
Abstract
Despite a plethora of publications on the murine model of cutaneous leishmaniasis and their contribution to our understanding of the factors that regulate the development of CD4+ T cell immunity in vivo, there is still no effective vaccine against the human disease. While recovery from natural or experimental infection with Leishmania major, the causative agent of human cutaneous leishmaniasis, results in persistence of parasites at the primary infection site and the development of long-lasting immunity to reinfection, vaccination with killed parasites or recombinant proteins induces only short-term protection. The reasons for the difference in protective immunity following recovery from live infection and vaccination with heat-killed parasites are not known. This may in part be related to persistence of live parasites following healing of primary cutaneous lesions, because complete clearance of parasites leads to rapid loss of infection-induced immunity. Recent reports indicate that in addition to persistent parasites, IL-10-producing natural regulatory T cells may also play critical roles in the maintenance and loss of infection-induced immunity. This review focuses on current understanding of the factors that regulate the development, maintenance and loss of anti-Leishmania memory responses and highlights the role of persistent parasites and regulatory T cells in this process. Understanding these factors is crucial for designing effective vaccines and vaccination strategies against cutaneous leishmaniasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immunologic memory is the capacity of the immune system to respond faster and better (both in quality and quantity) to a secondary antigenic challenge than the first encounter with the same antigen [1–3]. This is one of the hallmarks of the adaptive immunity and is the basis for vaccination against infectious diseases. There are mounting and undisputable evidence showing that strong memory responses develop following most viral and some bacterial infections [4–6]. In contrast, there is a continuing controversy about the nature and extent of immunologic memory that develops following parasitic infections. Although there is clear evidence that certain parasitic infections, such as leishmaniasis and toxoplasmosis, are known to induce excellent protection against a secondary challenge, some researchers believe that this is due to concomitant immunity rather than true memory [7]. Complicating this issue is the fact that most parasitic infections are chronic in nature resulting in continuous release of antigens and the consequent chronic stimulation of the immune system. Such chronic stimulation is known to favor the maintenance of effector cells and to impair their transition into memory cells [8–10]. Leishmania major infections in both human and mice result in persistence of parasites at the primary site of infection and solid immunity to reinfection. This observation is the basis for leishmanization, a practice that involves deliberate inoculation of live organisms to hidden parts of the body with the idea that healing from the resulting lesion provides solid protection against subsequent natural infections. Studies in mice indicate that memory CD4+ T cells mediate this infection-induced immunity. In this review, we focus on the role of persistent parasites and regulatory T cells in the maintenance and loss of anti-Leishmania memory cells, and discuss their implications in vaccination strategies against cutaneous leishmaniasis.
Cutaneous leishmaniasis: the disease
Leishmaniasis is a globally widespread group of parasitic diseases caused by different species of parasite in the genus Leishmania. The parasite is transmitted by the bite of infected female Phlebotomine sand fly. Most leishmaniases are zoonotic (animal to human transmission) and are transmitted to humans who are accidentally exposed to the natural transmission cycle. In the anthroponotic form (human to human transmission) humans are the sole reservoir. The disease is endemic in 88 countries: 72 are developing countries out of which 13 are among the least developed countries of the world [11]. Currently, it is estimated that about 12 million people are afflicted with the disease worldwide with 350 million people at risk [12, 13]. About 1.5–2 million new cases occur annually all over the world with majority of them occurring in the tropics and subtropics.
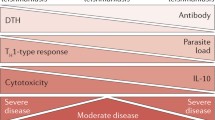
Leishmania infections can produce diverse symptoms in the mammalian host, depending on the species of the parasites and the host genetic makeup. L. donovani and L. infantum cause visceral leishmaniasis, the most severe form of the disease [11, 14]. L. braziliensis causes mucocutaneous leishmaniasis, infecting the mucous membranes of the host [11, 14, 15]. Lastly, L. tropica, L. major, and L. mexicana cause cutaneous leishmaniasis producing skin ulcers, which is the most common type of the disease. Cutaneous leishmaniasis can present as localized skin ulcers on the exposed parts of the body, (simple cutaneous leishmaniasis), or serious widespread lesions and ulcers all over the body (diffuse cutaneous leishmaniasis). While the simple cutaneous form is self-limiting (healing with strong immunity to reinfection), the diffuse cutaneous form never heals and tends to relapse after treatment. These differences in clinical disease presentation are related to the nature of the host immune response. A strong cell-mediated immune response is able to contain parasite proliferation resulting in healing, whereas non-healing disease progression and visceralization are related to inability to mount a strong cell-mediated immunity.
Primary immunity to Leishmania major
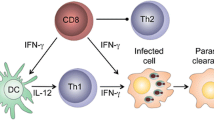
Resistance or susceptibility to L. major infections in mice is dependent on the type of CD4+ helper T cell (Th) subset that is induced [16–18]. Healing in resistant mice infected with L. major is associated with the development of IFN-γ-producing CD4+ Th1 cells. These cells activate macrophages to produce nitric oxide, an effector molecule for killing intracellular Leishmania parasites [16, 19–21]. In contrast, cells from susceptible mice produce early IL-4 that promotes the development and expansion of CD4+ Th2 cells. The expanded and activated Th2 cells produce IL-4 and IL-10, cytokines that deactivate macrophages and inhibit intracellular parasite killing [16, 20, 21]. A key factor that determines resistance or susceptibility to L. major in mice is the production of (and responsiveness) to IL-12 [22–24]. The highly susceptible BALB/c produce less and respond poorly to IL-12 due to downregulation of IL-12 receptor β chain [24–26]. In contrast, resistant mice produce more and maintain their IL-12 responsiveness [24, 26]. Another cytokine that regulates disease outcome is IL-10. IL-10 gene deficient mice are highly resistant to L. major [27], and over expression of IL-10 renders resistant mice susceptible [28]. In addition to deactivating macrophages and inhibiting intracellular parasite killing, IL-10 also directly inhibits the development of Th1 cells and their production of IFN-γ [29, 30]. Recently, IL-10 produced by natural CD4+CD25+ regulatory T cells has been shown to play an important role in disease chronicity [31, 32]. For comprehensive information on the factors that regulate the outcome of primary L. major infection, the reader is referred to the following current and detailed reviews [33–36].
