j.eplepsyres.2011.10.014
Higurashi N, Nakamura M, Sugai M et al (2013) PCDH19-related female-limited epilepsy: further details regarding early clinical features and therapeutic efficacy. Epilepsy Res 106:191–199. doi:10.1016/j.eplepsyres.2013.04.005
Hynes K, Tarpey P, Dibbens LM et al (2010) Epilepsy and mental retardation limited to females with PCDH19 mutations can present de novo or in single generation families. J Med Genet 47:211–216. doi:10.1136/jmg.2009.068817
Jamal SM, Basran RK, Newton S et al (2010) Novel de novo PCDH19 mutations in three unrelated females with epilepsy female restricted mental retardation syndrome. Am J Med Genet Part A 152(A):2475–2481. doi:10.1002/ajmg.a.33611
Kwong AKY, Ho ACC, Fung CW, Wong VCN (2015) Analysis of mutations in 7 genes associated with neuronal excitability and synaptic transmission in a cohort of children with non-syndromic infantile epileptic encephalopathy. PLoS One 10:e0126446. doi:10.1371/journal.pone.0126446
Leonardi E, Sartori S, Vecchi M et al (2014) Identification of four novel PCDH19 mutations and prediction of their functional impact. Ann Hum Genet 78:389–398. doi:10.1111/ahg.12082
Marini C, Darra F, Specchio N et al (2012) Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 53:2111–2119. doi:10.1111/j.1528-1167.2012.03649.x
Vincent AK, Noor A, Janson A et al (2012) Identification of genomic deletions spanning the PCDH19 gene in two unrelated girls with intellectual disability and seizures. Clin Genet 82:540–545. doi:10.1111/j.1399-0004.2011.01812.x
Liua A, Xu X, Yang X et al (2016) The clinical Spectrum of female epilepsy patients with PCDH19 mutations in a Chinese population. Clin Genet. doi:10.1111/cge.12846
Marini C, Mei D, Parmeggiani L et al (2010) Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology 75:646–653. doi:10.1212/WNL.0b013e3181ed9e67
Tan C, Shard C, Ranieri E et al (2015) Mutations of protocadherin 19 in female epilepsy (PCDH19-FE) lead to allopregnanolone deficiency. Hum Mol Genet 24:5250–5259. doi:10.1093/hmg/ddv245
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest. HAN and KLH are employed by Ambry Genetics; PCDH19 sequencing is among its commercially available tests. This study was supported by the “Stichting Vrienden WKZ” (project 1614054) on behalf of Stichting Panta Rhei. Funders had no involvement in the study design, the collection, analysis, and interpretation of data, the writing of the report and in the decision to submit the paper for publication.
Electronic supplementary material
ESM 1
(PDF 94 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
de Lange, I.M., Rump, P., Neuteboom, R.F. et al. Male patients affected by mosaic PCDH19 mutations: five new cases. Neurogenetics 18, 147–153 (2017). https://doi.org/10.1007/s10048-017-0517-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-017-0517-5