
Abstract
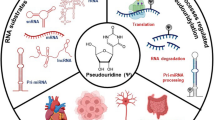
Pseudouridylation is one of the most abundant RNA modifications in eukaryotes, making pseudouridine known as the “fifth nucleoside.” This highly conserved alteration affects all non-coding and coding RNA types. Its role and importance have been increasingly widely researched, especially considering that its absence or damage leads to serious hereditary diseases. Here, we summarize the human genetic disorders described to date that are related to the participants of the pseudouridylation process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The variability of the phenotype is primarily explained by genomic sequence changes. Accordingly, Mendelian disorders are generally caused by single nucleotide variations of the coding sequence. Nevertheless, epigenetic modifications largely contribute to the phenotype variability, and its severe forms can be even disease-causing (Araki and Mimura 2018, Cullell, Soriano-Tárraga et al. 2022, Derakhshan et al. 2022, Ojaimi, Banimortada et al. 2022, Pierce and Black 2022). Besides DNA methylation and histone modifications, RNAs are also subjects to epigenetic modifications that have serious impact on their function, implying a potential role in disease development (Han and Phizicky 2018; Ontiveros et al. 2019; Barbieri and Kouzarides 2020; Haruehanroengra et al. 2020; Destefanis et al. 2021, Kumari, Groza et al. 2021). Almost 200 different types of RNA modifications associated with 175 human diseases have thus far been identified according to the MODOMICS database (Boccaletto et al. 2022).
All types of RNAs, including transfer RNAs (tRNA), ribosomal RNAs (rRNA), messenger RNAs (mRNA) and other non-coding RNAs, undergo modifications during their maturation (Boccaletto et al. 2022). Nuclear and mitochondrial tRNAs are the subjects of the most diverse modifications. The tRNA and rRNA modifications were studied most extensively, because of their highest expression rate. Nevertheless, the modifications of the mRNA are also of special importance as they may directly affect the translation (Schwartz 2016; Helm and Motorin 2017; Zhao et al. 2017; Linder and Jaffrey 2019).
One of the most abundant RNA modifications in eukaryotic cells is pseudouridylation: the transformation of uridine to its other isoform called pseudouridine (Ψ).(Fig. 1). Some specific pseudouridylation sites are already present in archaebacteria, eubacteria and in the organelles of eukaryotic cells, such as mitochondria and chloroplasts (Charette and Gray 2000). Based on the conservation of these sites, they seem to be essential modifications, but no lethal or severe phenotypes were found related to most of them in yeasts or eubacteria. In these species, their major role is supposed to be the fine tuning of the RNA structure (Charette and Gray 2000). In mammals, the role of RNA pseudouridylation is supposed to be more remarkable. Inducible pseudouridylation of mammalian mRNA has a stimuli-specific pattern, which may help the cell in adaptation to stress situations. Heat shock and H2O2 treatment activate hundreds of different inducible pseudouridylation sites in mRNAs that are implicated in transport- and telomere- or chromatin-related functions, respectively (Li et al. 2015).

Isomerisation of uridine, schematic figure
Most of the uridine-pseudouridine transformations occur in tRNAs, rRNAs and snRNAs (Charette and Gray 2000; Spenkuch et al. 2014), but as described recently, it can also be found in mRNAs (Carlile et al. 2014; Lovejoy et al. 2014; Schwartz et al. 2014; Li et al. 2015). More than 100 specific uridines are known to be pseudouridylated in rRNAs, which maintain the proper functioning, folding and conformational stability of the rRNAs, their associations with ribosomal proteins, and thus ensure the catalytic activity of the ribosome (Decatur and Fournier 2002; King et al. 2003; Liang et al. 2009; Natchiar et al. 2017; Abou Assi et al. 2020). The modified uridines are typically located in evolutionarily conserved regions close to functional domains (Natchiar et al. 2017). Alteration of rRNA pseudouridylation thus directly influences the interactions with tRNAs and mRNAs, modifying translational efficiency, gene expression patterns and levels (Jack et al. 2011; Bastide and David 2018).
Two main types of enzymes catalyze the pseudouridylation. Stand-alone pseudouridine synthases (PUSs) — that are classified into six families (TruA, TruB, TruD, RsuA, RluA, PUS10) (Hamma and Ferré-D'Amaré 2006; Roovers et al. 2006; Rintala-Dempsey and Kothe 2017) — can recognize directly, without a guide RNA, the target uridine(s), and implement the modification by themselves (Hamma and Ferré-D'Amaré 2006). While bacteria use only stand-alone PUSs, eukaryotes also possess H/ACA small nucleolar ribonucleoprotein complexes for this purpose. This latter type of pseudouridine synthase necessitates a unique guide RNA specific for the target uridine and four core proteins: non-histone protein 2 (NHP2), nucleolar protein 10 (NOP10), glycine-arginine-rich protein 1 (GAR1) and the catalytically active dyskerin (DKC1), the sequence of which is similar to that of the TruB PUS family members (Lafontaine et al. 1998; Ramamurthy et al. 1999; Spedaliere et al. 2000; Kiss et al. 2004; Khanna et al. 2006; Penzo and Montanaro 2018). The H/ACA box small-nucleolar RNAs (snoRNA, “SNORA”) guide the enzymatic protein complex to the substrate via site-specific complementary base-pairing at the target site (Ganot et al. 1997; Sumita et al. 2005; Henras et al. 2008; McMahon et al. 2015; Rintala-Dempsey and Kothe 2017; Czekay and Kothe 2021). The rRNA pseudouridylation is primarily mediated by the H/ACA snoRNP complex: Knockdown of dyskerin decreases it by more than half (Schwartz et al. 2014). Nevertheless, at least 76 so-called orphan snoRNAs are known in human that are not complementary to any rRNAs (Ono et al. 2010; Gong et al. 2017), suggesting that they guide the modifications of mRNAs or other RNA classes (Kishore and Stamm 2006; Chu et al. 2012; Kishore et al. 2013; Bieth et al. 2015; Falaleeva et al. 2016). Site-specific synthases that modify mRNAs are also found among stand-alone PUSs (TRUB1, PUS7 and PUS1) (Li et al. 2015; Safra et al. 2017; Carlile et al. 2019). PUS enzymes are encoded in humans by 13 genes, out of which four (PUS1, PUS3, PUS7 and DKC1) are associated with genetic diseases (Bykhovskaya et al. 2004; Shaheen et al. 2016; de Brouwer et al. 2018; Borghesi et al. 2022).
Besides pseudouridylation, these enzymes also mediate other processes. Stand-alone PUSs may function as RNA chaperons or as part of multiprotein complexes, as their absence causes more severe phenotypes in bacteria and yeasts than the lack of some pseudouridines per se (Grosshans et al. 2001; Ishitani et al. 2003; Hamma and Ferré-D'Amaré 2006). The H/ACA box small nucleolar ribonucleoprotein complexes also participate in the cleavage of the precursor of 18S rRNA and in maintaining telomerase activity (Heiss et al. 1998; Hamma and Ferré-D'Amaré 2006). Dyskerin is associated with — besides the H/ACA snoRNAs—telomerase RNA, which also contains a H/ACA RNA motif and serves as template to telomere elongation (Mitchell et al. 1999a, b).