Infection-induced immunity in leishmaniasis
It has since been known that recovery from active infection in humans and mice is associated with the development of strong and durable immunity to rechallenge infection. This is referred to as infection-induced immunity and is the first strong indication that memory T cells develop after recovery from Leishmaniasis. This realization that solid immunity develops after recovery from natural or experimental infection is the fundamental principle underlying leishmanization, a practice in which individuals are infected with live organisms to protect against more serious ulcers after natural infection [37, 38]. Leishmanization has been employed for centuries and is still currently practiced in some countries including Uzbekistan, Afghanistan, Iraq, and Iran [39–41]. Due to complications and lack of standardization (some 5–10% develop severe disease), there are calls for the practice to be discontinued, although there are recent efforts to standardize it and make it a useful tool for rapidly assessing the efficacy of new vaccines [41]. Understanding the factors that regulate and mediate infection-induced resistance is critically important for designing an effective vaccine and vaccination strategies against leishmaniasis.
Depletion and adoptive transfer studies indicate that IFN-γ-producing CD4+ T cells mediate infection-induced resistance in mice. For instance, depletion of CD4+ T cells or treatment with anti-IFN-γ neutralizing mAb abolishes this immunity [42, 43]. Similar to immunity after primary infection, the production of IFN-γ by memory CD4+ T cells and maintenance of cell-mediated immunity against L. major is dependent on IL-12 production by antigen presenting cells [44, 45]. Thus, the highly susceptible IL-12 deficient mice treated with rIL-12 develop Th1 response and resolve their lesion. However, in contrast to wild-type mice, these rIL-12-treated IL-12 deficient mice develop progressive disease and uncontrolled parasite replication upon rechallenge infection [44, 46]. In fact, lesion recrudescence (disease reactivation) occurred at the primary site of infection in most healed IL-12-deficient mice prior to secondary challenge, suggesting that exogenous administration of rIL-12 was able to promote only short-term resistance. Furthermore, while immune cells from healed wild-type mice protected RAG deficient and naïve wild-type mice from L. major challenge, cells from healed IL-12 deficient mice failed to confer protection to naïve IL-12 deficient mice although they perfectly protected naïve wild-type mice [44, 46]. Taken together, these studies indicate that IL-12 is necessary both for the development and maintenance of both anti-Leishmania effector and memory cells in vivo. There are several possible explanations as to why IL-12 may be necessary to maintain infection-induced resistance and anti-Leishmania memory responses. IL-12 may be required for optimal proliferation and differentiation of memory CD4+ T into IFN-γ-producing effector cells. Alternatively, IL-12 could be acting to enhance the development and survival of Leishmania-specific effector memory cells. These sub-populations of memory cells are important for mediating rapid secondary anti-Leishmania immunity [47–49] (Uzonna et al., unpublished data).
Under certain conditions, infection-induced immunity can be lost and previously immune animals become highly susceptible to rechallenge infections. This loss of resistance has been linked to complete parasite clearance [32, 50], suggesting that persistent parasites are important for the maintenance of anti-Leishmania memory responses (see below). Recent studies from our group show that infection-induced immunity can also be lost despite the presence of persistent parasites. Injection of autoclaved or freeze-thawed (killed) parasites into B6 mice that have healed their primary L. major infection results in rapid loss of infection-induced immunity. These mice (unlike those given PBS) became highly susceptible to virulent L. major challenge (Okwor et al., submitted). This loss of infection-induced immunity was associated with rapid expansion of IL-10-producing CD4+CD25+ regulatory T cells in the draining lymph nodes. Depletion of CD25+ cells or blockade of IL-10 signaling with anti-IL-10R mAb abolished this loss of immunity, strongly suggesting that these cells are responsible for this phenomenon. These regulatory cells have previously been shown to enhance disease in infected mice and to mediate reactivation of latent leishmaniasis [51]. Interestingly, this striking loss of immunity is not observed if healed mice were injected with avirulent (genetically attenuated) live parasites, suggesting that antigens from killed and live parasites may be presented differently to T cells, particularly CD4+CD25+ regulatory T cells.
Leishmaniasis and immunologic memory
The importance of immunologic memory to the development of vaccines cannot be over emphasized. An understanding of how immunologic memory is generated, maintained and lost is essential for the development of effective vaccines and vaccination strategies against infectious agents and their diseases. Since resistance to cutaneous leishmaniasis is primarily mediated by cell-mediated immunity (T cells), it is reasonable to assume that memory T cells mediate secondary anti-Leishmania immunity.
Memory T cells arise from the expansion and differentiation of antigen-specific T cells upon interaction with their cognate antigen in the secondary lymphoid organs. They confer protective immunity in peripheral tissues and mediate recall responses in secondary lymphoid organs. In order to provide efficient and effective protection, memory T cells must rapidly home to the lymph node draining the challenge site, proliferate and then migrate to the site of antigenic challenge in the periphery to mediate their effector function. This proliferation in the lymph node is important because the frequency of antigen-specific T cells in the memory pool, although higher than naïve cells, is not high enough to provide rapid protection against pathogens [2, 52]. Furthermore, pathogen-specific effector T cells may be short-lived and hence need to be continuously replenished from the memory T cell pool [53].
