
Abstract
Background
Chemotherapy-induced peripheral neuropathy (CIPN) is regarded as one of the most common dose-limiting adverse effects of several chemotherapeutic agents, such as platinum derivatives (oxaliplatin and cisplatin), taxanes, vinca alkaloids and bortezomib. CIPN affects more than 60% of patients receiving anticancer therapy and although it is a nonfatal condition, it significantly worsens patients’ quality of life. The number of analgesic drugs used to relieve pain symptoms in CIPN is very limited and their efficacy in CIPN is significantly lower than that observed in other neuropathic pain types. Importantly, there are currently no recommended options for effective prevention of CIPN, and strong evidence for the utility and clinical efficacy of some previously tested preventive therapies is still limited.
Methods
The present article is the second one in the two-part series of review articles focused on CIPN. It summarizes the most recent advances in the field of studies on CIPN caused by oxaliplatin, the third-generation platinum-based antitumor drug used to treat colorectal cancer. Pharmacological properties of oxaliplatin, genetic, molecular and clinical features of oxaliplatin-induced neuropathy are discussed.
Results
Available therapies, as well as results from clinical trials assessing drug candidates for the prevention of oxaliplatin-induced neuropathy are summarized.
Conclusion
Emerging novel chemical structures—potential future preventative pharmacotherapies for CIPN caused by oxaliplatin are reported.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Oxaliplatin as a neurotoxic drug
Oxaliplatin (trans-l-diaminocyclohexane oxalate platinum (II), Fig. 1), a third-generation platinum-based chemotherapeutic agent [1], has been placed on the World Health Organization’s List of Essential Medicines [2]. Similar to other platinum-based compounds (e.g., cisplatin, carboplatin), oxaliplatin interferes with tumor cell proliferation by forming deoxyribonucleic acid (DNA)-platinum adducts, which leads to cancer cell destruction [1].
Clinically, oxaliplatin is used in combination with folinic acid and 5-fluorouracil as a part of the FOLFOX regimen or with folinic acid, 5-fluorouracil and irinotecan (FOLFOXIRI) regimen as the first-line and as adjuvant colorectal cancer therapy [3].
Recent preclinical studies have shown that transporter-mediated uptake of oxaliplatin (and paclitaxel [4, 5]) into dorsal root ganglia neurons might trigger pathophysiological changes in sensory neurons, thus resulting in chemotherapy-induced peripheral neuropathy (CIPN) development [4].
The organic cation/carnitine transporters OCTN1 and OCTN2 belong to the organic cation transporter family. These proteins are responsible for transporting oxaliplatin and are regarded as key transporters maintaining the intracellular concentration of platinum derivatives via active uptake and efflux processes. OCTN1 and OCTN2 are also functionally expressed within dorsal root ganglia neurons, and OCTN1 is thought to be the main contributor to the neuronal accumulation of oxaliplatin and a mediator of its neurotoxicity [4, 6,7,8]. In addition, CTR1 (copper transporter 1), CTR2 (copper transporter 2), ATP7A (copper-transporting p-type adenosine triphosphatase 1) and ATP7B (copper-transporting p-type adenosine triphosphatase 2) are copper transporters that not only maintain homeostasis and copper metabolism but also are involved in cisplatin, carboplatin and oxaliplatin transport. A high CTR1 level is regarded as a predictor of prolonged survival and enhanced response to chemotherapy in cancer patients [9].
A unique feature of oxaliplatin is that it is rapidly and non-enzymatically transformed into several metabolites, including oxalate, monochloro-, dichloro- and diaquo-diaminocyclohexane (DACH) platinum metabolites (Fig. 1). This occurs via the replacement of the oxalate moiety with chloride ions and water in the blood plasma. The formation of these metabolites distinguishes oxaliplatin from cisplatin. Of note, oxalate is thought to be responsible for the cold-induced hypersensitivity observed in oxaliplatin-treated, but not cisplatin-treated, patients [10, 11], although additional mechanisms, including the binding of platinum complexes to cellular proteins, have also been suggested [12].

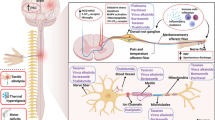
Chemical structure of oxaliplatin, its biotransformation pathways and a potential mechanism underlying the development of oxaliplatin-induced neuropathy: oxaliplatin (I) is rapidly hydrolyzed in vivo to bioactive derivatives through the displacement of the oxalate group by H2O and Cl− ions to produce oxalate (II) as well as reactive monochloro-diaminocyclohexane (DACH) (III), dichloro-DACH (IV) and diaquo-DACH platinum (V) metabolites. Oxalate, which reacts with Ca2+ ions, is the main contributor to neurotoxicity caused by oxaliplatin
Oxalate is a well-known calcium chelator [13], and oxalate-induced pain is regarded as a consequence of the chelation of extracellular calcium ions (i.e., the removal of extracellular calcium ions), which in turn leads to the increase in sodium conductance, neuronal depolarization and hyperexcitability (Fig. 1) [14, 15].
An increase in extracellular calcium concentration increases the probability of voltage-gated sodium (Nav) channel closure and results in decreased excitability of peripheral neurons. Hence, the administration of calcium and magnesium ions before oxaliplatin infusion was evaluated in several clinical trials as a strategy to prevent the development of oxaliplatin-induced peripheral neuropathy, although clinical benefits of this approach were not confirmed [16, 17].
Genetic polymorphisms associated with oxaliplatin-induced neuropathy
Data from mouse studies show that similarly to humans, various mouse strains exposed to oxaliplatin develop chemotherapy-induced peripheral neuropathy (CIPN). Significant and strain-dependent differences were observed in the severity and behavioral features of CIPN. This supports the hypothesis that, for oxaliplatin, a genetic component might be relevant for CIPN development, both in rodent models and in clinical settings. Importantly, these different neurotoxic phenotypes seem to be crucial in the choice of an appropriate mouse strain for CIPN studies [18].
Genetic risk factors associated with the development of CIPN have been studied recently [19]. As such, they can influence the absorption, distribution, metabolism or excretion of chemotherapeutic agents that are responsible for CIPN formation. It was suggested that polymorphisms of glutathione transferases, cytochrome P450 enzymes and ATP binding cassette transporters may be involved in the development of platinum-induced CIPN [5, 20,21,22]. The Ile105Val polymorphism of the GSTP1 gene, encoding glutathione S-transferase P1, has been associated with a decreased risk of oxaliplatin-related CIPN. This mutation increases the activity of glutathione S-transferase P1, resulting in the increased conjugation of hydrophobic and electrophilic compounds with glutathione, thus reducing their toxicity [23].
In addition, genetic or pharmacological knockout of OCT2 prevented the development of cold and mechanical hypersensitivity following oxaliplatin therapy, suggesting that the OCT2 transporter plays a key role in oxaliplatin-induced cytotoxicity [24].
A polymorphism in the ABCC2 (ATP binding cassette subfamily C member 2) gene coding for the multidrug resistance protein 2 (MRP2) that leads to higher oxaliplatin concentrations in neurons was associated with oxaliplatin-induced neurotoxicity [25]. Genetic variants of AGXT (alanine glyoxylate aminotransferase) gene coding for alanine glyoxylate aminotransferase (i.e., an enzyme involved in oxalate metabolism) have been correlated with the severity of CIPN in patients on oxaliplatin therapy [26]. The development of acute oxaliplatin-induced peripheral neuropathy was altered in patients with single-nucleotide polymorphisms in SCNA genes encoding selected Nav channels. A polymorphism (rs2302237) in the SCN4A gene, which encodes the Nav1.4 channel and a polymorphism (rs1263292) in the SCN10A coding for Nav1.8 channel have been associated with an increased incidence of acute oxaliplatin-induced CIPN [27], while a polymorphism (rs6746030) in the SCN9A gene, which encodes the Nav1.7 channel, protected against severe oxaliplatin-induced CIPN [28].
Clinical features of oxaliplatin-induced CIPN
In contrast to cisplatin, oxaliplatin is not nephrotoxic or ototoxic, but its use is still associated with various adverse effects, including myelotoxicity and CIPN. Neurotoxicity of oxaliplatin appears in two distinct forms, i.e., acute and chronic [29]. The acute, transient neuropathy develops in almost 90% of patients within hours of infusion and lasts a few days (usually 7 days), recurring with subsequent infusions. Acute neuropathy results from a transient impairment of ion channels and nerve hyperexcitability due to Nav channel activation [30, 31] and is characterized by dysesthesia and paresthesia of the hands and feet. These symptoms are often exacerbated by cold. Motor symptoms (e.g., tetanic spasms, fasciculations, prolonged muscular contractions) can also be present in patients on oxaliplatin therapy. The acute form of oxaliplatin-induced CIPN tends to weaken between treatment cycles, but prolonged exposure to this drug often leads to the development of a severe chronic form of CIPN.