Despite being an ubiquitous process, the deficiency in pseudouridylation leads to organ-specific defects. The description of the related human disorders (Summary in Table 1.) (Heiss et al. 1998; Ruggero et al. 2003; Montanaro et al. 2006; Jonkhout et al. 2017) and unraveling their pathophysiology may help to understand what makes these organs more susceptible than others to the defect of the most abundant RNA modification.
Human diseases related to DKC1, dyskerin
Secondary to the Xq28 localization of the DKC1 gene, its mutations cause X-linked disorders (Devriendt et al. 1997; Heiss et al. 1998; Mitchell et al. 1999a, b). Most of the related disorders are recessive: Males and heterozygous females with extremely skewed X-inactivation are affected (Alder et al. 2013). There are, however, exceptions: the DKC1 p.E206K-related cataracts and hearing impairment are transmitted in a dominant fashion: Heterozygous females are typically affected (Balogh et al. 2020).
As dyskerin contributes to various processes, such as pseudouridylation of different types of RNA, telomere lengthening and RNA maturation, the molecular mechanisms of most of the associated disorders are challenging to differentiate.
Dyskeratosis congenita (DC) (Mitchell et al. 1999a, b; Vulliamy et al. 2001; Alder et al. 2013; AlSabbagh 2020), the disease that gave dyskerin its name, results from telomere shortening. Mutations of several genes, some of which are implicated only in telomere maintenance but not in pseudouridylation, cause DC, indicating that the pathophysiology is independent of the pseudouridylation defect.
Telomeres, located at the end of chromosomes, protect their integrity and structure. Their length and stability are maintained by complexes of various proteins and RNAs. The main participants of the telomerase complex are: TERT: the reverse transcriptase enzyme; TERC: the RNA template for the telomere elongation; and dyskerin, NHP2, NOP10 and GAR1: the proteins responsible for the stability of the telomerase complex. Several additional regulators, protectors, and repair molecules are not detailed here, but the failure of either of them can lead to DC with various severity and different inheritance manner (autosomal dominant, autosomal recessive, X-linked) (Tummala et al. 2018; Garus and Autexier 2021; Dorgaleleh et al. 2022). Though it is genetically heterogeneous, mutations of DKC1, with X-linked inheritance fashion, are its most common causes. The DC-related DKC1 mutations are C- and N-terminal missense mutations which affect less conserved regions and are implicated in guide RNA binding, but some missense variants in the TruB domain were also described (Aalfs, van den Berg et al. 1995; Knight et al. 1999a, b; Dokal 2000; Kiss et al. 2004; Trahan et al. 2010; Balogh et al. 2020). Without any alteration in its coding sequence, DC can also develop as a result of a promoter mutation and decreased dyskerin expression (Knight et al. 1999a, b; Salowsky et al. 2002; Parry et al. 2011).
In general, the early symptoms of DC represent the triad of mucocutaneous features (reticular skin hyperpigmentation, oral leukoplakia and nail dystrophy) typically not presenting before late childhood. Bone marrow failure develops later and gives rise to opportunistic infections, anemia, thrombocytopenia, and, as a consequence, internal bleeding events. There is a high risk of pulmonary fibrosis, which, together with malignancies and opportunistic infections, are responsible for the early mortality. Life expectancy is 20–50 years. Most of the severely affected organs and tissues (skin, nail, bone marrow) require a high proliferation rate (Dokal 2000; Gu et al. 2008; Dorgaleleh et al. 2022).
The p.S121G substitution in the TruB domain (pseudouridine synthase motif) of dyskerin was found in a 15-year-old patient with metachronous rectal cancer and bone marrow failure without other typical DC symptoms (Watanabe et al. 2019). The same variant of dyskerin was previously reported in HH (Hoyeraal-Hreidarsson) syndrome, a severe form of DC characterized by intrauterine growth retardation, microcephaly, cerebellar hypoplasia, and occasionally, enteropathy (Aalfs, van den Berg et al. 1995; Knight et al. 1999a, b). Along this line, another TruB domain substitution (p.R158W) was also described in HH syndrome (Knight et al. 2001; Vulliamy et al. 2006), suggesting that TruB domain substitutions cause more severe DC phenotypes than the N- and C-terminal substitutions.
DKC1 and NOP10-related nephrotic syndrome, cataracts, hearing impairment and enterocolitis
Dyskeratosis and HH syndrome already represented a significant pleiotropy of DKC1 mutations. Nevertheless, a novel syndrome was recently described in two families with either a DKC1 mutation (p.E206K) or a NOP10 homozygous mutation (p.T16M) (Balogh et al. 2020). The affected children are asymptomatic during the first months of life, but stop growing in late infancy, develop cataracts, hearing impairment, diarrhea, nephrotic syndrome and later bone marrow failure. The majority of patients died during the first three years of life due to opportunistic infections, long before the potential appearance of DC-related mucocutaneous features. Some organ involvements are transmitted in a dominant fashion: Heterozygous females also develop cataracts and hearing impairment, typically in the second decade of life. The phenotype of a girl with a highly skewed X inactivation was similarly severe to that of the males, but no bone marrow failure or diarrhea was associated, indicating the rescue effect of cells expressing the wild type X and allowing her to reach adulthood.
Intriguingly, the two affected amino acids, DKC1 E206 and NOP10 T16, are known to interact with each other in the ribonucleoprotein complex (Rashid et al. 2006), and both pathogenic substitutions disrupt the catalytic pseudouridylation pocket, detaching the catalytic D125 of dyskerin from the uridine of the substrate RNA (Balogh et al. 2020). Though the telomeres of the affected patients are shortened, similarly to DC and HH syndrome, the highly different clinical presentation suggests different underlying pathophysiology. Accordingly, decreased pseudouridine levels were found in the patients’ rRNA. Zebrafish dkc1 mutants recapitulated the human phenotype and showed reduced 18S pseudouridylation, ribosomal dysregulation and a cell-cycle defect in the absence of telomere attrition. This novel disorder is thus the consequence of defective snoRNP pseudouridylation and ribosomal dysfunction (Balogh et al. 2020).
Tumor predisposition
Patients with DC are susceptible to malignancies (Bellodi et al. 2010). Loss of dyskerin has a negative impact on the translation of specific mRNAs with IRES (internal ribosomal entry site) elements. Many antiapoptotic proteins belong to this group, where the IRES elements foster their translation under stress conditions, and help the survival of the cell (Holcik and Sonenberg 2005). The p27 tumor suppressor is one of them: its inappropriate expression predisposes to pituitary tumor (Slingerland and Pagano 2000; Bellodi et al. 2010). The DKC1 p.S485G somatic mutation was identified in a patient with recurring pituitary tumor. The p.S485G dyskerin was found less stable and active, the 18S rRNA pseudouridylation as well as the p27 quantity decreased with a preserved expression at the mRNA level (Bellodi et al. 2010). The p.H259P variant of dyskerin was also identified in pituitary adenoma with lower P27 protein levels (Martins et al. 2016). A similar telomere independent mechanism was described in breast cancer, affecting the translation of the p53 tumor suppressor (Montanaro et al. 2010). These findings together suggest a tumor suppressor role for dyskerin. Even though, DKC1 variants are rarely found in sporadic malignancies (Penzo et al. 2013).