On the basis of their homing characteristics and effector function, two types of memory T cells have been distinguished in humans [54, 55]. Central memory T cells (Tcm) express high levels of CCR7 and CD62L, molecules that are important for extravasation of T cells through the high endothelial venules and homing to the secondary lymphoid organs [56, 57]. In contrast, effector memory T cells (Tem) do not express significant levels of these molecules and home preferentially to non-lymphoid tissue where they exert effector functions [56, 57]. Upon antigenic recall stimulation, Tcm cells produce only IL-2 and do not make effector cytokines such as IFN-γ. In contrast, tissue homing Tem cells produce copious amounts of effector cytokines (IL-4 and IFN-γ) upon antigenic challenge and may constitutively express other cytotoxic effector molecules such as perforins and granzymes (CD8+ cells). As in humans, several reports in mice have corroborated these findings and further suggest that the memory T cell pool is heterogeneous, containing cells with different migratory and effector capacities [1, 58–60].
The relative contributions of these subsets of memory cells in secondary immunity to Leishmania major is still poorly defined but is a subject of intense investigation by many labs including ours (see below). We found that in healed C57BL/6 mice, Leishmania-specific memory cells proliferating in response to secondary challenge differentially express CD62L depending on their location [47]. In the draining lymph node, most of the proliferating cells expressed high levels of CD62L whereas in the footpad, 98% of the proliferating cells were CD62L low [47]. This dichotomy in phenotype was also reflected in their effector function such that CD62L high cells produced predominantly IL-2 while CD62L low cells in the periphery (footpad) produced high levels of IL-2 and IFN-γ [47]. Functional studies revealed that although both subsets of anti-Leishmania memory cells are protective, their efficiency of protection is dramatically different. The CD62L low (Tem) cells mediate rapid protection (within 2 weeks), whereas the protection mediated by CD62L high (Tcm) cells is delayed (≥6 weeks for observable significant protection) [47]. Thus, as seen in other systems, both Tcm and Tem cells develop in Leishmania-infected mice.
Parasite persistence and maintenance of immunologic memory in Leishmaniasis
Following recovery from natural or deliberate infection with Leishmania sp, a small number of viable parasites persist in the immune host at the primary site of infection and its draining lymph node [61, 62]. This has led to the dogma that Leishmania organisms persist forever and are never completely eliminated from infected host. Under certain conditions including malnutrition and immunosuppression, recrudescence (reactivation leishmaniasis) can occur from persisting parasites. Indeed, reactivation leishmaniasis is a major complication of AIDS in the sub-Saharan Africa and India [63–65].
It has been debated whether “reactivation disease” is due to a new infection or truly a reactivation of persistent parasites. The same strain of parasite that caused initial disease was isolated from 50% of individuals with recurrent cutaneous leishmaniasis due to L. braziliensis [66]. However, one might argue that this may have been from a new infection with the same strain. The strongest evidence linking persistent parasites to reactivation disease comes from murine studies. Healed mice treated with L-NIL, the competitive inhibitor of nitric oxide synthase (important for nitric oxide production), develop recrudescence and progressive disease [67]. Similarly, adoptive transfer of CD4+CD25+ cells isolated from infected mice into healed mice also led to recrudescence at the primary site of infection [51, 68]. Furthermore, administration of rIL-12 to IL-12 deficient mice promotes control of L. major but recrudescence occurs following cessation of cytokine treatment [44]. We have also found that administration of anti-IFN-γ to healed mice results in reactivation disease (Uzonna, unpublished data). Collectively, these murine studies suggest that some cases of reactivation disease in humans could arise from persistent parasites.
The factors and conditions that favor parasite persistence are not clearly understood but are intensely investigated by many labs. We showed that the generation of an exclusive Th1 immune response (with no detectable Th2 component) in BALB/c mice leads to resistance that is associated with complete parasite clearance [50, 69]. In this model, parasite persistence occurred in an infection initiated with intermediate dose, which results in the development of weak protective immune response with a substantial Th2 component. Others have shown that the extent of IL-10 production during infection influences parasite persistence. For instance, IL-10 receptor deficient mice or healed WT mice treated with anti-IL-10R mAb mount strong Th1 response and completely clear parasite [32]. In this model, natural CD4+CD25+Foxp3+ regulatory T cells were implicated as the major source of IL-10 and hence the key cells that mediate parasite persistence in mice [51, 68]. Unlike murine studies, the factors responsible for parasite persistence and development of subclinical disease in humans are not known and have not been properly investigated. Given that CD4+CD25+ T cells isolated from lesions of infected patients mediate suppressive activities in vitro [70], and the presence of high levels of IL-10 in the plasma of human patients [71, 72], it is conceivable that as in mice, both CD4+CD25+ T cells and IL-10 may also play important roles in the persistence of parasites in humans.
Why few hundreds to thousand parasites persist at the primary site of infection when such mouse can control a secondary challenge with millions of parasites injected at different site is still unknown. One plausible explanation is that persistent parasites reside in cells that are resistant to IFN-γ-mediated activation for parasite killing. These so-called “safe heavens” [73] would allow minimal parasite replication without eliciting significant inflammatory responses, and upon lysis would release parasites that can infect permissive macrophages. Another mechanism may involve the role of local IL-10-producing regulatory T cells that act to downregulate the effect of IFN-γ at the infection site. Consistent with this, depletion of CD4+CD25+ T cells in healed mice prevented reactivation disease caused by secondary challenge, whereas transfer of CD4+CD25+ T cells purified from infected mice lead to disease recrudescence [51, 68]. Interestingly, healed and resistant mice manipulated to completely clear parasites also lost their immunity suggesting that persistent live parasites are required for the maintenance of anti-Leishmania memory [32, 50]. Thus, persistence of parasites may have provided an evolutionary balance and advantage for both the host and the parasite; providing a source of continuous antigenic stimuli required for maintaining protective memory cells while permitting a readily available pool of parasites for transmission by the vector to a new host.