The incidence of chronic peripheral neuropathy following oxaliplatin treatment has been estimated as approximately 70% [32, 33]. Clinically, the symptoms of chronic oxaliplatin-induced neuropathy closely resemble those of the acute form and include paresthesia, hypoesthesia and dysesthesia of the hands and feet. Additionally, changed proprioception negatively affecting normal daily activities that require fine motor coordination might also occur [34, 35]. The main mechanism responsible for the observed permanent distal sensory loss is associated with the death of sensory neurons (i.e., nerve cell necrosis) due to the binding of oxaliplatin to mitochondrial DNA [36].
It has been estimated that the development of CIPN due to oxaliplatin administration is likely to occur at cumulative doses exceeding 780–850 mg/m2 [29, 35, 37, 38]. The symptoms that occur may worsen or appear weeks to several months after the completion of therapy (‘coasting’ phenomenon), and the estimated prevalence of these persistent symptoms is approximately 60% of patients; these patients report long-lasting neuropathic symptoms that negatively interfere with their daily functions and significantly impair their quality of life [29, 34]. Importantly, overall recovery from CIPN symptoms is usually incomplete and might last up to five years following oxaliplatin chemotherapy cessation [39].
Symptoms of acute and chronic neuropathy were evaluated in patients receiving an adjuvant FOLFOX regimen in a phase III clinical trial (ClinicalTrials.gov: NCT00316914). Acute neuropathy was assessed for 6 days with each cycle of FOLFOX. Chronic neuropathy was evaluated until 18 months after chemotherapy cessation. Acute neuropathy symptoms included sensitivity to touching cold items, sensitivity to swallowing cold items, throat discomfort, and muscle cramps. Acute symptoms peaked at day 3 and improved, but they tended not to resolve completely between treatments. For chronic neurotoxicity, tingling was the most severe symptom, followed by numbness and then pain. Patients with more severe acute neuropathy during the first cycle of therapy experienced more severe chronic sensory neurotoxicity [40].
Available preventative therapies for CIPN caused by oxaliplatin
Considering the severity of CIPN and its irreversibility in a large proportion of patients, for many years efforts have been being made to alleviate this pharmacoresistant clinical condition. Therefore, the National Cancer Institute's Symptom Management and Health-Related Quality of Life Steering Committee has announced that CIPN is a priority area of translational research in cancer care [4]. Even the latest literature data clearly indicate that although numerous preventative therapies were tested for their potential utility for CIPN alleviation, this clinical entity currently is still not preventable [41,42,43], and many strategies tested were found to be ineffective (Table 1). Therefore, based on meta-analyses of clinical trials, at present, no drug can be proposed as a gold standard to either prevent CIPN or treat its symptoms, and chemotherapeutic drug dose modification remains the only preventative strategy [41].
Several clinical trials aimed to examine potentially neuroprotective therapies to prevent CIPN. Based on the data obtained, only duloxetine is moderately recommended by the American Society of Clinical Oncology (ASCO) for the prevention of oxaliplatin-induced CIPN (Table 1) [44, 45]. These guidelines undoubtedly show that there are no clear algorithms for CIPN prevention because high-quality and strong evidence for the preventative action of agents tested is not available.
It should also be noted that many other drugs have been assessed, but their utility for the prevention of oxaliplatin-induced CIPN is still controversial (Table 1). For example, venlafaxine and neurotropin are also not recommended for routine use because of insufficient information on their clinical efficacy. A pilot randomized, placebo-controlled, double-blinded phase III trial (ClinicalTrials.gov: NCT01611155) tested the use of venlafaxine to prevent oxaliplatin neurotoxicity in 50 patients with stage II-IV colon cancer. The results of this study supported neither the use of venlafaxine for the prevention of oxaliplatin-induced neuropathy in clinical practice nor the initiation of a further phase III trial for an investigation of venlafaxine in this setting [51].
Pregabalin was also assessed in a phase III, randomized, double-blind, placebo-controlled clinical trial for its efficacy and safety in the prevention and reduction of oxaliplatin-induced painful neuropathy (PreOx; ClinicalTrials.gov: NCT01450163). Patients received either pregabalin or placebo for 3 days before and 3 days after each oxaliplatin infusion and were followed for up to 6 months. The preemptive use of pregabalin during oxaliplatin infusions was safe, but it did not decrease the incidence of CIPN (Table 1) [41, 52].
Carbamazepine, oxcarbazepine, glutamate and glutamine, goshajinkigan, oral alpha-lipoic acid, omega-3 fatty acids and oral vitamin B complex are not recommended as CIPN-preventing drugs because of their inconclusive data, low level of evidence and low strength of available evidence (Table 1) [41, 45, 48, 50, 53,54,55,56,57].
Other trials on potentially promising drug candidates for CIPN prevention, such as a fatty acid amide hydrolase inhibitor (BIA 10-2474, Table 1), were prematurely terminated due to serious adverse events noted in participants (death, severe neurological complications) [44].
Duloxetine
Evidence from preclinical [58, 59] and clinical studies [4] suggests that duloxetine, an antidepressant drug acting as a serotonin-noradrenaline reuptake inhibitor (SNRI), may be effective in the prevention of CIPN without reducing the antitumor activity of oxaliplatin [49]. As shown in Table 1, ASCO recommends duloxetine as the only potential treatment for CIPN, but importantly, this guidance still lacks the support of sufficient evidence. Moreover, since in clinical trials duloxetine was not fully effective and it did not work in every patient enrolled, the identification of strong predictors for duloxetine response is a priority area of research. Results from a phase III double-blind trial of oral duloxetine for the treatment of pain associated with chemotherapy-induced peripheral neuropathy (ClinicalTrials.gov: NCT00489411) revealed that oxaliplatin-treated patients with high emotional functioning are more likely to experience pain reduction resulting from duloxetine administration [14].
Since patients with oxaliplatin-induced painful CIPN are more likely to benefit from the use of duloxetine than those with paclitaxel-induced CIPN, this also suggests that duloxetine’s mechanism of action may be closely associated with very specific molecular mechanisms underlying oxaliplatin-induced neurotoxicity [14, 60]. This possibility has not been fully explained, so far, and it seems to be even more exciting in terms of a distinct clinical efficacy of duloxetine and venlafaxine, both being representatives of SNRI antidepressants. Their efficacy was directly compared in cancer patients who were suffering from CIPN. The administration of duloxetine was more effective than that of venlafaxine in decreasing the symptoms of peripheral neuropathy due to chemotherapy. Duloxetine had a better effect on reducing the grade of motor neuropathy and neuropathic pain severity [61]. A potential explanation for this observed diversity in effectiveness is the difference in the pharmacological profile of both drugs. Compared to other SNRIs, venlafaxine has a high affinity for the serotonin transporter (SERT) but a relatively lower affinity for the norepinephrine transporter (NET), so at lower doses, it may act as a selective serotonin reuptake inhibitor (SSRI) and may not have the same activity against painful neuropathy as duloxetine [51].
Drug candidates for the prevention of CIPN caused by oxaliplatin
Since CIPN is not preventable using currently available drugs, there is a strong medical demand to search for novel therapeutic options for this clinical entity. Our understanding of the complexity of neurobiological mechanisms implicated in CIPN symptomatology (altered ion channel expression, neuronal hyperexcitability, mitochondrial dysfunction, impaired immune cell signaling and axon degeneration) has constituted a basis for extended research on pharmacological prevention of CIPN. In this context, not only novel chemical compounds but also repurposed drugs are being investigated at preclinical and clinical stages of research. Considering the risk factors and causes of CIPN development, several potential approaches have been proposed (Fig. 2) [44].

Potential preventative therapies for CIPN caused by oxaliplatin: clinically tested drug candidates, repurposed drugs and preclinically tested lead compounds. APE apyrimidinic endonuclease/redox effector factor, GAT-1 γ-aminobutyric acid (GABA) transporter isoform 1, Glu glutamate, GLT-1 glutamate transporter 1, GLAST GLutamate and ASpartate Transporter, GM1 monosialotetrahexosylganglioside, ROS reactive oxygen species, TLR4 toll-like receptor 4, Nav voltage-gated sodium channels, Cav voltage-gated calcium channels, TREK-1 two-pore-domain background potassium channel, TRPA1 Transient Receptor Potential Ankyrin-repeat 1 channel, IL interleukin, TNFα tumor necrosis factor α, SARM-1 sterile alpha and TIR motif-containing protein 1, ATP adenosine triphosphate, Bclw Bcl2 family member, NOP nociceptin opioid peptide receptor, S1P1 sphingosine-1-phosphate receptor type 1, OATP1B2 solute carrier organic anion-transporting polypeptide B2, OCTN2 Na+-dependent organic cation/carnitine transporter, MSC mesenchynmal stem cell therapy
An ‘ideal’ CIPN-preventing drug should have the following features: (i) it should act as a multitarget agent, as this might increase its protective efficacy; (ii) it should not diminish the antitumor efficacy of the chemotherapeutic used; and (iii) it should be introduced into therapy relying on an individual patient’s reported outcomes to increase its efficacy in preventing the symptoms and causes of CIPN. Several experimental therapies for CIPN prevention have reached the clinical stage of testing. These emerging drug candidates are shown in Table 2 and Fig. 2.