Nevertheless, the role of dyskerin in tumor development is contradictory. Dyskerin overexpression was described in breast- (Montanaro et al. 2006, 2008; Elsharawy et al. 2020), and prostate cancer (Sieron et al. 2009; Stockert et al. 2019, 2021) and malignant glioma (Miao et al. 2019). High DKC1 expression levels are associated with worse prognosis in these malignancies (Miao et al. 2019; Elsharawy et al. 2020). In vitro knock-down experiments in glioma cell lines indicate the role of DKC1 in proliferation, migration and invasion (Miao et al. 2019). In addition, dyskerin expression was found to be regulated by N-Myc and c-Myc oncogenes, and its downregulation resulted in reduced proliferation of neuroblastoma cells, which process was independent from the telomere length and p53 level (O'Brien et al. 2016). Dyskerin is often regarded also as an oncogene, but its role and the underlying molecular mechanisms are still to be elucidated.
Along this line, elevated pseudouridine level in blood or urine was suggested to be a potential biomarker of several malignancies (Seidel et al. 2006; Patejko et al. 2018): breast (Zheng et al. 2005), colorectal (Feng et al. 2005), oesophageal (Masuda et al. 1993), gallbladder (Jiao et al. 2014), prostate (Stockert et al. 2021), ovarian (Chen et al. 2012; Zeleznik et al. 2020), small cell lung cancer (Tamura et al. 1986a, b; Tamura et al. 1987), hepatocellular carcinoma (Tamura et al. 1986a, b; Amuro et al. 1988), leukemia (Li et al. 1992) and lymphoma (Rasmuson and Björk 1983; Masaki et al. 2006).
The role of dyskerin in tumor development seems to depend on the specific tumor type.
Plasma and urine pseudouridine levels as potential biomarkers were also suggested in cardiovascular diseases, such as heart failure or cardiac hypertrophy. The role of pseudouridines in these conditions is not yet elucidated, but presumably the mitochondrial function and oxidative phosphorylation may be impaired via mitochondrial RNA modifications (Razavi et al. 2020; Wu et al. 2021).
The double-edged role of dyskerin, acting potentially both as tumor suppressor or oncogene, is intriguing. Sustained telomerase activity due to dyskerin overexpression or altered expression of pro- and anti-apoptotic factors secondary to dyskerin dysfunction can contribute to tumor progression. A minor isoform of dyskerin (Iso3) with cytoplasmic localization was found to have a role in oxidative metabolism. Its overexpression might also contribute to cancer progression by protecting cells from oxidative stress and apoptosis (Angrisani et al. 2018). On the other hand, loss-of-function of dyskerin seems to inhibit cell proliferation and tumor progression. Accordingly, cells expressing the mutant allele in heterozygous females with p.E206K undergo natural selection and X-inactivation tends to be skewed toward the mutant allele by the second decade of life (Balogh et al. 2020). Furthermore, the immortalized cell line established from the leukocytes of heterozygous females expressed exclusively the wild-type allele (our unpublished data). Autophagy and heat shock response have been similarly found to have such a dual role in cancer (Santagata et al. 2011; Chavez-Dominguez et al. 2020; Cyran and Zhitkovich 2022). As the stress response pathways are also associated with dyskerin functions (Li et al. 2015) and tumor progression (Santagata et al. 2011; Chavez-Dominguez et al. 2020; Cyran and Zhitkovich 2022), their causality remains to be explored in the double-edged relationship between dyskerin in cancer.
Human diseases related to stand-alone pseudouridine synthases
PUS1
MLASA (mitochondrial myopathy, lactic acidosis, sideroblastic anemia)(Inbal et al. 1995) is a rare autosomal recessive, oxidative phosphorylation disorder, characterized primarily by muscle and bone marrow defects leading to exercise intolerance and anemia. Cognitive impairment, skeletal and dental abnormalities, delayed motor milestones, cardiomyopathy, dysphagia and respiratory insufficiency can be associated. Loss of function of the YARS2 gene, encoding the mitochondrial tyrosyl-tRNA synthase, results in a similar phenotype. It remains to be explored how the defect of the pseudouridine synthase PUS1 disrupts the oxidative phosphorylation. PUS1, a member of the TruA stand-alone pseudouridylation synthase family, is necessary for the pseudouridylation of cytoplasmic and mitochondrial tRNAs. Its R116W substitution in the highly conserved catalytic center of the protein was the first reported causal variant in MLASA (Bykhovskaya et al. 2004; Zeharia et al. 2005). Since then, several other loss of function mutations have been described (listed in Table 1.) resulting the same (Fernandez-Vizarra et al. 2007; Cao et al. 2016, Kasapkara Ç, Tümer et al. 2017), or a similar disorder. (Metodiev et al. 2015) In a mouse model, it was shown that the rate of the muscle fibers expressing myosin heavy chain IIB and IIA is altered in the PUS1 null mutants, resulting in an altered muscle metabolism, and causing a very similar phenotype to the human (Mangum et al. 2016, Kasapkara Ç, Tümer et al. 2017).
Recently, PUS1 overexpression was found in breast cancer, and its knockdown was proved to suppress tumor proliferation and invasion in breast cancer cell lines (Fang et al. 2022).
PUS3
Intellectual disability is usually caused by chromosomal rearrangements or single gene mutations. Several tRNA modification enzymes were found defected indicating that brain development is especially sensitive to tRNA dysfunction (Ropers 2010; Torres et al. 2014; Shaheen et al. 2016; Abdelrahman et al. 2018; de Paiva et al. 2019; Borghesi et al. 2022). During their maturation tRNAs undergo several post-transcriptional modifications, which stabilize their structure and function and prevent translational frameshifting via stabilizing the codon-anticodon base pairing. Hypomodified tRNAs are often degraded, and the imbalances of tRNA pool may affect protein synthesis (Phizicky and Hopper 2010; Torres et al. 2014, Pereira, Francisco et al. 2018).
PUS3, a TruA family member is a general pseudouridine synthase of tRNAs, the alterations of which cause global developmental delay/intellectual disability (GDD/ID), microcephaly, short stature, severe hypotonia, gray sclera and severe syndromic features. The p.R435* mutation truncates the protein in the C-terminal region which is highly conserved in mammals. The mutation abolishes the isomerization of the U39 at least in six different tRNAs (Shaheen et al. 2016). Along this line, the p.S394Cfs*18 mutation was also described to be disease causing and triggering complete degradation of the mRNA by nonsense-mediated decay (Abdelrahman et al. 2018). The p.R166Q and p.L366P variants were reported to cause also renal involvement (de Paiva et al. 2019). The reported variants are listed in Table 1.
PUS7
PUS7, similarly to PUS3, targets several tRNAs and mRNAs. The catalytic domain of the TruD family member enzyme is located to its C-terminal. The phenotype of patients with PUS7 loss of function mutations is very similar to those with PUS3 variants. Intellectual disability, short stature, and microcephaly are common. Aggressive behavior was reported in most of the cases. The altered enzymes seem to lose the isomerization capacity of the U13 of at least ten cytosolic tRNAs. In addition, dysregulation of general protein translation also follows (de Brouwer et al. 2018; Shaheen et al. 2019; Han et al. 2022). The reported mutations can be found in Table 1.