The findings that complete parasite clearance results in loss of established immunity is surprising given that several reports show that memory cells can persist in the absence of their cognate antigens [2, 74, 75]. However, this is consistent with other reports that show the requirement of persistent antigens for the maintenance of immunologic memory [53, 76, 77]. If immunity induced by live parasites is lost upon parasite clearance, it might imply that as with other parasitic infections [9], true memory cells do not develop following Leishmania infection. This might be particularly an attractive proposal since leishmaniasis is a chronic disease and some studies suggest that continued antigenic stimulation inhibits memory T cell development [1, 78]. In this regard, several investigators suggest that concomitant immunity and not true memory cells is responsible for secondary anti-parasitic immunity [7, 79–82]. Consistent with this, infected mice presenting non-healing lesion in one foot or ear have been shown to resist virulent contralateral challenges [83]. Recently, the induction of local natural regulatory T cells has been suggested to be responsible for this inability to clear parasites at the primary infection site [32, 51].
We do not think that concomitant immunity is totally responsible for secondary anti-Leishmania immunity because we recently showed that both effector and central memory T cells are induced following L. major infection in mice [47–49]. These cells mediate protection against secondary L. major challenge when adoptively transferred into naïve mice [47]. Interestingly, we also found that while Leishmania-specific effector memory cells decline in the absence of parasites, the maintenance of central memory cells appears to be independent of live parasites [47–49]. A closer look at the reports that showed complete loss of immunity following clearance of parasites reveal that these studies were performed using ultra low dose (100–1,000 parasites) infection of BALB/c [50] or IL-10R deficient C57BL/6 [32] mice. It is possible that the low dose infection used in these studies was insufficient to generate a strong pathogen-independent memory T cell population since it is known that antigen dose influences both the quantity (magnitude) and quality of T cell memory response [78]. Furthermore, in the absence of IL-10, activated antigen-specific T cells may progress to become Tem cells, and thus fail to generate any Tcm cells whose maintenance are independent of parasites [47].
To circumvent these problems, we have used a high dose infection with a mutant parasite to investigate the influence of persistent parasite on the maintenance of anti-Leishmania memory cells. Leishmania major parasites lacking the gene dihydrofolate reductase-thymidilate synthetase (termed dhfr-ts), which is important for the synthesis of thymidine, are non pathogenic in mice and are completely cleared by 8 weeks post-infection [84]. Mice infected with high dose dhfr-ts parasites show early immune response that is comparable in quality to those infected with wild-type parasites, and are resistant to early virulent challenge (Uzonna et al., submitted). However, from 12 weeks post-infection (when dhfr-ts are completely cleared), cells from dhfr-ts-infected mice (unlike those from WT) failed to proliferate or produce IFN-γ upon short-term (72 h) in vitro stimulated with SLA. Furthermore, dhfr-ts-infected mice did not exhibit antigen-specific delayed type hypersensitivity (DTH) response upon rechallenge and were unable to rapidly control parasite proliferation after secondary challenge. Interestingly, these mice showed delayed but significant protection over their naïve age-matched controls [47]. These results indicate that anti-Leishmania memory cells develop after infection and are maintained in the absence of live parasites. However, they strongly suggest that the nature and quality of memory cells maintained in the presence and absence of live parasites are different. These findings, if shown to be true in humans, must be considered in vaccine designs and strategies against leishmaniasis.
Recently, we showed that vaccination of BALB/c mice with a mutant parasite induces striking protection in the absence of a DTH and significant IFN-γ production [85]. Leishmania major parasites with targeted deletion of the gene involved in transporting GDP-mannose into the Golgi for synthesis of glycophosphate conjugate repeats, termed lpg2-, are unable to make lipophosphoglycan (LPG) and other phosphoglycans and are highly attenuated in vivo [73]. Remarkably, these parasites persist in BALB/c mice without causing any pathology [73] and do not induce significant effector T cell responses several weeks after infection. Nevertheless, when challenged with virulent L. major, lpg2-infected mice were resistant [85]. We speculate that the protective immune cells bear the hallmarks of Tcm cells we described in mice infected with high dose dhfr-ts L. major. If it is shown to be true, these studies strongly suggest that Tcm cells might exist in infected mice even in the face of persistent parasites, and further corroborate our findings that both Tem and Tcm cells are induced following L. major infection, but the requirements of these different memory T cell subsets are different [48, 49].
Vaccines and vaccination strategies
Although the factors that regulate the development of protective immune response in Leishmaniasis are very well known, there is still no effective vaccine against the disease. Several vaccination trials have been conducted in several parts of the world using heat-killed whole Leishmania promastigotes as immunogen [86–90]. In some trials, these crude antigen preparations have been used with or without Bacillus Calmette Guerin (BCG) [91–93]. Overall, these trials have yielded very poor and disappointing results. This is predictable given that similar murine studies show that although heat-killed Leishmania or recombinant Leishmania immunogens induces an early Th1 response and protection, this immunity is not sustained and wanes with time [22, 94, 95]. Like heat-killed vaccines, immunization with purified parasite fractions [96, 97] or recombinant proteins [98–100] also do not induce significant and reproducible protection against virulent challenges in humans.
In contrast to vaccination with killed parasites or purified Leishmania proteins, recovery from deliberate (leishmanization in humans) or active infections in both human and mice is associated with life-long immunity. These results strongly suggest that there are fundamental differences in memory responses induced by killed or recombinant proteins and live infections. We hypothesize that the failure of heat-killed or protein-based vaccines to induce long-term immunity may be related to their failure to induce sustained effector-like memory cells, which mediate DTH response and rapid anti-Leishmania immunity following secondary challenge. If this is the case, it implies that an effective anti-Leishmania vaccine must be able to induce both population of memory T cells, akin to what is seen following healing of live infections [47]. In this regard, we recently found that repeated injection (boosting) of killed parasites into naïve mice lead to the induction of both Tem and Tcm cells resulting in prolonged (9 months after the last boost) protection against virulent challenge (Okwor and Uzonna, unpublished data).