Targeting chemotherapeutic drug uptake transporters: tyrosine kinase inhibitors
In vitro and in vivo studies have shown that two tyrosine kinase inhibitors, nilotinib (an OATP1B2 inhibitor) and dasatinib (an OCTN2 inhibitor), might offer a potential neuroprotective strategy for CIPN caused by paclitaxel and oxaliplatin (Fig. 2) without negatively impacting their systemic clearance or antitumor efficacy. As repurposed drugs, nilotinib and dasatinib are currently being tested in an ongoing phase Ib trial and in double-blinded, placebo-controlled, randomized phase II studies (Table 2) [4].
Targeting mitochondrial dysfunction and oxidative stress
Oxidative stress caused by chemotherapy is a significant contributor to mitochondrial damage, and reactive oxygen species (ROS) neutralization is a potential preventative strategy for CIPN. Manganese superoxide dismutase (MnSOD) catalyzes the dismutation of superoxide and forms hydrogen peroxide and molecular oxygen. Hence, this enzyme limits the reaction of superoxide with nitric oxide, thus preventing the formation of the reactive and highly toxic nitrogen species peroxynitrite, which is able to nitrate the tyrosine residue of tissue proteins [64]. Recently, targeting MnSOD has emerged as a very promising strategy to prevent CIPN caused by oxaliplatin [83,84,85,86].
Calmangafodipir
Calmangafodipir (Ca4Mn(DPDP)5, PledOxVR), a derivative of mangafodipir, a magnetic resonance imaging contrast and a cytoprotectant agent [87, 88], is a mitochondrial MnSOD mimetic that reduces ROS tissue levels (Fig. 2). Although preclinical and clinical data have confirmed that such compounds have significant preventative neuroprotective activity in oxaliplatin-induced CIPN [83,84,85,86], it is hypothesized that neuroprotection from calmangafodipir may also be possible with other chemotherapeutics [4, 64].
In a placebo-controlled, double-blinded, randomized, phase II study in patients with metastatic colorectal cancer (Table 2), calmangafodipir reduced cold allodynia and other sensory symptoms during and after treatment with oxaliplatin. The neuroprotective effects seen were clinically meaningful, as a delay in both the onset and the reduction of the intensity of CIPN symptoms were noted both in the acute and in the chronic phase of neuropathy. In this trial, progression-free and overall survival outcomes were not negatively influenced by calmangafodipir [64].
Two international trials, POLAR A and POLAR M are currently evaluating the efficacy of calmangafodipir for the prevention of oxaliplatin-induced neuropathy in colorectal cancer patients. Results are expected in the years 2020/2021 (POLAR A) or 2021–2023 (POLAR M) [4, 62].
Apart from CIPN, calmangafodipir has also shown efficacy in severe paracetamol overdose and, based on results from a clinical trial (POP Trial, Clinicaltrials.gov: NCT03177395), might be regarded as an adjuvant or alternative to the N-acetylcysteine antidote for paracetamol overdose [89, 90]. In this trial, the most frequent adverse effect was gastrointestinal toxicity (nausea, vomiting, diarrhea, dyspepsia) [90,91,92].
Targeting apurinic/apyrimidinic endonuclease function
Another mechanism of nerve injury that underlies CIPN formation is associated with impaired DNA repair within the sensory nervous system. The pathway for base excision repair is the primary means for the repair of oxidative DNA damage. In this pathway, the enzyme apyrimidinic endonuclease/redox effector factor (APE1) is an important molecule for the removal of the damaged bases. Decreased APE1 levels in sensory neurons increase neurotoxicity and targeting APE1 by the small molecules APX3330 and APX2009 (Fig. 2) provided neuroprotection against cisplatin- and oxaliplatin-induced neurotoxicity. APX2009 also demonstrated a strong tumor cell-killing effect. These data suggest that such compounds might be effective in preventing or reversing platinum-induced CIPN without reducing the anticancer activity of platinum-based chemotherapeutics [93]. Currently, APX3330 is entering clinical trials as an antineoplastic agent as well for the prevention of CIPN (Table 2) [94].
Targeting the inhibition of neuronal apoptosis and astrocyte activation
Impaired sphingolipid metabolism is also considered a mechanism contributing to CIPN development. Certain chemotherapeutics, including paclitaxel [66], oxaliplatin [95] and bortezomib [96], induce the development of CIPN by dysregulating sphingolipid metabolism, leading to increased formation of sphingosine-1-phosphate (S1P), which binds and stimulates sphingosine-1-phosphate receptor 1 (S1PR1). In rodent models of CIPN, the ceramide metabolic pathway was activated in the spinal cord, and blocking the formation of S1P with sphingosine kinase inhibitors reversed CIPN symptoms, mainly allodynia and hyperalgesia [66].
Fingolimod (FTY720; Table 2, Fig. 2) acts as a nonselective agonist of sphingosine-1-phosphate receptors (S1P1R, S1P3R, S1P4R, and S1P5R but not S1P2R). Fingolimod phosphate activates S1P1Rs with high potency (KD = 0.3 nM) and with higher efficacy than S1P. The overactivation of S1P1R caused by fingolimod results in rapid receptor desensitization and internalization. Thus, fingolimod behaves as a selective functional antagonist of the S1P1R subtype through the induction of receptor downregulation. The downstream effect of S1P1R blockade is the inhibition of the NF-κB pathway [97]. At present, fingolimod is commercially available as a marketed drug for multiple sclerosis, but its daily injections also inhibited the development of mechanical allodynia and hyperalgesia induced by paclitaxel, oxaliplatin, and bortezomib. Similar effects were also noted with other S1P1R antagonists. Moreover, a prolonged treatment with fingolimod or other S1P1R functional antagonists did not induce tolerance to their analgesic effects. S1P1R antagonists inhibited the development of CIPN, but they were also able to induce a sustained reversal of paclitaxel-induced neuropathic pain. Apart from fingolimod, novel S1PR modulators are being developed, including ponesimod [66], siponimod [98], and CYM5442, as well as selective S1P1R antagonists, including W146, NIBR-14/15, and TASP0251078, as novel drug candidates for the treatment of various diseases, including multiple sclerosis, rheumatoid arthritis, colitis and cancer [99,100,101]. Importantly, the S1PR1 antagonists developed to date are not expected to reduce the anticancer activity of chemotherapeutic agents, and they may act synergistically [97, 102], making fingolimod and its analogs attractive agents for CIPN prevention [101]. Transient cardiovascular adverse effects of fingolimod result from a transient activation of S1P1R—a phenomenon that precedes its functional antagonism. These cardiovascular adverse effects include first-dose bradycardia, atrioventricular conduction block and hypotension, and they must be cautiously considered when utilizing this drug for CIPN prevention and treatment [4].
Monosialotetrahexosylganglioside (GM1) is a glycosphingolipid located in the outer layer of the plasma membrane (Fig. 2). It is critical for nerve development, differentiation and repair after the injury as it has neuroprotective, neurotrophic-factor-like activity by activating the Trk neurotrophin receptors [103, 104]. GM1 can also prevent seizures and oxidative stress [105]. Initially, GM1 was approved for the treatment of vasculogenic or traumatic central nervous impairments and Parkinson’s disease, and now its potential procognitive activity is also suggested [103].
In a clinical trial conducted in colon cancer patients on FOLFOX therapy (Table 2), GM1 attenuated symptoms of acute neuropathy (cold sensitivity, throat discomfort, muscle cramps), but its use to prevent the neurotoxicity of oxaliplatin is currently not recommended, and additional placebo-controlled studies are needed to better assess the utility of this agent in this clinical condition [4, 67].
Sodium channel inhibition
Nav channels are an important target for analgesic drugs used in neuropathic pain conditions. Most of the available Nav inhibitors nonselectively block Nav channels within the sensory nervous system. From these drugs, lidocaine (Table 2, Fig. 2), a local anesthetic agent, will be assessed in a pilot study to determine the tolerability and the efficacy of its intravenous administration for the prevention of oxaliplatin-induced cold hypersensitivity and spontaneous pain in the setting of mFOLFOX6 chemotherapy for advanced colorectal cancer [72].
Among Nav channels, Nav1.7 plays an important role in multiple neuropathic pain states. It was shown that the expression and function of Nav1.7 are increased in preclinical models of CIPN [106, 107]. Data from a randomized, double-blind, placebo-controlled pilot study investigating the efficacy of a Nav1.7 antagonist, XEN402, indicate that it is effective in patients with inherited erythromelalgia, but so far, this compound has not been assessed in CIPN [4].