Mutation of the target U in a human disease
Besides enzyme function loss, the substitution of the target uridine may also inhibit the process, which, in case of a cardinal uridine site, can result in a phenotype by itself. Wang et al. found that the substitution of the U55 in the mitochondrial tRNAGlu by cytosine (m.14692A > G) results in maternally inherited diabetes and deafness (MIDD). U55 pseudouridylation is a very conserved and essential step in the maturation of mitochondrial tRNAGlu. In absence of the Ψ55, the tRNAGlu becomes unstable and due to its structural alterations it cannot bind properly to the components of the translational machinery. As a consequence, the mitochondrial protein translation becomes hampered, and the ATP synthesis and the mitochondrial membrane potential will be reduced (Wang et al. 2016).
Hypothetical therapeutic usage
Due to the methodological development, hundreds of mRNA pseudouridylation sites were recently detected (Cerneckis et al. 2022). A modified stop codon (UAA, UAG, UGA) to ΨAA, ΨAG or ΨGA results in readthrough in yeast. It is tempting to speculate about its potential use in human nonsense mutation-caused disorders, i.e., to pass the premature termination codon in a directed way and thus translate the whole peptide (Mort et al. 2008; Karijolich and Yu 2011; Fernandez et al. 2013; Adachi and Yu 2020). Targeted pseudouridylation with designed guide RNAs would change the STOP codon to serine or threonine (ΨAA, ΨAG); tyrosine or phenylalanine (ΨGA) (Cerneckis et al. 2022).
This hypothetical method was studied by Nir et al. (2022) and they made the following conclusions: snoRNA-mediated pseudouridylation can occur on mRNA targets, but at very low levels, the snoRNA complementary region should be longer than required in rRNAs and the natural intron cleveage is an important part of the process, that should be considered at the experimental design. Very recently two parallel methods were published (Adachi, Pan et al. 2023; Song et al. 2023), where the pseudouridylation of premature termination codons restored the translation at a low but promising level.
In vivo studies have been performed only in yeast to date (Karijolich and Yu 2011; Huang et al. 2012; Fernandez et al. 2013).
Conclusion
Pseudouridylation, the most abundant modification of RNAs, has been found implicated in several human disorders. While the underlying pathophysiology is being unraveled, its potential in therapeutic interventions is to be explored.
References
Aalfs CM, van den Berg H, Barth PG, Hennekam RC (1995) The Hoyeraal-Hreidarsson syndrome: the fourth case of a separate entity with prenatal growth retardation, progressive pancytopenia and cerebellar hypoplasia. Eur J Pediatr 154(4):304–308
Abdelrahman HA, Al-Shamsi AM, Ali BR, Al-Gazali L (2018) A null variant in PUS3 confirms its involvement in intellectual disability and further delineates the associated neurodevelopmental disease. Clin Genet 94(6):586–587
Abou Assi H, Rangadurai AK, Shi H, Liu B, Clay MC, Erharter K, Kreutz C, Holley CL, Al-Hashimi HM (2020) 2’-O-Methylation can increase the abundance and lifetime of alternative RNA conformational states. Nucleic Acids Res 48(21):12365–12379
Adachi H, Yu YT (2020) Pseudouridine-mediated stop codon readthrough in S. cerevisiae is sequence context-independent. RNA 26(9):1247–1256
Adachi H, Pan Y, He X, Chen JL, Klein B, Platenburg G, Morais P, Boutz P, Yu YT (2023) Targeted pseudouridylation: an approach for suppressing nonsense mutations in disease genes. Mol Cell 83(4):637–651
Alder JK, Parry EM, Yegnasubramanian S, Wagner CL, Lieblich LM, Auerbach R, Auerbach AD, Wheelan SJ, Armanios M (2013) Telomere phenotypes in females with heterozygous mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum Mutat 34(11):1481–1485
Alfares A, Alfadhel M, Wani T, Alsahli S, Alluhaydan I, Al Mutairi F, Alothaim A, Albalwi M, Al Subaie L, Alturki S, Al-Twaijri W, Alrifai M, Al-Rumayya A, Alameer S, Faqeeh E, Alasmari A, Alsamman A, Tashkandia S, Alghamdi A, Alhashem A, Tabarki B, AlShahwan S, Hundallah K, Wali S, Al-Hebbi H, Babiker A, Mohamed S, Eyaid W, Zada AAP (2017) A multicenter clinical exome study in unselected cohorts from a consanguineous population of Saudi Arabia demonstrated a high diagnostic yield. Mol Genet Metab 121(2):91–95
AlSabbagh MM (2020) Dyskeratosis congenita: a literature review. J Dtsch Dermatol Ges 18(9):943–967
Amuro Y, Nakaoka H, Shimomura S, Fujikura M, Yamamoto T, Tamura S, Hada T, Higashino K (1988) Serum pseudouridine as a biochemical marker in patients with hepatocellular carcinoma. Clin Chim Acta 178(2):151–158
Angrisani A, Matrone N, Belli V, Vicidomini R, Di Maio N, Turano M, Scialò F, Netti PA, Porcellini A, Furia M (2018) A functional connection between dyskerin and energy metabolism. Redox Biol 14:557–565
Araki Y, Mimura T (2018) Epigenetic basis of autoimmune disorders in humans translational epigenetics. T O. Academic Press, Tollefsbol, pp 353–385
Balogh E, Chandler JC, Varga M, Tahoun M, Menyhárd DK, Schay G, Goncalves T, Hamar R, Légrádi R, Szekeres Á, Gribouval O, Kleta R, Stanescu H, Bockenhauer D, Kerti A, Williams H, Kinsler V, Di WL, Curtis D, Kolatsi-Joannou M, Hammid H, Szőcs A, Perczel K, Maka E, Toldi G, Sava F, Arrondel C, Kardos M, Fintha A, Hossain A, D’Arco F, Kaliakatsos M, Koeglmeier J, Mifsud W, Moosajee M, Faro A, Jávorszky E, Rudas G, Saied MH, Marzouk S, Kelen K, Götze J, Reusz G, Tulassay T, Dragon F, Mollet G, Motameny S, Thiele H, Dorval G, Nürnberg P, Perczel A, Szabó AJ, Long DA, Tomita K, Antignac C, Waters AM, Tory K (2020) Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc Natl Acad Sci U S A 117(26):15137–15147
Barbieri I, Kouzarides T (2020) Role of RNA modifications in cancer. Nat Rev Cancer 20(6):303–322
Bastide A, David A (2018) Interaction of rRNA with mRNA and tRNA in translating mammalian ribosome: functional implications in health and disease. Biomolecules 8(4):100
Bellodi C, Krasnykh O, Haynes N, Theodoropoulou M, Peng G, Montanaro L, Ruggero D (2010) Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res 70(14):6026–6035
Bieth E, Eddiry S, Gaston V, Lorenzini F, Buffet A, Conte Auriol F, Molinas C, Cailley D, Rooryck C, Arveiler B, Cavaille J, Salles JP, Tauber M (2015) Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet 23(2):252–255
Boccaletto P, Stefaniak F, Ray A, Cappannini A, Mukherjee S, Purta E, Kurkowska M, Shirvanizadeh N, Destefanis E, Groza P, Avşar G, Romitelli A, Pir P, Dassi E, Conticello SG, Aguilo F, Bujnicki JM (2022) MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res 50(D1):D231-d235
Borghesi A, Plumari M, Rossi E, Viganò C, Cerbo RM, Codazzi AC, Valente EM, Gana S (2022) PUS3-related disorder: report of a novel patient and delineation of the phenotypic spectrum. Am J Med Genet A 188(2):635–641
Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N (2004) Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 74(6):1303–1308
Cao M, Donà M, Valentino ML, Valentino L, Semplicini C, Maresca A, Cassina M, Torraco A, Galletta E, Manfioli V, Sorarù G, Carelli V, Stramare R, Bertini E, Carrozzo R, Salviati L, Pegoraro E (2016) Clinical and molecular study in a long-surviving patient with MLASA syndrome due to novel PUS1 mutations. Neurogenetics 17(1):65–70
Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515(7525):143–146
Carlile TM, Martinez NM, Schaening C, Su A, Bell TA, Zinshteyn B, Gilbert WV (2019) mRNA structure determines modification by pseudouridine synthase 1. Nat Chem Biol 15(10):966–974
Cerneckis J, Cui Q, He C, Yi C, Shi Y (2022) Decoding pseudouridine: an emerging target for therapeutic development. Trends Pharmacol Sci 43(6):522–535
Charette M, Gray MW (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB Life 49(5):341–351
Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D (2020) The double-edge sword of autophagy in cancer: from tumor suppression to pro-tumor activity. Front Oncol 10:578418
Chen J, Zhou L, Zhang X, Lu X, Cao R, Xu C, Xu G (2012) Urinary hydrophilic and hydrophobic metabolic profiling based on liquid chromatography-mass spectrometry methods: differential metabolite discovery specific to ovarian cancer. Electrophoresis 33(22):3361–3369
Chu L, Su MY, Maggi LB Jr, Lu L, Mullins C, Crosby S, Huang G, Chng WJ, Vij R, Tomasson MH (2012) Multiple myeloma-associated chromosomal translocation activates orphan snoRNA ACA11 to suppress oxidative stress. J Clin Invest 122(8):2793–2806
Copyright © 1993–2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.