Implications for vaccine designs and vaccination strategies
Both Tem and Tcm cells develop in mice infected with L. major. While Tem cells mediate DTH response and rapid secondary anti-Leishmania immunity, the protection mediated by Tcm cells is delayed and weak. These findings have important implications for vaccine development and effective vaccination strategy against leishmaniasis and perhaps other chronic infectious diseases caused by intracellular pathogens. Mice with persistent parasites have fast-responding effector memory cells that mediate rapid secondary immunity. However, these mice develop huge inflammatory DTH response upon secondary challenge and under adverse conditions such as immunosuppression, aging, malnutrition, etc., may develop reactivation leishmaniasis [61, 64, 101]. In contrast, mice that completely clear parasite have memory cells, which do not elicit DTH response but are capable of mediating delayed and weak but appreciably significant protection against secondary challenge. It is not yet known whether complete parasite clearance occurs in humans, and what impact such clearance would have on resistance. If it is shown that the maintenance of fast-acting effector memory cells in humans (as in mice) requires persistent parasites while central memory cells do not, these must be considered in vaccination strategies against cutaneous leishmaniasis. Due to their replicating nature, it is conceivable that live parasite-based vaccines would preferentially induce both effector and central memory cells [47]. In contrast, subunit and killed parasites-based vaccines may preferentially induce central memory cells. Thus, determining the factors induced by live parasites that drive the generation of fast-protecting effector memory cells is necessary in order to enhance the immunity induced by killed antigens.
Should we therefore aim to generate, by vaccination, a state leading to persistent infection or complete parasite elimination? Given the potential hazard associated with persistent infection (i.e., reactivation disease), we would favor the generation of a response that can completely eliminate the pathogen or the attenuated pathogen employed for vaccination, with the maintenance of the appropriate resistance by natural exposure to parasites and/or deliberate booster immunizations.
Concluding remarks
The immune response to Leishmania is complex. The outcome of infection is a product of many factors, including the host genetics and the infecting specie. Resolution of primary infection with Leishmania results in solid immunity to reinfection. Both Tem and Tcm CD4+ T cells mediate this infection-induced immunity. The requirement of persistent parasites for the maintenance of these sub-populations of memory cells is different: Tem cells are rapidly lost in the absence of live parasites while the maintenance of Tcm cells is independent of live parasites. Understanding the factors that regulate parasite persistent and its role in maintenance of immunologic memory in leishmaniasis is critical for proper design and development of effective vaccine and vaccination strategies against the disease. The promising results (protection) obtained in mice vaccinated with mutant parasites like lpg2 KO [85, 102] and dhfr-ts KO [84, 103] suggest the development of an anti-Leishmania vaccine is feasible. However, some questions still remain unanswered. Are persistent parasites required to maintain anti-Leishmania immunity in humans? If so, are there differences (function, phenotype, and migratory properties) in CD4+ (Tcm and Tem) memory cell population following infection with Leishmania or vaccination in humans? How long can Tcm cells persist in the absence of live parasites? Can non-replicating parasites maintain memory (Tcm and Tem) cells induced by vaccination with non-replicating avirulent live parasites? What is the nature of memory T cell subsets induced following vaccination with heat-killed Leishmania vaccine? How do Tcm cells mediate secondary anti-Leishmania immunity? Do they have to first convert to Tem? Does different Leishmania sp require different conditions for memory cell maintenance? A good anti-Leishmania vaccine should protect against most of the human infection by the different Leishmania species in the absence of persisting parasites in order to reduce the risk of reactivation leishmaniasis.
References
Swain SL, Agrewala JN, Brown DM, Roman E. Regulation of memory CD4 T cells: generation, localization and persistence. Adv Exp Med Biol. 2002;512:113–20.
Sprent J, Surh CD. T cell memory. Annu Rev Immunol. 2002;20:551–79.
Gourley TS, Wherry EJ, Masopust D, Ahmed R. Generation and maintenance of immunological memory. Semin Immunol. 2004;16:323–33.
Whitmire JK, Murali-Krishna K, Altman J, Ahmed R. Antiviral CD4 and CD8 T-cell memory: differences in the size of the response and activation requirements. Philos Trans R Soc Lond B Biol Sci. 2000;355:373–9.
Slifka MK. Immunological memory to viral infection. Curr Opin Immunol. 2004;16:443–50.
Zinkernagel RM, Hengartner H. On immunity against infections and vaccines: credo 2004. Scand J Immunol. 2004;60:9–13.
Brown SP, Grenfell BT. An unlikely partnership: parasites, concomitant immunity and host defence. Proc Biol Sci. 2001;268:2543–9.
Manfras BJ, Reuter S, Wendland T, Boehm BO, Kern P. Impeded Th1 CD4 memory T cell generation in chronic-persisting liver infection with Echinococcus multilocularis. Int Immunol. 2004;16:43–50.
Brake DA. Parasites and immune responses: memory illusion? DNA Cell Biol. 2003;22:405–19.
Albareda MC, Laucella SA, Alvarez MG, Armenti AH, Bertochi G, Tarleton RL, Postan M. Trypanosoma cruzi modulates the profile of memory CD8+ T cells in chronic Chagas’ disease patients. Int Immunol. 2006;18:465–71.
Desjeux P. Leishmaniasis. Nat Rev Microbiol. 2004;2:692.
Desjeux P. Human leishmaniases: epidemiology and public health aspects. World Health Stat Q. 1992;45:267–75.
Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27:305–18.