Sigma-1 receptor antagonism
Selective sigma-1 receptor antagonists (e.g., MR309) attenuated oxaliplatin-induced CIPN [76], and it was suggested that sigma-1 receptors might constitute a novel drug target for CIPN-preventing drugs [77]. Currently, MR309 (400 mg/day, 5 days per cycle) has completed a phase II randomized, double-blind and placebo-controlled clinical trial in patients with colorectal cancer receiving FOLFOX, showing efficacy and a good safety profile in patients on oxaliplatin therapy [76]. Compared to placebo, MR309 significantly reduced the cold pain threshold temperature and lowered the proportion of patients with severe chronic neuropathy. MR309 increased the total amount of oxaliplatin delivered and it attenuated motor hyperexcitability symptoms in patients with colorectal cancer treated with oxaliplatin. A potential neuroprotective role of MR309 for acute and chronic CIPN induced by oxaliplatin was demonstrated. Taken together, it has been suggested that MR309 might be a first-in-class drug with a novel mechanism of action to improve some signs and symptoms of acute CIPN (e.g., cold pain and motor symptoms) caused by oxaliplatin. However, it has to be noted that further studies should be focused on testing different dose regimens of MR309 administration and continuous dosing during the full chemotherapy period [76].
Angiotensin II type 2 receptor antagonism
EMA401 (olodanrigan) is an angiotensin II type 2 (AT2) receptor antagonist. In clinical trials, this orally active agent was tested in patients with postherpetic neuralgia, painful diabetic neuropathy [79,80,81,82] and CIPN [78]. Although EMA401 showed antineuropathic properties in the postherpetic neuralgia and painful diabetic neuropathy patient populations, these trials were withdrawn or terminated due to the side effects observed [108,109,110,111].
A phase II clinical trial of EMA401 in patients suffering from CIPN was conducted as an open-label biomarker study designed to provide a proof of concept of the use of EMA401 in CIPN due to taxanes or platinum derivatives. In this trial, the primary endpoint was the change (i.e., a decrease of at least 30%) in mean spontaneous pain intensity score between baseline and the last week of 28 days of EMA401 dosing. A number of secondary endpoints were also evaluated, including changes in nerve characteristics in skin biopsies taken from the calf, change in evoked (light touch) mean pain intensity score from baseline over time, change in evoked (cold touch) mean pain intensity score over time and patient global impression of change [109]. It has to be noted that the results of this trial are inconclusive and difficult for interpretation because statistical analyses have not been specified as only one arm was reported [78].
Future outlook: data from preclinical studies
Although the mechanisms underlying CIPN are relatively well understood, the available results of clinical trials clearly indicate that the success rate for the introduction of novel drugs that are able to prevent CIPN is low [50], the progress in this area is slow and, at present, there is insufficient evidence to recommend any specific agent for CIPN prevention. Hence, animal research is a strong scientific and practical demand, because it enables the discovery and development of novel interventions for this clinical entity. Since mechanism-based therapies are urgently needed [112], novel targets are being explored to develop potential drug candidates. However, the drug development process is both time- and cost-consuming. Therefore, drug repurposing, i.e., the identification of new clinical applications for registered drugs, is also a valuable alternative approach to drug discovery and development research [113].
In this context, targeting sterile alpha and TIR motif-containing protein 1 (SARM-1), an injury-inducible nicotinamide adenine dinucleotide glycohydrolase (NADase) that triggers axon loss, is an emerging strategy for the prevention of CIPN-related axon degeneration (Fig. 2), and some SARM-1-targeting agents are currently in development. In addition to small-molecule inhibitors, a very potent, dominant-negative version of SARM-1 was developed. It acts by blocking the activation of wild-type SARM-1, and this protects axons from damage. Gene therapy using AAV-mediated delivery of this SARM-1 dominant-negative molecule provided long-lasting axonal protection [114]. In addition, SARM1-mediated NADH dehydrogenase subunit I (NAD1) destruction and loss is compensated by the development of NAD1 precursors, which have the potential ability to protect axons. These NAD1 precursors are natural products that have potential utility in CIPN prevention [114]. In addition to the direct influence on SARM-1, targeting the upstream pathways regulating SARM-1 to prevent CIPN is of interest. Efforts have been made to boost the function and increase the expression of nicotinamide nucleotide adenylyltransferase 2 (NMNAT2). Targeting the degradation of NMNAT2 might also be an alternative for neuropathy [114,115,116].
Ethoxyquin was recently identified as a neuroprotective compound showing potential benefits in toxic neuropathies. Its efficacy without compromising antitumor efficacy was demonstrated against paclitaxel-induced CIPN in vivo. Ethoxyquin also prevented cisplatin-induced neurotoxicity in vitro. In vivo, chronic administration of ethoxyquin partially attenuated cisplatin-induced abnormalities. The neuroprotective effect of ethoxyquin is thought to result from the inhibition of the chaperone activity of heat shock protein, Hsp90 [117, 118].
Bclw is a death-protecting member of the Bcl2 family of apoptosis-regulating proteins. The increased levels or activity of Bclw might also represent a novel therapeutic target for the prevention of CIPN (Fig. 2). Bclw was shown to play a role in paclitaxel-induced CIPN. Paclitaxel reduced axonal Bclw synthesis, altered intracellular calcium flux and activated calpain proteases. It also selectively impaired axonal trafficking of RNA granules and initiated inositol trisphosphate (IP3)-receptor-dependent axon degeneration [119]. Thus, the modulation of Bclw levels [101], as well as the design of Bclw mimetics [119], might represent another novel therapeutic strategy for CIPN prevention.
Since oxidative stress in the nervous system is one of triggering factors for CIPN, and an impaired mitochondrial antioxidant response after paclitaxel, oxaliplatin and bortezomib is thought to underlie CIPN [120], novel mitochondria-targeting antioxidants have also been evaluated. In mice, SS-31 prevented symptoms of acute neuropathy caused by oxaliplatin [121].
In CIPN, for the protection of mitochondria, the inhibition of nitro-oxidative stress with novel potential drugs might also include the use of the mitochondrial protectant pifithrin-µ, which acts by inhibiting mitochondrial p53 accumulation [122, 123], histone deacetylase 6 inhibitors [124], metformin [44, 74, 125], peroxynitrite decomposition catalysts [126], and antioxidants and anti-inflammatory cytokines, such as interleukin (IL) 10 [125, 127] (Fig. 2).
In addition, the targeting of inflammation and its markers in glial cells and peripheral immune system might contribute to CIPN prevention. Etanercept, a tumor necrosis factor-α (TNFα)-targeting antibody, decreased levels of IL-1α, IL-1β, IL-6, TNFα, interferon γ (INF-γ) and monocyte chemoattractant protein-1 (MCP-1), (Fig. 2) and reduced painful symptoms caused by paclitaxel, showing a potential utility of etanercept as a CIPN-preventing drug [128].
The neuroprotective potential of niclosamide in CIPN induced by oxaliplatin was confirmed in mice, normal neuron-like cells and cancer cells. In neuron-like cells, niclosamide reduced the production of oxaliplatin-mediated hydrogenium peroxide, thereby preventing cell death. In colon cancer cells, niclosamide enhanced oxaliplatin-mediated cell death through increased hydrogenium peroxide production. In neuropathic mice, niclosamide prevented tactile hypoesthesia, thermal hyperalgesia and decreased neuronal hyperexcitability. Niclosamide prevented oxaliplatin-induced increased levels of IL-6 and TNFα (Fig. 2) and reduced oxidative stress. Taken together, niclosamide displayed antitumor and neuroprotective effects without reducing the antitumor efficacy of oxaliplatin [129].
As mentioned above, peripheral immune system cells such as macrophages and monocytes have been shown to be targets of CIPN mechanisms. In this context, another potential strategy to prevent CIPN is the targeting of chemokines and their receptors. For instance, blocking MCP-1 (CCR2) signaling might be an effective therapeutic strategy to prevent or reverse CIPN caused by paclitaxel [130].
Chemokine receptor CXCR1/CXCR2 ligands also show some promising CIPN-preventing properties. Brandolini and colleagues investigated the effect of reparixin, an inhibitor of CXCR1/CXCR2, in the suppression of the development of paclitaxel-induced neuropathic pain in rats. Reparixin attenuated paclitaxel-induced mechanical and cold allodynia in rats. This study suggested that the inhibition of CXCR1/CXCR2 combined with standard taxane therapy, in addition to potentiating the taxane antitumor activity, can reduce paclitaxel-induced neurotoxicity, thus giving some insight for the development of novel treatments [131]. Dual inhibitors of CXCR1/CXCR2 were also tested in the oxaliplatin-induced neuropathic pain model. One of such compounds, DF2726A, reduced pain behavior in oxaliplatin-treated rats. This study confirmed that IL-8 (CXCL8) and its receptors CXCR1/CXCR2 [132] are involved in the development of CIPN caused by oxaliplatin, and blocking the CXCL8 pathway by DF2726A can significantly counteract neurotoxic effects resulting from oxaliplatin administration [133].