Cullell N, Soriano-Tárraga C, Gallego-Fábrega C, Cárcel-Márquez J, Torres-Águila NP, Muiño E, Lledós M, Llucià-Carol L, Esteller M, Castro de Moura M, Montaner J, Fernández-Sanlés A, Elosua R, Delgado P, Martí-Fábregas J, Krupinski J, Roquer J, Jiménez-Conde J, Fernández-Cadenas I (2022) DNA methylation and ischemic stroke risk: an epigenome-wide association study. Thromb Haemost 122(10):1767–1778
Cyran AM, Zhitkovich A (2022) Heat shock proteins and HSF1 in cancer. Front Oncol 12:860320
Czekay DP, Kothe U (2021) H/ACA small ribonucleoproteins: structural and functional comparison between archaea and eukaryotes. Front Microbiol 12:654370
de Brouwer APM, Abou Jamra R, Körtel N, Soyris C, Polla DL, Safra M, Zisso A, Powell CA, Rebelo-Guiomar P, Dinges N, Morin V, Stock M, Hussain M, Shahzad M, Riazuddin S, Ahmed ZM, Pfundt R, Schwarz F, de Boer L, Reis A, Grozeva D, Raymond FL, Riazuddin S, Koolen DA, Minczuk M, Roignant JY, van Bokhoven H, Schwartz S (2018) Variants in PUS7 cause intellectual disability with speech delay, microcephaly, short stature, and aggressive behavior. Am J Hum Genet 103(6):1045–1052
de Paiva ARB, Lynch DS, Melo US, Lucato LT, Freua F, de Assis BDR, Barcelos I, Listik C, de Castro Dos D, Santos LI, Macedo-Souza HH, Kok F (2019) PUS3 mutations are associated with intellectual disability, leukoencephalopathy, and nephropathy. Neurol Genet 5(1):e306
Decatur WA, Fournier MJ (2002) rRNA modifications and ribosome function. Trends Biochem Sci 27(7):344–351
Derakhshan M, Kessler NJ, Ishida M, Demetriou C, Brucato N, Moore GE, Fall CHD, Chandak GR, Ricaut FX, Prentice AM, Hellenthal G, Silver MJ (2022) Tissue- and ethnicity-independent hypervariable DNA methylation states show evidence of establishment in the early human embryo. Nucleic Acids Res 50(12):6735–6752
Destefanis E, Avsar G, Groza P, Romitelli A, Torrini S, Pir P, Conticello SG, Aguilo F, Dassi E (2021) A mark of disease: how mRNA modifications shape genetic and acquired pathologies. RNA 27(4):367–389
Devriendt K, Matthijs G, Legius E, Schollen E, Blockmans D, van Geet C, Degreef H, Cassiman JJ, Fryns JP (1997) Skewed X-chromosome inactivation in female carriers of dyskeratosis congenita. Am J Hum Genet 60(3):581–587
Dokal I (2000) Dyskeratosis congenita in all its forms. Br J Haematol 110(4):768–779
Dorgaleleh S, Naghipoor K, Hajimohammadi Z, Dastaviz F, Oladnabi M (2022) Molecular insight of dyskeratosis congenita: Defects in telomere length homeostasis. J Clin Transl Res 8(1):20–30
Elsharawy KA, Mohammed OJ, Aleskandarany MA, Hyder A, El-Gammal HL, Abou-Dobara MI, Green AR, Dalton LW, Rakha EA (2020) The nucleolar-related protein Dyskerin pseudouridine synthase 1 (DKC1) predicts poor prognosis in breast cancer. Br J Cancer 123(10):1543–1552
Falaleeva M, Pages A, Matuszek Z, Hidmi S, Agranat-Tamir L, Korotkov K, Nevo Y, Eyras E, Sperling R, Stamm S (2016) Dual function of C/D box small nucleolar RNAs in rRNA modification and alternative pre-mRNA splicing. Proc Natl Acad Sci U S A 113(12):E1625-1634
Fang H, Zhang L, Xiao B, Long H, Yang L (2020) Compound heterozygous mutations in PUS3 gene identified in a Chinese infant with severe epileptic encephalopathy and multiple malformations. Neurol Sci 41(2):465–467
Fang Z, Shen HY, Xu Q, Zhou HL, Li L, Yang SY, Zhu Z, Tang JH (2022) PUS1 is a novel biomarker for predicting poor outcomes and triple-negative status in breast cancer. Front Oncol 12:1030571
Feng B, Zheng MH, Zheng YF, Lu AG, Li JW, Wang ML, Ma JJ, Xu GW, Liu BY, Zhu ZG (2005) Normal and modified urinary nucleosides represent novel biomarkers for colorectal cancer diagnosis and surgery monitoring. J Gastroenterol Hepatol 20(12):1913–1919
Fernandez IS, Ng CL, Kelley AC, Wu G, Yu YT, Ramakrishnan V (2013) Unusual base pairing during the decoding of a stop codon by the ribosome. Nature 500(7460):107–110
Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M (2007) Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J Med Genet 44(3):173–180
Froukh T, Nafie O, Al Hait SAS, Laugwitz L, Sommerfeld J, Sturm M, Baraghiti A, Issa T, Al-Nazer A, Koch PA, Hanselmann J, Kootz B, Bauer P, Al-Ameri W, Abou Jamra R, Alfrook AJ, Hamadallah M, Sofan L, Riess A, Haack TB, Riess O, Buchert R (2020) Genetic basis of neurodevelopmental disorders in 103 Jordanian families. Clin Genet 97(4):621–627
Ganot P, Bortolin ML, Kiss T (1997) Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell 89(5):799–809
Garus A, Autexier C (2021) Dyskerin: an essential pseudouridine synthase with multifaceted roles in ribosome biogenesis, splicing, and telomere maintenance. RNA 27(12):1441–1458
Gong J, Li Y, Liu CJ, Xiang Y, Li C, Ye Y, Zhang Z, Hawke DH, Park PK, Diao L, Putkey JA, Yang L, Guo AY, Lin C, Han L (2017) A Pan-cancer analysis of the expression and clinical relevance of small nucleolar RNAs in human cancer. Cell Rep 21(7):1968–1981
Grosshans H, Lecointe F, Grosjean H, Hurt E, Simos G (2001) Pus1p-dependent tRNA pseudouridinylation becomes essential when tRNA biogenesis is compromised in yeast. J Biol Chem 276(49):46333–46339
Gu BW, Bessler M, Mason PJ (2008) A pathogenic dyskerin mutation impairs proliferation and activates a DNA damage response independent of telomere length in mice. Proc Natl Acad Sci U S A 105(29):10173–10178
Guo W, Lai Y, Yan Z, Wang Y, Nie Y, Guan S, Kuo Y, Zhang W, Zhu X, Peng M, Zhi X, Wei Y, Yan L, Qiao J (2020) Trio-whole-exome sequencing and preimplantation genetic diagnosis for unexplained recurrent fetal malformations. Hum Mutat 41(2):432–448
Hamma T, Ferré-D’Amaré AR (2006) Pseudouridine synthases. Chem Biol 13(11):1125–1135
Han L, Phizicky EM (2018) A rationale for tRNA modification circuits in the anticodon loop. RNA 24(10):1277–1284