Berman J. Current treatment approaches to leishmaniasis. Curr Opin Infect Dis. 2003;16:397–401.
Vanloubbeeck Y., Jones DE. The immunology of Leishmania infection and the implications for vaccine development. Ann N Y Acad Sci. 2004;1026:267–72.
Reiner SL, Locksley RM. The regulation of immunity to Leishmania major. Annu Rev Immunol. 1995;13:151–77.
Scott P. Th cell development and regulation in experimental cutaneous leishmaniasis. Chem Immunol. 1996;63:98–114.
Locksley RM, Heinzel FP, Holaday BJ, Mutha SS, Reiner SL, Sadick MD. Induction of Th1 and Th2 CD4+ subsets during murine Leishmania major infection. Res Immunol. 1991;142:28–32.
Scharton-Kersten T, Afonso LC, Wysocka M, Trinchieri G, Scott P. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J Immunol. 1995;154:5320–30.
Scott P, Pearce E, Cheever AW, Coffman RL, Sher A. Role of cytokines and CD4+ T-cell subsets in the regulation of parasite immunity and disease. Immunol Rev. 1989;112:161–82.
Scott P. The role of Th1 and Th2 cells in experimental cutaneous leishmaniasis. Exp Parasitol. 1989;68:369–72.
Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science. 1994;263:235–7.
Park AY, Scott P. Il-12: keeping cell-mediated immunity alive. Scand J Immunol. 2001;53:529–32.
Jones D, Elloso MM, Showe L, Williams D, Trinchieri G, Scott P. Differential regulation of the interleukin-12 receptor during the innate immune response to Leishmania major. Infect Immun. 1998;66:3818–24.
Himmelrich H, Parra-Lopez C, Tacchini-Cottier F, Louis JA, Launois P. The IL-4 rapidly produced in BALB/c mice after infection with Leishmania major down-regulates IL-12 receptor beta 2-chain expression on CD4+ T cells resulting in a state of unresponsiveness to IL-12. J Immunol. 1998;161:6156–63.
Louis J, Himmelrich H, Parra-Lopez C, Tacchini-Cottier F, Launois P. Regulation of protective immunity against Leishmania major in mice. Curr Opin Immunol. 1998;10:459–64.
Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol. 2001;166:1141–7.
Groux H, Cottrez F, Rouleau M, Mauze S, Antonenko S, Hurst S, McNeil T, Bigler M, Roncarolo MG, Coffman RL. A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J Immunol. 1999;162:1723–9.
Mosmann TR, Moore KW. The role of IL-10 in cross-regulation of Th1 and Th2 responses. Immunol Today. 1991;12:A49–53.
Chatelain R, Mauze S, Coffman RL. Experimental Leishmania major infection in mice: role of IL-10. Parasite Immunol. 1999;21:211–8.
Belkaid Y. The role of CD4+CD25+ regulatory T cells in Leishmania infection. Expert Opin Biol Ther. 2003;3:875–85.
Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–7.
Hondowicz B, Scott P. Influence of host and parasite factors on the innate immune response and Th2 stability following infection with Leishmania major. Microbes Infect. 1999;1:65–71.
Gumy A, Louis JA, Launois P. The murine model of infection with Leishmania major and its importance for the deciphering of mechanisms underlying differences in Th cell differentiation in mice from different genetic backgrounds. Int J Parasitol. 2004;34:433–44.
von Stebut E, Udey MC. Requirements for Th1-dependent immunity against infection with Leishmania major. Microbes Infect. 2004;6:1102–9.
Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol. 2002;2:845–58.
Nadim A, Javadian E, Tahvildar-Bidruni G, Ghorbani M. Effectiveness of leishmanization in the control of cutaneous leishmaniasis. Bull Soc Pathol Exot Filiales. 1983;76:377–83.
Modabber F. Experiences with vaccines against cutaneous leishmaniasis: of men and mice. Parasitology. 1989;98 Suppl:S49–60.
Shuikina EE, Sergiev VP, Triers II, Shcherbakov VA, Diveev S. [Experience of antileishmaniasis vaccinations with cultures of Leishmania tropica major grown in various types of media]. Med Parazitol (Mosk). 1968;37:648–51.
Sergiev PG, Beislekhem RI, Moshkovskii, ShD, Demina NA, Kellina OI. Results of massive vaccination against zoonotic cutaneous leishmaniasis. Med Parazitol (Mosk). 1970;39:541–51.
Khamesipour A, Dowlati Y, Asilian A, Hashemi-Fesharki R, Javadi A, Noazin S, Modabber F. Leishmanization: use of an old method for evaluation of candidate vaccines against leishmaniasis. Vaccine. 2005;23:3642–8.
Muller I. Role of T cell subsets during the recall of immunologic memory to Leishmania major. Eur J Immunol. 1992;22:3063–9.
Muller I, Kropf P, Etges RJ, Louis JA. Gamma interferon response in secondary Leishmania major infection: role of CD8+ T cells. Infect Immun. 1993;61:3730–8.
Park AY, Hondowicz BD, Scott P. IL-12 is required to maintain a Th1 response during Leishmania major infection. J Immunol. 2000;165:896–902.
Stobie L, Gurunathan S, Prussin C, Sacks DL, Glaichenhaus N, Wu CY, Seder RA. The role of antigen and IL-12 in sustaining Th1 memory cells in vivo: IL-12 is required to maintain memory/effector Th1 cells sufficient to mediate protection to an infectious parasite challenge. Proc Natl Acad Sci USA. 2000;97:8427–32.
Park AY, Hondowicz B, Kopf M, Scott P. The role of IL-12 in maintaining resistance to Leishmania major. J Immunol. 2002;168:5771–7.
Zaph C, Uzonna J, Beverley SM, Scott P. Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat Med. 2004;10:1104–10.