Novel therapies based on chemokine CX3CL1 (fractalkine) and its receptor expressed on monocytes, [134] have also emerged as potential options to alleviate CIPN symptoms [135]. It has been shown that gastrodin relieved vincristine-induced CIPN in rats by inhibiting the activation of spinal microglia through CX3CL1 and its receptor, CX3CR1 [136].
Using the oxaliplatin model of CIPN, it has also been demonstrated that the increased expression of C–C chemokine ligand 2 (CCL2) and its receptor, CCR2, in the dorsal root ganglia was lowered after intrathecal administration of anti-CCL2 antibodies, which prevented the development of oxaliplatin-induced mechanical hypersensitivity [137].
In a mouse model of oxaliplatin-induced CIPN, it has also been demonstrated that thrombomodulin α, a recombinant human soluble thrombomodulin, prevented allodynia, most likely via a thrombin-dependent antineuropathic action. The oral anticoagulants warfarin, dabigatran and rivaroxaban suppressed this beneficial antineuropathic effect of thrombomodulin α [138].
Matrix metalloproteinases (MMPs) are also a class of enzymes that have emerged as potential targets for CIPN-preventing drugs. An increase in MMP2 and MMP9 and a decrease in metallopeptidase inhibitor 1 (TIMP1), a strong endogenous MMP9 inhibitor, were observed in a paclitaxel mouse model of CIPN.
Intrathecal injections of exogenous TIMP1 or a monoclonal antibody targeting MMP9 (MMP9 mAb) significantly prevented and reversed paclitaxel-induced mechanical allodynia in mice. This antibody significantly decreased oxidative stress and levels of neuroinflammatory IL-6 and TNFα. MMP9 mAb prevented paclitaxel-induced intraepidermal nerve fiber loss. Hence, it was suggested that MMP9 mAb has therapeutic potential for the treatment of CIPN. Importantly, a humanized MMP9 antibody (andecaliximab) has reached advanced clinical trials for the treatment of colitis (ClinicalTrials.gov: NCT02405442) [139, 140] and alimentary tract cancer (ClinicalTrials.gov: NCT01803282) [141, 142]; hence, it may also be repurposed for CIPN [143].
Our unpublished studies, as well as research results from other laboratories, indicate that minocycline, a tetracycline antibiotic, a microglia inhibitor and a MMP9 blocker, alleviates the development and symptoms of CIPN caused by oxaliplatin, cisplatin, paclitaxel and bortezomib [144,145,146,147,148].
Animal models have also indicated that the targeting of receptors expressed within pain pathways might be an important pharmacological approach for CIPN-preventing drugs. In mice, the blockade of muscarinic receptors with the use of benztropine prevented both acute and chronic symptoms of peripheral neuropathy caused by oxaliplatin. In in vitro studies, benztropine also attenuated electrophysiological alterations due to CIPN and prevented the decrease in neuronal density in the paws of mice that were injected with oxaliplatin. A lower tumor growth was observed in mice receiving benztropine when compared to untreated animals, and benztropine acted synergistically with oxaliplatin by increasing its anticancer effect. This activity was attributed to the influence of benztropine on ROS imbalance in tumor cells [149].
Nicotine is under investigation as another potential therapeutic strategy within the cholinergic system for both the prevention and the treatment of paclitaxel-induced CIPN. Available data suggest that the targeting of the nicotinic-acetylcholine-receptor-mediated pathway may be a promising strategy for the prevention and treatment of CIPN-induced symptoms, in particular mechanical allodynia and intraepidermal nerve fiber loss [150]. A primary concern is that nicotine may also stimulate tumor growth, and smoking history has been classified as a risk factor for CIPN. The research is focused on nicotinic receptor agonists as novel strategies for the prevention and treatment of CIPN [150, 151].
R-47, an α7 nAChR silent agonist, showed promising activity for the prevention and treatment of CIPN caused by paclitaxel. Its activity did not interfere with the antitumor activity of paclitaxel. No tolerance developed following repeated administration of R-47, and R-47 lacked intrinsic rewarding effects. Additionally, R-47 did not influence the rewarding effects of nicotine in the conditioned place preference test in mice, and it did not enhance mecamylamine-precipitated withdrawal [151].
Cebranopadol (a.k.a. GRT-6005) is a first-in-class potent analgesic compound with agonistic activity at nociceptin/orphanin FQ and opioid receptors [152]. It has been recently developed in phase II clinical trials for the treatment of painful diabetic neuropathy or cancer pain. Subcutaneously administered cebranopadol reduced cold allodynia in oxaliplatin-treated mice, and this effect was observed both in the acute and the late phase of CIPN [153].
In rodent models, the administration of carvedilol, a β-adrenolytic drug with cardioprotective properties, reduced levels of nitrotyrosine and subsequently increased the expression of mitochondrial SOD in both sciatic nerves and dorsal root ganglia. Based on this mechanism of action, it was suggested that this drug might have the potential for preventing an alteration in mitochondrial membrane potential in nerves and the loss of intraepidermal nerve fiber density. Several ongoing studies evaluating carvedilol for a variety of clinical conditions, but, at present, there is no clinical study for the prevention of CIPN [4].
A series of novel amide derivatives of 1,3-dimethyl-2,6-dioxopurin-7-yl-alkylcarboxylic acids, analogs of a TRPA1 antagonist, i.e., HC-030031, were assessed in vitro and in vivo. These compounds were identified as dual TRPA1 channel antagonists and phosphodiesterase 4B/7A inhibitors. They reduced tactile allodynia in oxaliplatin-treated mice and showed TNFα-lowering properties; therefore, they are regarded as interesting drug candidates for CIPN [154].
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are overexpressed in various inflammatory and neuropathic pain states, and a novel HCN1 inhibitor, MEL57A, has recently demonstrated antiallodynic and antihyperalgesic properties in oxaliplatin-treated rats. HCN1 channels were proposed as a new drug target in CIPN, and HCN1-selective inhibitors might be a new class of medications for CIPN [155].
Many mechanisms underlying CIPN overlap and can reinforce each other. Thus, a combination drug therapy might also be an interesting and more efficacious (compared to monotherapy) approach to CIPN prevention [120]. Although there are no clear algorithms for the clinical use of combined drugs in CIPN patients, data from preclinical research indicate that certain drug combinations might effectively attenuate symptoms of CIPN [156]. In our recent study, a hyperadditive effect of combined subanalgesic doses of ambroxol and pregabalin on cold allodynia was demonstrated in oxaliplatin-treated mice. This effect was particularly strong when both drugs were given simultaneously or sequentially, i.e., 4 h apart. These results point out a synergistic reaction between Nav and Cav channel inhibitors in CIPN caused by oxaliplatin [156].
Mesenchymal stem cell (MSC) therapy is also an emerging attractive approach for CIPN. Evidence has demonstrated that MSCs reduce oxidative stress, neuroinflammation, and neuronal cell apoptosis and stimulate axon regeneration after nerve damage. Hence, MSC-based therapies may provide a new therapeutic strategy for patients suffering from CIPN. Combined therapy using ‘traditional’ pharmacological agents and MSCs might also be an interesting approach for CIPN prevention and treatment [157].
Several studies have also been conducted to assess whether targeting pathways associated with gut microbiota might be a way to overcome CIPN [158]. Probiotics show therapeutic effects in various diseases, such as travelers’ diarrhea or irritable bowel syndrome, and previous studies have also confirmed their protective role in various nociceptive pain states [159,160,161], but, at present, little is known about their potential effect on CIPN.
Using F11 cells, the probiotic DSF was tested for its ability to attenuate paclitaxel-induced neuropathic pain. The results obtained suggested its potential use as an adjuvant agent for CIPN [162]. The following mechanisms explain a beneficial effect of probiotics through which they might inhibit CIPN: a reduction in proinflammatory cytokine levels, the regulation of the balance between anti-inflammatory and proinflammatory cytokines, the inhibition of neutrophil infiltration and increased intestinal permeability, augmented production of short-chain fatty acids, the inhibition of ammonia formation, changes in the expression levels of toll-like receptors: TLR2 and TLR4, the maintenance of mucosal integrity through mucin secretion and the activation of suppressive T-cell populations [158]. Here, it should be strongly emphasized that the research on the gut microbiome in CIPN is still at an exploratory stage, and potential therapeutic methods based on microbiota manipulation require further preclinical and clinical research. In particular, clinical trials assessing the use of probiotics to prevent or treat CIPN are a strong medical demand [158].