Han ST, Kim AC, Garcia K, Schimmenti LA, Macnamara E, Network UD, Gahl WA, Malicdan MC, Tifft CJ (2022) PUS7 deficiency in human patients causes profound neurodevelopmental phenotype by dysregulating protein translation. Mol Genet Metab 135(3):221–229
Haruehanroengra P, Zheng YY, Zhou Y, Huang Y, Sheng J (2020) RNA modifications and cancer. RNA Biol 17(11):1560–1575
Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I (1998) X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet 19(1):32–38
Helm M, Motorin Y (2017) Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 18(5):275–291
Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y (2008) The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65(15):2334–2359
Holcik M, Sonenberg N (2005) Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6(4):318–327
Huang C, Wu G, Yu YT (2012) Inducing nonsense suppression by targeted pseudouridylation. Nat Protoc 7(4):789–800
Inbal A, Avissar N, Shaklai M, Kuritzky A, Schejter A, Ben-David E, Shanske S, Garty BZ (1995) Myopathy, lactic acidosis, and sideroblastic anemia: a new syndrome. Am J Med Genet 55(3):372–378
Ishitani R, Nureki O, Nameki N, Okada N, Nishimura S, Yokoyama S (2003) Alternative tertiary structure of tRNA for recognition by a posttranscriptional modification enzyme. Cell 113(3):383–394
Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, Kopmar N, Krasnykh O, Dean AM, Thompson SR, Ruggero D, Dinman JD (2011) rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell 44(4):660–666
Jiao X, Mo Y, Wu Y, He J, Zhang P, Hu R, Luo C, Du J, Fu J, Shi J, Zhou L, Li D (2014) Upregulated plasma and urinary levels of nucleosides as biological markers in the diagnosis of primary gallbladder cancer. J Sep Sci 37(21):3033–3044
Jonkhout N, Tran J, Smith MA, Schonrock N, Mattick JS, Novoa EM (2017) The RNA modification landscape in human disease. RNA 23(12):1754–1769
Karijolich J, Yu YT (2011) Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 474(7351):395–398
Kasapkara ÇS, Tümer L, Zanetti N, Ezgü F, Lamantea E, Zeviani M (2017) A Myopathy, lactic acidosis, sideroblastic anemia (MLASA) case due to a novel PUS1 mutation. Turk J Haematol 34(4):376–377
Khanna M, Wu H, Johansson C, Caizergues-Ferrer M, Feigon J (2006) Structural study of the H/ACA snoRNP components Nop10p and the 3’ hairpin of U65 snoRNA. RNA 12(1):40–52
King TH, Liu B, McCully RR, Fournier MJ (2003) Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol Cell 11(2):425–435
Kishore S, Stamm S (2006) The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 311(5758):230–232
Kishore S, Gruber AR, Jedlinski DJ, Syed AP, Jorjani H, Zavolan M (2013) Insights into snoRNA biogenesis and processing from PAR-CLIP of snoRNA core proteins and small RNA sequencing. Genome Biol 14(5):R45
Kiss AM, Jády BE, Bertrand E, Kiss T (2004) Human box H/ACA pseudouridylation guide RNA machinery. Mol Cell Biol 24(13):5797–5807
Knight SW, Heiss NS, Vulliamy TJ, Aalfs CM, McMahon C, Richmond P, Jones A, Hennekam RC, Poustka A, Mason PJ, Dokal I (1999a) Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol 107(2):335–339
Knight SW, Heiss NS, Vulliamy TJ, Greschner S, Stavrides G, Pai GS, Lestringant G, Varma N, Mason PJ, Dokal I, Poustka A (1999b) X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am J Hum Genet 65(1):50–58
Knight SW, Vulliamy TJ, Morgan B, Devriendt K, Mason PJ, Dokal I (2001) Identification of novel DKC1 mutations in patients with dyskeratosis congenita: implications for pathophysiology and diagnosis. Hum Genet 108(4):299–303
Koziel JE, Fox MJ, Steding CE, Sprouse AA, Herbert BS (2011) Medical genetics and epigenetics of telomerase. J Cell Mol Med 15(3):457–467
Kumari K, Groza P, Aguilo F (2021) Regulatory roles of RNA modifications in breast cancer. NAR Cancer 3(3):zcab036
Lafontaine DL, Bousquet-Antonelli C, Henry Y, Caizergues-Ferrer M, Tollervey D (1998) The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev 12(4):527–537
Li Y, Wang S, Zhong N (1992) Simultaneous determination of pseudouridine and creatinine in urine of normal children and patients with leukaemia by high performance liquid chromatography. Biomed Chromatogr 6(4):191–193
Li X, Zhu P, Ma S, Song J, Bai J, Sun F, Yi C (2015) Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol 11(8):592–597
Liang XH, Liu Q, Fournier MJ (2009) Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA 15(9):1716–1728
Lim BC, Yoo SK, Lee S, Shin JY, Hwang H, Chae JH, Hwang YS, Seo JS, Kim JI, Kim KJ (2014) Hoyeraal-Hreidarsson syndrome with a DKC1 mutation identified by whole-exome sequencing. Gene 546(2):425–429
Linder B, Jaffrey SR (2019) Discovering and mapping the modified nucleotides that comprise the epitranscriptome of mRNA. Cold Spring Harb Perspect Biol 11(6):a032201
Lovejoy AF, Riordan DP, Brown PO (2014) Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE 9(10):e110799
Mangum JE, Hardee JP, Fix DK, Puppa MJ, Elkes J, Altomare D, Bykhovskaya Y, Campagna DR, Schmidt PJ, Sendamarai AK, Lidov HG, Barlow SC, Fischel-Ghodsian N, Fleming MD, Carson JA, Patton JR (2016) Pseudouridine synthase 1 deficient mice, a model for mitochondrial myopathy with sideroblastic anemia, exhibit muscle morphology and physiology alterations. Sci Rep 6:26202
Martins CS, Camargo RC, Saggioro FP, Neder L, Machado HR, Moreira AC, de Castro M (2016) P27/CDKN1B translational regulators in pituitary tumorigenesis. Horm Metab Res 48(12):840–846
Masaki Y, Itoh K, Sawaki T, Karasawa H, Kawanami T, Fukushima T, Kawabata H, Wano Y, Hirose Y, Suzuki T, Sugai S, Umehara H (2006) Urinary pseudouridine in patients with lymphoma: comparison with other clinical parameters. Clin Chim Acta 371(1–2):148–151
Masuda M, Nishihira T, Itoh K, Mizugaki M, Ishida N, Mori S (1993) An immunohistochemical analysis for cancer of the esophagus using monoclonal antibodies specific for modified nucleosides. Cancer 72(12):3571–3578