Scott P. Immunologic memory in cutaneous leishmaniasis. Cell Microbiol. 2005;7:1707–13.
Scott P, Artis D, Uzonna J, Zaph C. The development of effector and memory T cells in cutaneous leishmaniasis: the implications for vaccine development. Immunol Rev. 2004;201:318–38.
Uzonna JE, Wei G, Yurkowski D, Bretscher P. Immune elimination of Leishmania major in mice: implications for immune memory, vaccination, and reactivation disease. J Immunol. 2001;167:6967–74.
Suffia IJ, Reckling SK, Piccirillo CA, Goldszmid RS, Belkaid Y. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J Exp Med. 2006;203:777–88.
Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev Immunol. 1998;16:201–23.
Gray D, Matzinger P. T cell memory is short-lived in the absence of antigen. J Exp Med. 1991;174:969–74.
Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63.
Tough DF. Deciphering the relationship between central and effector memory CD8+ T cells. Trends Immunol. 2003;24:404–7.
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12.
Sallusto F, Lanzavecchia A. Exploring pathways for memory T cell generation. J Clin Invest. 2001;108:805–6.
Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–34.
Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–22.
Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410:101–5.
Aebischer T, Moody SF, Handman E. Persistence of virulent Leishmania major in murine cutaneous leishmaniasis: a possible hazard for the host. Infect Immun. 1993;61:220–6.
Aebischer T. Recurrent cutaneous leishmaniasis: a role for persistent parasites? Parasitol Today. 1994;10:25–8.
Berhe N, Hailu A, Wolday D, Negesse Y, Cenini P, Frommel D. Ethiopian visceral leishmaniasis patients co-infected with human immunodeficiency virus. Trans R Soc Trop Med Hyg. 1995;89:205–7.
Wolday D., Berhe N, Akuffo H, Britton S. Leishmania-HIV interaction: immunopathogenic mechanisms. Parasitol Today. 1999;15:182–7.
Wolday D, Berhe N, Akuffo H, Desjeux P, Britton S. Emerging Leishmania/HIV co-infection in Africa. Med Microbiol Immunol. (Berl) 2001;190:65–7.
Saravia NG, Weigle K, Segura I, Giannini SH, Pacheco R, Labrada LA, Goncalves A. Recurrent lesions in human Leishmania braziliensis infection-reactivation or reinfection? Lancet. 1990;336:398–402.
Stenger S, Donhauser N, Thuring H, Rollinghoff M, Bodgan C. Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J Exp Med. 1996;183:1501–14.
Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J Exp Med. 2004;200:201–10.
Uzonna JE, Bretscher PA. Anti-IL-4 antibody therapy causes regression of chronic lesions caused by medium-dose Leishmania major infection in BALB/c mice. Eur J Immunol. 2001;31:3175–84.
Campanelli AP, Roselino AM, Cavassani KA, Pereira MS, Mortara RA, Brodskyn CI, Goncalves HS, Belkaid Y, Barral-Netto M, Barral A, Silva JS. CD4+CD25+ T cells in skin lesions of patients with cutaneous leishmaniasis exhibit phenotypic and functional characteristics of natural regulatory T cells. J Infect Dis. 2006;193:1313–22.
Bourreau E, Prevot G, Gardon J, Pradinaud R, Launois P. High intralesional interleukin-10 messenger RNA expression in localized cutaneous leishmaniasis is associated with unresponsiveness to treatment. J Infect Dis. 2001;184:1628–30.
Habibi GR, Khamesipour A, McMaster WR, Mahboudi F. Cytokine gene expression in healing and non-healing cases of cutaneous leishmaniasis in response to in vitro stimulation with recombinant gp63 using semi-quantitative RT-PCR. Scand J Immunol. 2001;54:414–20.
Spath GF, Lye LF, Segawa H, Sacks DL, Turco SJ, Beverley SM. Persistence Without Pathology in Phosphoglycan-Deficient Leishmania major. Science. 2003;301:1241–3.
Lau LL, Jamieson BD, Somasundaram T, Ahmed R. Cytotoxic T-cell memory without antigen. Nature. 1994;369:648–52.
Swain SL. CD4 T-cell memory can persist in the absence of class II. Philos Trans R Soc Lond B Biol Sci. 2000;355:407–11.
Gray D. A role for antigen in the maintenance of immunological memory. Nat Rev Immunol. 2002;2:60–5.
Zinkernagel RM. On differences between immunity and immunological memory. Curr Opin Immunol. 2002;14:523–36.
Wherry EJ, McElhaugh MJ, Eisenlohr LC. Generation of CD8(+) T cell memory in response to low, high, and excessive levels of epitope. J Immunol. 2002;168:4455–61.
Brown GV. Progress in the development of malaria vaccines: context and constraints. Parassitologia. 1999;41:429–32.
Andreassen J. Immunity to adult cestodes: basic knowledge and vaccination problems. A review. Parassitologia. 1991;33:45–53.
Rajakumar S, Bleiss W, Hartmann S, Schierack P, Marko A, Lucius R. Concomitant immunity in a rodent model of filariasis: the infection of Meriones unguiculatus with Acanthocheilonema viteae. J Parasitol. 2006;92:41–5.
Struik SS, Riley EM. Does malaria suffer from lack of memory? Immunol Rev. 2004;201:268–90.
Anderson CF, Mendez S, Sacks DL. Nonhealing infection despite Th1 polarization produced by a strain of Leishmania major in C57BL/6 mice. J Immunol. 2005;174:2934–41.
Titus RG, Gueiros-Filho FJ, de Freitas LA, Beverley SM. Development of a safe live Leishmania vaccine line by gene replacement. Proc Natl Acad Sci USA. 1995;92:10267–71.