The efficacy of complementary therapies in the prevention and treatment of CIPN has also been a subject of research. Herbal medicinal therapies (e.g., Acorus calamus, Cannabis species, Matricaria chamomilla, Ginkgo biloba, Curcuma longa, Camellia sinensis) counteract phenomena underlying CIPN, i.e., they reduce oxidative stress and attenuate inflammation in animals [163], but their utility for CIPN prevention requires further investigation to confirm their efficacy and safety [164,165,166,167].
In addition to novel pharmacological targets for the prevention and/or treatment of CIPN, nonpharmacological approaches were also assessed. Interestingly, a growing body of literature suggests that exercise can prevent CIPN. In rodent models of CIPN, volitional wheel running reduced both the development and the maintenance of cold and mechanical allodynia. This finding was confirmed in patients assigned to the exercise group during chemotherapy who reported less severe thermal and sensory symptoms of CIPN than patients who received chemotherapy alone [94].
Preclinical data regarding potential future preventative therapies for CIPN are summarized in Table 3.
Conclusions
Although preclinical models reflecting CIPN have provided a deeper insight into mechanisms that initiate and maintain antitumor drug-induced neurotoxicity, this clinical entity currently cannot be prevented. New therapeutic approaches and drug targets should be assessed, and improved clinical trials need to be designed to search for drugs for CIPN prevention. This research should address genotypic profiles of patients suffering from CIPN, and the knowledge regarding genetic CIPN susceptibility needs to be incorporated into clinical trials. This research not only would contribute to recent advances in basic and translational research for CIPN prevention [168] but also would enable the prediction of patients’ susceptibility to CIPN development prior to starting chemotherapy based on CIPN-inducing drugs. In addition, pharmacogenomics studies could be useful in the identification of mutations in genes influencing antitumor drug metabolism, and this could also be an important source of data for optimal dosing of these drugs in patients [101].
References
Riddell IA, Lippard SJ. 1. Cisplatin and oxaliplatin: our current understanding of their actions. In: Sigel A, Sigel H, Freisinger E, Sigel RKO, editors. Met. Dev. Action Anticancer Agents. Berlin: De Gruyter; 2018. p. 1–42. https://doi.org/10.1515/9783110470734-001.
WHO | WHO Model Lists of Essential Medicines. WHO; 2019.
Marques RP, Duarte GS, Sterrantino C, Pais HL, Quintela A, Martins AP, et al. Triplet (FOLFOXIRI) versus doublet (FOLFOX or FOLFIRI) backbone chemotherapy as first-line treatment of metastatic colorectal cancer: A systematic review and meta-analysis. Crit Rev Oncol Hematol. 2017;118:54–62. https://doi.org/10.1016/j.critrevonc.2017.08.006.
Hu S, Huang KM, Adams EJ, Loprinzi CL, Lustberg MB. Recent developments of novel pharmacologic therapeutics for prevention of chemotherapy-induced peripheral neuropathy. Clin Cancer Res. 2019;25:6295–301. https://doi.org/10.1158/1078-0432.CCR-18-2152.
Leblanc AF, Sprowl JA, Alberti P, Chiorazzi A, Arnold WD, Gibson AA, et al. OATP1B2 deficiency protects against paclitaxel-induced neurotoxicity. J Clin Invest. 2018;128:816–25. https://doi.org/10.1172/JCI96160.
Nishida K, Takeuchi K, Hosoda A, Sugano S, Morisaki E, Ohishi A, et al. Ergothioneine ameliorates oxaliplatin-induced peripheral neuropathy in rats. Life Sci. 2018;207:516–24. https://doi.org/10.1016/j.lfs.2018.07.006.
Fujita S, Hirota T, Sakiyama R, Baba M, Ieiri I. Identification of drug transporters contributing to oxaliplatin-induced peripheral neuropathy. J Neurochem. 2019;148:373–85. https://doi.org/10.1111/jnc.14607.
Jong NN, Nakanishi T, Liu JJ, Tamai I, McKeage MJ. Oxaliplatin transport mediated by organic cation/carnitine transporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 2011;338:537–47. https://doi.org/10.1124/jpet.111.181297.
Sun S, Cai J, Yang Q, Zhao S, Wang Z. The association between copper transporters and the prognosis of cancer patients undergoing chemotherapy: a meta-analysis of literatures and datasets. Oncotarget. 2017. https://doi.org/10.18632/oncotarget.13917.
Sakurai M, Egashira N, Kawashiri T, Yano T, Ikesue H, Oishi R. Oxaliplatin-induced neuropathy in the rat: involvement of oxalate in cold hyperalgesia but not mechanical allodynia. Pain. 2009;147:165–74. https://doi.org/10.1016/j.pain.2009.09.003.
Pereira AF, de Oliveira FFB, de Freitas Alves BW, de Menezes KLS, de Mesquita AKV, Lisboa MRP, et al. Neurotoxic effect of oxaliplatin: comparison with its oxalate-free analogue cis-[PtII(1R,2R-DACH)(3-acetoxy-1,1-cyclobutanedicarboxylato)](LLC-1402) in mice. Toxicol Appl Pharmacol. 2018;340:77–84. https://doi.org/10.1016/j.taap.2018.01.001.
Starobova H, Vetter I. Pathophysiology of chemotherapy-induced peripheral neuropathy. Front Mol Neurosci. 2017. https://doi.org/10.3389/fnmol.2017.00174.
Embi A, Scherlag BJ, Embi PJ, Menes M, Po SS. Targeted cellular ionic calcium chelation by oxalates: implications for the treatment of tumor cells. Cancer Cell Int. 2012;12:51. https://doi.org/10.1186/1475-2867-12-51.
Smith EML, Pang H, Ye C, Cirrincione C, Fleishman S, Paskett ED, et al. Predictors of duloxetine response in patients with oxaliplatin-induced painful chemotherapy-induced peripheral neuropathy (CIPN): a secondary analysis of randomised controlled trial—CALGB/alliance 170601. Eur J Cancer Care (Engl). 2017;26:e12421. https://doi.org/10.1111/ecc.12421.
Deuis JR, Zimmermann K, Romanovsky AA, Possani LD, Cabot PJ, Lewis RJ, et al. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Nav1.6 in peripheral pain pathways. Pain. 2013;154:1749–57. https://doi.org/10.1016/j.pain.2013.05.032.
Loprinzi CL, Qin R, Dakhil SR, Fehrenbacher L, Flynn KA, Atherton P, et al. Phase III randomized, placebo-controlled, double-blind study of intravenous calcium and magnesium to prevent oxaliplatin-induced sensory neurotoxicity (N08CB/Alliance). J Clin Oncol. 2014;32:997–1005. https://doi.org/10.1200/JCO.2013.52.0536.
Piccolo J, Kolesar JM. Prevention and treatment of chemotherapy-induced peripheral neuropathy. Am J Health Pharm. 2014;71:19–25. https://doi.org/10.2146/ajhp130126.
Marmiroli P, Riva B, Pozzi E, Ballarini E, Lim D, Chiorazzi A, et al. Susceptibility of different mouse strains to oxaliplatin peripheral neurotoxicity: phenotypic and genotypic insights. PLoS ONE. 2017;12:e0186250. https://doi.org/10.1371/journal.pone.0186250.
Cliff J, Jorgensen AL, Lord R, Azam F, Cossar L, Carr DF, et al. The molecular genetics of chemotherapy–induced peripheral neuropathy: a systematic review and meta-analysis. Crit Rev Oncol Hematol. 2017;120:127–40. https://doi.org/10.1016/j.critrevonc.2017.09.009.
Lee K-H, Chang HJ, Han S-W, Oh D-Y, Im S-A, Bang Y-J, et al. Pharmacogenetic analysis of adjuvant FOLFOX for Korean patients with colon cancer. Cancer Chemother Pharmacol. 2013;71:843–51. https://doi.org/10.1007/s00280-013-2075-3.
Johnson C, Pankratz VS, Velazquez AI, Aakre JA, Loprinzi CL, Staff NP, et al. Candidate pathway-based genetic association study of platinum and platinum–taxane related toxicity in a cohort of primary lung cancer patients. J Neurol Sci. 2015;349:124–8. https://doi.org/10.1016/j.jns.2014.12.041.
Qian C-Y, Zheng Y, Wang Y, Chen J, Liu J-Y, Zhou H-H, et al. Associations of genetic polymorphisms of the transporters organic cation transporter 2 (OCT2), multidrug and toxin extrusion 1 (MATE1), and ATP-binding cassette subfamily C member 2 (ABCC2) with platinum-based chemotherapy response and toxicity in non-sma. Chin J Cancer. 2016;35:85. https://doi.org/10.1186/s40880-016-0145-8.
Lecomte T. Glutathione S-transferase P1 polymorphism (Ile105Val) predicts cumulative neuropathy in patients receiving oxaliplatin-based chemotherapy. Clin Cancer Res. 2006;12:3050–6. https://doi.org/10.1158/1078-0432.CCR-05-2076.