McMahon M, Contreras A, Ruggero D (2015) Small RNAs with big implications: new insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip Rev RNA 6(2):173–189
Metodiev MD, Assouline Z, Landrieu P, Chretien D, Bader-Meunier B, Guitton C, Munnich A, Rötig A (2015) Unusual clinical expression and long survival of a pseudouridylate synthase (PUS1) mutation into adulthood. Eur J Hum Genet 23(6):880–882
Miao FA, Chu K, Chen HR, Zhang M, Shi PC, Bai J, You YP (2019) Increased DKC1 expression in glioma and its significance in tumor cell proliferation, migration and invasion. Invest New Drugs 37(6):1177–1186
Mitchell JR, Cheng J, Collins K (1999a) A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3’ end. Mol Cell Biol 19(1):567–576
Mitchell JR, Wood E, Collins K (1999b) A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402(6761):551–555
Montanaro L, Brigotti M, Clohessy J, Barbieri S, Ceccarelli C, Santini D, Taffurelli M, Calienni M, Teruya-Feldstein J, Trere D, Pandolfi PP, Derenzini M (2006) Dyskerin expression influences the level of ribosomal RNA pseudo-uridylation and telomerase RNA component in human breast cancer. J Pathol 210(1):10–18
Montanaro L, Calienni M, Ceccarelli C, Santini D, Taffurelli M, Pileri S, Treré D, Derenzini M (2008) Relationship between dyskerin expression and telomerase activity in human breast cancer. Cell Oncol 30(6):483–490
Montanaro L, Calienni M, Bertoni S, Rocchi L, Sansone P, Storci G, Santini D, Ceccarelli C, Taffurelli M, Carnicelli D, Brigotti M, Bonafè M, Treré D, Derenzini M (2010) Novel dyskerin-mediated mechanism of p53 inactivation through defective mRNA translation. Cancer Res 70(11):4767–4777
Mort M, Ivanov D, Cooper DN, Chuzhanova NA (2008) A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat 29(8):1037–1047
Natchiar SK, Myasnikov AG, Kratzat H, Hazemann I, Klaholz BP (2017) Visualization of chemical modifications in the human 80S ribosome structure. Nature 551(7681):472–477
Nir R, Hoernes TP, Muramatsu H, Faserl K, Kariko K, Erlacher MD, Sas-Chen A, Schwartz S (2022) A systematic dissection of determinants and consequences of snoRNA-guided pseudouridylation of human mRNA. Nucleic Acids Res 50(9):4900–4916
O’Brien R, Tran SL, Maritz MF, Liu B, Kong CF, Purgato S, Yang C, Murray J, Russell AJ, Flemming CL, von Jonquieres G, Pickett HA, London WB, Haber M, Gunaratne PH, Norris MD, Perini G, Fletcher JI, MacKenzie KL (2016) MYC-Driven neuroblastomas are addicted to a telomerase-independent function of dyskerin. Cancer Res 76(12):3604–3617
Ojaimi MA, Banimortada BJ, Othman A, Riedhammer KM, Almannai M, El-Hattab AW (2022) Disorders of histone methylation: molecular basis and clinical syndromes. Clin Genet 102(3):169–181
Ono M, Yamada K, Avolio F, Scott MS, van Koningsbruggen S, Barton GJ, Lamond AI (2010) Analysis of human small nucleolar RNAs (snoRNA) and the development of snoRNA modulator of gene expression vectors. Mol Biol Cell 21(9):1569–1584
Ontiveros RJ, Stoute J, Liu KF (2019) The chemical diversity of RNA modifications. Biochem J 476(8):1227–1245
Parry EM, Alder JK, Lee SS, Phillips JA 3rd, Loyd JE, Duggal P, Armanios M (2011) Decreased dyskerin levels as a mechanism of telomere shortening in X-linked dyskeratosis congenita. J Med Genet 48(5):327–333
Patejko M, Struck-Lewicka W, Siluk D, Waszczuk-Jankowska M, Markuszewski MJ (2018) Urinary nucleosides and deoxynucleosides. Adv Clin Chem 83:1–51
Penzo M, Montanaro L (2018) Turning Uridines around: Role of rRNA Pseudouridylation in ribosome biogenesis and ribosomal function. Biomolecules 8(2):38
Penzo M, Casoli L, Ceccarelli C, Treré D, Ludovini V, Crinò L, Montanaro L (2013) DKC1 gene mutations in human sporadic cancer. Histol Histopathol 28(3):365–372
Pereira M, Francisco S, Varanda AS, Santos M, Santos MAS, Soares AR (2018) Impact of tRNA modifications and tRNA-modifying enzymes on proteostasis and human disease. Int J Mol Sci 19(12):3738
Phizicky EM, Hopper AK (2010) tRNA biology charges to the front. Genes Dev 24(17):1832–1860
Pierce ZP, Black JM (2022) Stress and susceptibility: a systematic review of prenatal epigenetic risks for developing post-traumatic stress disorder. Trauma Violence Abuse. https://doi.org/10.1177/15248380221109792
Ramamurthy V, Swann SL, Paulson JL, Spedaliere CJ, Mueller EG (1999) Critical aspartic acid residues in pseudouridine synthases. J Biol Chem 274(32):22225–22230
Rashid R, Liang B, Baker DL, Youssef OA, He Y, Phipps K, Terns RM, Terns MP, Li H (2006) Crystal structure of a Cbf5-Nop10-Gar1 complex and implications in RNA-guided pseudouridylation and dyskeratosis congenita. Mol Cell 21(2):249–260
Rasmuson T, Björk GR (1983) Pseudouridine: a modified nucleoside as biological marker in malignant lymphomas. Cancer Detect Prev 6(1–2):293–296
Razavi AC, Bazzano LA, He J, Li S, Fernandez C, Whelton SP, Krousel-Wood M, Nierenberg JL, Shi M, Li C, Mi X, Kinchen J, Kelly TN (2020) Pseudouridine and N-formylmethionine associate with left ventricular mass index: metabolome-wide association analysis of cardiac remodeling. J Mol Cell Cardiol 140:22–29
Rintala-Dempsey AC, Kothe U (2017) Eukaryotic stand-alone pseudouridine synthases: RNA modifying enzymes and emerging regulators of gene expression? RNA Biol 14(9):1185–1196
Roovers M, Hale C, Tricot C, Terns MP, Terns RM, Grosjean H, Droogmans L (2006) Formation of the conserved pseudouridine at position 55 in archaeal tRNA. Nucleic Acids Res 34(15):4293–4301
Ropers HH (2010) Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 11:161–187
Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP (2003) Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 299(5604):259–262
Safra M, Nir R, Farouq D, Vainberg Slutskin I, Schwartz S (2017) TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code. Genome Res 27(3):393–406