Uzonna JE, Spath GF, Beverley SM, Scott P. Vaccination with phosphoglycan-deficient Leishmania major protects highly susceptible mice from virulent challenge without inducing a strong Th1 response. J Immunol. 2004;172:3793–7.
Modabber F. Vaccines against leishmaniasis. Ann Trop Med Parasitol. 1995;89(Suppl 1):83–8.
Marzochi KB, Marzochi MA, Silva AF, Grativol N, Duarte R, Confort EM, Modabber F. Phase 1 study of an inactivated vaccine against American tegumentary leishmaniasis in normal volunteers in Brazil. Mem Inst Oswaldo Cruz. 1998;93:205–12.
Sharifi I, FeKri AR, Aflatonian MR, Khamesipour A, Nadim A, Mousavi MR, Momeni AZ, Dowlati Y, Godal T, Zicker F, Smith PG, Modabber F. Randomised vaccine trial of single dose of killed Leishmania major plus BCG against anthroponotic cutaneous leishmaniasis in Bam, Iran. Lancet. 1998;351:1540–3.
Momeni AZ, Jalayer T, Emamjomeh M, Khamesipour A, Zicker F, Ghassemi RL, Dowlati Y, Sharifi I, Aminjavaheri M, Shafiei A, Alimohammadian MH, Hashemi-Fesharki R, Nasseri K, Godal T, Smith PG, Modabber F. A randomised, double-blind, controlled trial of a killed L. major vaccine plus BCG against zoonotic cutaneous leishmaniasis in Iran. Vaccine. 1999;17:466–72.
Velez ID, del Pilar Agudelo S, Arbelaez MP, Gilchrist K, Robledo SM, Puerta JA, Zicker F, Berman J, Modabber F. Safety and immunogenicity of a killed Leishmania (L.) amazonensis vaccine against cutaneous leishmaniasis in Colombia: a randomized controlled trial. Trans R Soc Trop Med Hyg. 2000;94:698–703.
Khalil EA, El Hassan AM, Zijlstra EE, Mukhtar MM, Ghalib HW, Musa B, Ibrahim ME, Kamil AA, Elsheikh M, Babiker A, Modabber F. Autoclaved Leishmania major vaccine for prevention of visceral leishmaniasis: a randomised, double-blind, BCG-controlled trial in Sudan. Lancet. 2000;356:1565–9.
Mahmoodi M, Khamesipour A, Dowlati Y, Rafati S, Momeni AZ, Emamjomeh M, Hejazi H, Modabber F. Immune response measured in human volunteers vaccinated with autoclaved Leishmania major vaccine mixed with low dose of BCG. Clin Exp Immunol. 2003;134:303–8.
Armijos RX, Weigel MM, Calvopina M, Hidalgo A, Cevallos W, Correa J. Safety, immunogenecity, and efficacy of an autoclaved Leishmania amazonensis vaccine plus BCG adjuvant against New World cutaneous leishmaniasis. Vaccine. 2004;22:1320–6.
Gurunathan S, Sacks DL, Brown DR, Reiner SL, Charest H, Glaichenhaus N, Seder RA. Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major. J Exp Med. 1997;186:1137–47.
Mitchell GF, Handman E. Leishmania tropica major in mice: vaccination against cutaneous leishmaniasis in mice of high genetic susceptibility. Aust J Exp Biol Med Sci. 1983;61:11–25.
Handman E, Noormohammadi AH, Curtis JM, Baldwin T, Sjolander A. Therapy of murine cutaneous leishmaniasis by DNA vaccination. Vaccine. 2000;18:3011–7.
da Silva VO, Borja-Cabrera GP, Correia Pontes NN, de Souza EP, Luz KG, Palatnik M, Palatnik de Sousa CB. A phase III trial of efficacy of the FML-vaccine against canine kala-azar in an endemic area of Brazil (Sao Goncalo do Amaranto, RN). Vaccine. 2000;19:1082–92.
Aebischer T, Wolfram M, Patzer SI, Ilg T, Wiese M, Overath P. Subunit vaccination of mice against new world cutaneous leishmaniasis: comparison of three proteins expressed in amastigotes and six adjuvants. Infect Immun. 2000;68:1328–36.
Stager S, Smith DF, Kaye PM. Immunization with a recombinant stage-regulated surface protein from Leishmania donovani induces protection against visceral leishmaniasis. J Immunol. 2000;165:7064–71.
Ahmed SB, Bahloul C, Robbana C, Askri S, Dellagi K. A comparative evaluation of different DNA vaccine candidates against experimental murine leishmaniasis due to L. major. Vaccine. 2004;22:1631–9.
Vardy DA, Cohen A, Kachko L, Zvulunov A, Frankenburg S. Relapse of cutaneous leishmaniasis in a patient with an infected subcutaneous rheumatoid nodule. Br J Dermatol. 1999;141:914–7.
Kebaier C, Uzonna JE, Beverley SM, Scott P. Immunization with persistent attenuated Delta lpg2 Leishmania major parasites requires adjuvant to provide protective immunity in C57BL/6 mice. Infect Immun. 2006;74:777–80.
Veras P, Brodskyn C, Balestieri F, Ld F, Ramos A, Queiroz A, Barral A, Beverley S, Barral-Netto M. A dhfr-ts- leishmania major knockout mutant cross-protects against leishmania amazonensis [In Process Citation]. Mem Inst Oswaldo Cruz. 1999;94:491–6.
Acknowledgments
This work is supported by grants from The Canadian Institutes of Health Research (CIHR), Manitoba Health Research Council (MHRC), and Manitoba Medical Service Foundation (MMSF).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Okwor, I., Uzonna, J. Persistent parasites and immunologic memory in cutaneous leishmaniasis: implications for vaccine designs and vaccination strategies. Immunol Res 41, 123–136 (2008). https://doi.org/10.1007/s12026-008-8016-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-008-8016-2