Sprowl JA, Ciarimboli G, Lancaster CS, Giovinazzo H, Gibson AA, Du G, et al. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc Natl Acad Sci. 2013;110:11199–204. https://doi.org/10.1073/pnas.1305321110.
Mori Y, Katsumata K, Tsuchida A, Aoki T. Single nucleotide polymorphism analysis in the GSTP1 and ABCC2 genes about neuropathy by the oxaliplatin. Gan To Kagaku Ryoho. 2008;35:2377–81.
Gamelin L, Capitain O, Morel A, Dumont A, Traore S, Anne LB, et al. Predictive factors of oxaliplatin neurotoxicity: the involvement of the oxalate outcome pathway. Clin Cancer Res. 2007;13:6359–68. https://doi.org/10.1158/1078-0432.CCR-07-0660.
Argyriou AA, Cavaletti G, Antonacopoulou A, Genazzani AA, Briani C, Bruna J, et al. Voltage-gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin-induced peripheral neurotoxicity: Results from a prospective multicenter study. Cancer. 2013;119:3570–7. https://doi.org/10.1002/cncr.28234.
Sereno M, Gutiérrez-Gutiérrez G, Rubio JM, Apellániz-Ruiz M, Sánchez-Barroso L, Casado E, et al. Genetic polymorphisms of SCN9A are associated with oxaliplatin-induced neuropathy. BMC Cancer. 2017;17:63. https://doi.org/10.1186/s12885-016-3031-5.
Ewertz M, Qvortrup C, Eckhoff L. Chemotherapy-induced peripheral neuropathy in patients treated with taxanes and platinum derivatives. Acta Oncol (Madr). 2015;54:587–91. https://doi.org/10.3109/0284186X.2014.995775.
Lehky TJ, Leonard GD, Wilson RH, Grem JL, Floeter MK. Oxaliplatin-induced neurotoxicity: acute hyperexcitability and chronic neuropathy. Muscle Nerve. 2004;29:387–92. https://doi.org/10.1002/mus.10559.
Alberti P, Canta A, Chiorazzi A, Fumagalli G, Meregalli C, Monza L, et al. Topiramate prevents oxaliplatin-related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology. 2020;164:107905. https://doi.org/10.1016/J.NEUROPHARM.2019.107905.
Molassiotis A, Cheng HL, Lopez V, Au JSK, Chan A, Bandla A, et al. Are we mis-estimating chemotherapy-induced peripheral neuropathy? Analysis of assessment methodologies from a prospective, multinational, longitudinal cohort study of patients receiving neurotoxic chemotherapy. BMC Cancer. 2019;19:132. https://doi.org/10.1186/s12885-019-5302-4.
Kandula T, Farrar MA, Kiernan MC, Krishnan AV, Goldstein D, Horvath L, et al. Neurophysiological and clinical outcomes in chemotherapy-induced neuropathy in cancer. Clin Neurophysiol. 2017;128:1166–75. https://doi.org/10.1016/j.clinph.2017.04.009.
Miaskowski C, Mastick J, Paul SM, Topp K, Smoot B, Abrams G, et al. Chemotherapy-induced neuropathy in cancer survivors. J Pain Symptom Manag. 2017;54(204–218):e2. https://doi.org/10.1016/j.jpainsymman.2016.12.342.
Griffith KA, Zhu S, Johantgen M, Kessler MD, Renn C, Beutler AS, et al. Oxaliplatin-induced peripheral neuropathy and identification of unique severity groups in colorectal cancer. J Pain Symptom Manag. 2017;54(701–706):e1. https://doi.org/10.1016/j.jpainsymman.2017.07.033.
Kanat O, Ertas H, Caner B. Platinum-induced neurotoxicity: a review of possible mechanisms. World J Clin Oncol. 2017;8:329. https://doi.org/10.5306/wjco.v8.i4.329.
Balayssac D, Ferrier J, Pereira B, Gillet B, Petorin C, Vein J, et al. Prevention of oxaliplatin-induced peripheral neuropathy by a polyamine-reduced diet–NEUROXAPOL: protocol of a prospective, randomised, controlled, single-blind and monocentric trial. BMJ Open. 2015;5:e007479–e00747907479. https://doi.org/10.1136/bmjopen-2014-007479.
Ventzel L, Jensen AB, Jensen AR, Jensen TS, Finnerup NB. Chemotherapy-induced pain and neuropathy. Pain. 2016;157:560–8. https://doi.org/10.1097/j.pain.0000000000000404.
Kokotis P, Schmelz M, Kostouros E, Karandreas N, Dimopoulos M-A. Oxaliplatin-induced neuropathy: a long-term clinical and neurophysiologic follow-up study. Clin Colorectal Cancer. 2016;15:e133–e140140. https://doi.org/10.1016/j.clcc.2016.02.009.
Pachman DR, Qin R, Seisler DK, Smith EML, Beutler AS, Ta LE, et al. Clinical course of oxaliplatin-induced neuropathy: results from the randomized phase III Trial N08CB (Alliance). J Clin Oncol. 2015;33:3416–22. https://doi.org/10.1200/JCO.2014.58.8533.
Poupon L, Kerckhove N, Vein J, Lamoine S, Authier N, Busserolles J, et al. Minimizing chemotherapy-induced peripheral neuropathy: preclinical and clinical development of new perspectives. Expert Opin Drug Saf. 2015;14:1269–82. https://doi.org/10.1517/14740338.2015.1056777.
Beijers AJM, Bonhof CS, Mols F, Ophorst J, de Vos-Geelen J, Jacobs EMG, et al. Multicenter randomized controlled trial to evaluate the efficacy and tolerability of frozen gloves for the prevention of chemotherapy-induced peripheral neuropathy. Ann Oncol. 2020;31:131–6. https://doi.org/10.1016/J.ANNONC.2019.09.006.
Eldridge S, Guo L, Hamre J. A comparative review of chemotherapy-induced peripheral neuropathy in in vivo and in vitro models. Toxicol Pathol. 2020;48:190–201. https://doi.org/10.1177/0192623319861937.
Colvin LA. Chemotherapy-induced peripheral neuropathy. Pain. 2019;160:S1–. https://doi.org/10.1097/j.pain.0000000000001540.
ASCO Releases Guideline on Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers—The ASCO Post n.d. https://www.ascopost.com/News/16118. Accessed 4 Mar 2020
Kottschade LA, Sloan JA, Mazurczak MA, Johnson DB, Murphy BP, Rowland KM, et al. The use of vitamin E for the prevention of chemotherapy-induced peripheral neuropathy: results of a randomized phase III clinical trial. Support Care Cancer. 2011;19:1769–77. https://doi.org/10.1007/s00520-010-1018-3.
de Afonseca SO, Cruz FM, Cubero DIG, Lera AT, Schindler F, Okawara M, et al. Vitamin E for prevention of oxaliplatin-induced peripheral neuropathy: a pilot randomized clinical trial. Sao Paulo Med J. 2013;131:35–8. https://doi.org/10.1590/S1516-31802013000100006.
Majithia N, Loprinzi CL, Smith TJ. New practical approaches to chemotherapy-induced neuropathic pain: prevention, assessment, and treatment. Oncology (Williston Park). 2016;30:1020–9.
Meng J, Zhang Q, Yang C, Xiao L, Xue Z, Zhu J. Duloxetine, a balanced serotonin-norepinephrine reuptake inhibitor, improves painful chemotherapy-induced peripheral neuropathy by inhibiting activation of p38 MAPK and NF-κB. Front Pharmacol. 2019. https://doi.org/10.3389/fphar.2019.00365.
Hu L-Y, Mi W-L, Wu G-C, Wang Y-Q, Mao-Ying Q-L. Prevention and treatment for chemotherapy-induced peripheral neuropathy: therapies based on CIPN mechanisms. Curr Neuropharmacol. 2019;17:184–96. https://doi.org/10.2174/1570159X15666170915143217.
Zimmerman C, Atherton PJ, Pachman D, Seisler D, Wagner-Johnston N, Dakhil S, et al. MC11C4: a pilot randomized, placebo-controlled, double-blind study of venlafaxine to prevent oxaliplatin-induced neuropathy. Support Care Cancer. 2016;24:1071–8. https://doi.org/10.1007/s00520-015-2876-5.
de Andrade DC, Jacobsen Teixeira M, Galhardoni R, Ferreira KSL, Braz Mileno P, Scisci N, et al. Pregabalin for the prevention of oxaliplatin-induced painful neuropathy: a randomized double-blind trial. Oncologist. 2017;22:1154–e10505. https://doi.org/10.1634/theoncologist.2017-0235.
Oki E, Emi Y, Kojima H, Higashijima J, Kato T, Miyake Y, et al. Preventive effect of Goshajinkigan on peripheral neurotoxicity of FOLFOX therapy (GENIUS trial): a placebo-controlled, double-blind, randomized phase III study. Int J Clin Oncol. 2015;20:767–75. https://doi.org/10.1007/s10147-015-0784-9.