Salowsky R, Heiss NS, Benner A, Wittig R, Poustka A (2002) Basal transcription activity of the dyskeratosis congenita gene is mediated by Sp1 and Sp3 and a patient mutation in a Sp1 binding site is associated with decreased promoter activity. Gene 293(1–2):9–19
Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S, Ince TA (2011) High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A 108(45):18378–18383
Savage SA, Niewisch MR (1993). Dyskeratosis congenita and related telomere biology disorders. GeneReviews(®). Adam MP, Mirzaa GM, Pagon RA, et al. Seattle (WA), University of Washington, Seattle. Copyright © 1993–2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle
Schwartz S (2016) Cracking the epitranscriptome. RNA 22(2):169–174
Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, Fink G, Regev A (2014) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159(1):148–162
Seidel A, Brunner S, Seidel P, Fritz GI, Herbarth O (2006) Modified nucleosides: an accurate tumour marker for clinical diagnosis of cancer, early detection and therapy control. Br J Cancer 94(11):1726–1733
Shaheen R, Han L, Faqeih E, Ewida N, Alobeid E, Phizicky EM, Alkuraya FS (2016) A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum Genet 135(7):707–713
Shaheen R, Tasak M, Maddirevula S, Abdel-Salam GMH, Sayed ISM, Alazami AM, Al-Sheddi T, Alobeid E, Phizicky EM, Alkuraya FS (2019) PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum Genet 138(3):231–239
Sieron P, Hader C, Hatina J, Engers R, Wlazlinski A, Müller M, Schulz WA (2009) DKC1 overexpression associated with prostate cancer progression. Br J Cancer 101(8):1410–1416
Slingerland J, Pagano M (2000) Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 183(1):10–17
Song J, Dong L, Sun H, Luo N, Huang Q, Li K, Shen X, Jiang Z, Lv Z, Peng L, Zhang M, Wang K, Liu K, Hong J, Yi C (2023) CRISPR-free, programmable RNA pseudouridylation to suppress premature termination codons. Mol Cell 83(1):139-155.e139
Spedaliere CJ, Hamilton CS, Mueller EG (2000) Functional importance of motif I of pseudouridine synthases: mutagenesis of aligned lysine and proline residues. Biochemistry 39(31):9459–9465
Spenkuch F, Motorin Y, Helm M (2014) Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol 11(12):1540–1554
Stockert JA, Gupta A, Herzog B, Yadav SS, Tewari AK, Yadav KK (2019) Predictive value of pseudouridine in prostate cancer. Am J Clin Exp Urol 7(4):262–272
Stockert JA, Weil R, Yadav KK, Kyprianou N, Tewari AK (2021) Pseudouridine as a novel biomarker in prostate cancer. Urol Oncol 39(1):63–71
Sumita M, Desaulniers JP, Chang YC, Chui HM, Clos L 2nd, Chow CS (2005) Effects of nucleotide substitution and modification on the stability and structure of helix 69 from 28S rRNA. RNA 11(9):1420–1429
Tamura S, Amuro Y, Nakano T, Fujii J, Moriwaki Y, Yamamoto T, Hada T, Higashino K (1986a) Urinary excretion of pseudouridine in patients with hepatocellular carcinoma. Cancer 57(8):1571–1575
Tamura S, Fujii J, Nakano T, Hada T, Higashino K (1986b) Urinary pseudouridine as a tumor marker in patients with small cell lung cancer. Clin Chim Acta 154(2):125–132
Tamura S, Fujioka H, Nakano T, Hada T, Higashino K (1987) Serum pseudouridine as a biochemical marker in small cell lung cancer. Cancer Res 47(22):6138–6141
Torres AG, Batlle E, Ribas de Pouplana L (2014) Role of tRNA modifications in human diseases. Trends Mol Med 20(6):306–314
Trahan C, Martel C, Dragon F (2010) Effects of dyskeratosis congenita mutations in dyskerin, NHP2 and NOP10 on assembly of H/ACA pre-RNPs. Hum Mol Genet 19(5):825–836
Tummala H, Collopy LC, Walne AJ, Ellison A, Cardoso S, Aksu T, Yarali N, Aslan D, Fikret Akata R, Teo J, Songyang Z, Pontikos N, Fitzgibbon J, Tomita K, Vulliamy T, Dokal I (2018) Homozygous OB-fold variants in telomere protein TPP1 are associated with dyskeratosis congenita-like phenotypes. Blood 132(12):1349–1353
Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I (2001) The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413(6854):432–435
Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I (2006) Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 107(7):2680–2685
Wang M, Liu H, Zheng J, Chen B, Zhou M, Fan W, Wang H, Liang X, Zhou X, Eriani G, Jiang P, Guan MX (2016) A deafness- and diabetes-associated tRNA mutation causes deficient pseudouridinylation at position 55 in tRNAGlu and mitochondrial dysfunction. J Biol Chem 291(40):21029–21041
Watanabe M, Yamamoto G, Fujiyoshi K, Akagi Y, Kakuta M, Nishimura Y, Akagi K (2019) Development of metachronous rectal cancers in a young man with dyskeratosis congenita: a case report. J Med Case Rep 13(1):117
Wu Y, Zhan S, Xu Y, Gao X (2021) RNA modifications in cardiovascular diseases, the potential therapeutic targets. Life Sci 278:119565
Zeharia A, Fischel-Ghodsian N, Casas K, Bykhocskaya Y, Tamari H, Lev D, Mimouni M, Lerman-Sagie T (2005) Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J Child Neurol 20(5):449–452
Zeleznik OA, Eliassen AH, Kraft P, Poole EM, Rosner BA, Jeanfavre S, Deik AA, Bullock K, Hitchcock DS, Avila-Pacheco J, Clish CB, Tworoger SS (2020) A prospective analysis of circulating plasma metabolites associated with ovarian cancer risk. Cancer Res 80(6):1357–1367
Zhao BS, Roundtree IA, He C (2017) Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18(1):31–42
Zheng YF, Kong HW, Xiong JH, Lv S, Xu GW (2005) Clinical significance and prognostic value of urinary nucleosides in breast cancer patients. Clin Biochem 38(1):24–30
Acknowledgements
Project no. TKP2021-EGA-24 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.
Funding
Open access funding provided by Semmelweis University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Keszthelyi, T.M., Tory, K. The importance of pseudouridylation: human disorders related to the fifth nucleoside. BIOLOGIA FUTURA 74, 3–15 (2023). https://doi.org/10.1007/s42977-023-00158-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42977-023-00158-3