Kuriyama A, Endo K. Goshajinkigan for prevention of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Support Care Cancer. 2018;26:1051–9. https://doi.org/10.1007/s00520-017-4028-6.
Schloss JM, Colosimo M, Airey C, Masci P, Linnane AW, Vitetta L. A randomised, placebo-controlled trial assessing the efficacy of an oral B group vitamin in preventing the development of chemotherapy-induced peripheral neuropathy (CIPN). Support Care Cancer. 2017;25:195–204. https://doi.org/10.1007/s00520-016-3404-y.
Guo Y, Jones D, Palmer JL, Forman A, Dakhil SR, Velasco MR, et al. Oral alpha-lipoic acid to prevent chemotherapy-induced peripheral neuropathy: a randomized, double-blind, placebo-controlled trial. Support Care Cancer. 2014;22:1223–31. https://doi.org/10.1007/s00520-013-2075-1.
Hirayama Y, Ishitani K, Sato Y, Iyama S, Takada K, Murase K, et al. Effect of duloxetine in Japanese patients with chemotherapy-induced peripheral neuropathy: a pilot randomized trial. Int J Clin Oncol. 2015;20:866–71. https://doi.org/10.1007/s10147-015-0810-y.
Sałat K, Furgała A, Malikowska-Racia N. Searching for analgesic drug candidates alleviating oxaliplatin-induced cold hypersensitivity in mice. Chem Biol Drug Des. 2019. https://doi.org/10.1111/cbdd.13507.
Toyama S, Shimoyama N, Shimoyama M. The analgesic effect of orexin-A in a murine model of chemotherapy-induced neuropathic pain. Neuropeptides. 2017;61:95–100. https://doi.org/10.1016/j.npep.2016.12.007.
Smith EML, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy. JAMA. 2013;309:1359. https://doi.org/10.1001/jama.2013.2813.
Farshchian N, Alavi A, Heydarheydari S, Moradian N. Comparative study of the effects of venlafaxine and duloxetine on chemotherapy-induced peripheral neuropathy. Cancer Chemother Pharmacol. 2018;82:787–93. https://doi.org/10.1007/s00280-018-3664-y.
Home—ClinicalTrials.gov n.d. https://www.clinicaltrials.gov/. Accessed 22 Jan 2020
Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10:531–9. https://doi.org/10.1158/1535-7163.MCT-10-0731.
Glimelius B, Manojlovic N, Pfeiffer P, Mosidze B, Kurteva G, Karlberg M, et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx®): a placebo-controlled randomised phase II study (PLIANT). Acta Oncol (Madr). 2018;57:393–402. https://doi.org/10.1080/0284186X.2017.1398836.
Doolen S, Iannitti T, Donahue RR, Shaw BC, Grachen CM, Taylor BK. Fingolimod reduces neuropathic pain behaviors in a mouse model of multiple sclerosis by a sphingosine-1 phosphate receptor 1-dependent inhibition of central sensitization in the dorsal horn. Pain. 2018;159:224–38. https://doi.org/10.1097/j.pain.0000000000001106.
Janes K, Little JW, Li C, Bryant L, Chen C, Chen Z, et al. The development and maintenance of paclitaxel-induced neuropathic pain require activation of the sphingosine 1-phosphate receptor subtype 1. J Biol Chem. 2014;289:21082–97. https://doi.org/10.1074/jbc.M114.569574.
Wang D, Wang Z, Chen G, Peng J, Wang W, Deng Y, et al. Phase III randomized, placebo-controlled, double-blind study of monosialotetrahexosylganglioside for the prevention of oxaliplatin-induced peripheral neurotoxicity in stage II/III colorectal cancer. Cancer Med. 2020;9:151–9. https://doi.org/10.1002/cam4.2693.
Yehia R, Saleh S, El Abhar H, Saad AS, Schaalan M. l-Carnosine protects against Oxaliplatin-induced peripheral neuropathy in colorectal cancer patients: a perspective on targeting Nrf-2 and NF-κB pathways. Toxicol Appl Pharmacol. 2019;365:41–50. https://doi.org/10.1016/j.taap.2018.12.015.
Banerjee S, Sinha K, Chowdhury S, Sil PC. Unfolding the mechanism of cisplatin induced pathophysiology in spleen and its amelioration by carnosine. Chem Biol Interact. 2018;279:159–70. https://doi.org/10.1016/j.cbi.2017.11.019.
Kerckhove N, Busserolles J, Stanbury T, Pereira B, Plence V, Bonnetain F, et al. Effectiveness assessment of riluzole in the prevention of oxaliplatin-induced peripheral neuropathy: RILUZOX-01: protocol of a randomised, parallel, controlled, double-blind and multicentre study by the UNICANCER-AFSOS Supportive Care intergroup. BMJ Open. 2019;9:e027770. https://doi.org/10.1136/bmjopen-2018-027770.
Poupon L, Lamoine S, Pereira V, Barriere DA, Lolignier S, Giraudet F, et al. Targeting the TREK-1 potassium channel via riluzole to eliminate the neuropathic and depressive-like effects of oxaliplatin. Neuropharmacology. 2018;140:43–61. https://doi.org/10.1016/j.neuropharm.2018.07.026.
Lidocaine for Oxaliplatin-induced Neuropathy—Full Text View—ClinicalTrials.gov n.d. https://clinicaltrials.gov/ct2/show/NCT03254394?term=oxaliplatin+neuropathy&draw=2. Accessed 22 Jan 2020.
Metformin for Reduction of Paclitaxel-Related Neuropathy in Patients with Breast Cancer—Full Text View—ClinicalTrials.gov n.d. https://clinicaltrials.gov/ct2/show/NCT02360059?term=metformin+and+neuropathy&draw=2. Accessed 22 Jan 2020.
Inyang KE, McDougal TA, Ramirez ED, Williams M, Laumet G, Kavelaars A, et al. Alleviation of paclitaxel-induced mechanical hypersensitivity and hyperalgesic priming with AMPK activators in male and female mice. Neurobiol Pain. 2019;6:100037. https://doi.org/10.1016/j.ynpai.2019.100037.
EudraCT Number 2012-000398-21—Clinical trial results—EU Clinical Trials Register n.d. https://www.clinicaltrialsregister.eu/ctr-search/trial/2012-000398-21/results. Accessed 1 Apr 2020.
Bruna J, Videla S, Argyriou AA, Velasco R, Villoria J, Santos C, et al. Efficacy of a novel sigma-1 receptor antagonist for oxaliplatin-induced neuropathy: a randomized, double-blind placebo-controlled phase IIa clinical trial. Neurotherapeutics. 2018;15:178–89. https://doi.org/10.1007/s13311-017-0572-5.
Bruna J, Velasco R. Sigma-1 receptor: a new player in neuroprotection against chemotherapy-induced peripheral neuropathy. Neural Regen Res. 2018;13:775. https://doi.org/10.4103/1673-5374.232459.
EudraCT Number 2011-004033-13—Clinical trial results—EU Clinical Trials Register n.d. https://www.clinicaltrialsregister.eu/ctr-search/trial/2011-004033-13/results. Accessed 1 Apr 2020.
Keppel Hesselink J, Schatman ME. EMA401: an old antagonist of the AT2R for a new indication in neuropathic pain. J Pain Res. 2017;10:439–43. https://doi.org/10.2147/JPR.S128520.
Smith MT, Anand P, Rice ASC. Selective small molecule angiotensin II type 2 receptor antagonists for neuropathic pain. Pain. 2016;157:S33–41. https://doi.org/10.1097/j.pain.0000000000000369.
Sałat K, Kowalczyk P, Gryzło B, Jakubowska A, Kulig K. New investigational drugs for the treatment of neuropathic pain. Expert Opin Investig Drugs. 2014;23:1093–104. https://doi.org/10.1517/13543784.2014.916688.
Smith MT, Muralidharan A. Targeting angiotensin II type 2 receptor pathways to treat neuropathic pain and inflammatory pain. Expert Opin Ther Targets. 2015;19:25–35. https://doi.org/10.1517/14728222.2014.957673.
Guillaumot M-A, Cerles O, Bertrand HC, Benoit E, Nicco C, Chouzenoux S, et al. Oxaliplatin-induced neuropathy: the preventive effect of a new super-oxide dismutase modulator. Oncotarget. 2019;10:6418–31. https://doi.org/10.18632/oncotarget.27248.
Qin Y, Iwase A, Murase T, Ishida C, Kato N, et al. Protective effects of mangafodipir against chemotherapy-induced ovarian damage in mice. Reprod Biol Endocrinol. 2018;16:106. https://doi.org/10.1186/s12958-018-0426-y.
Karlsson J-E, El-Saadi W, Ali M, Puskar W, Skogvard P, Engvall JE, et al. Mangafodipir as a cardioprotective adjunct to reperfusion therapy: a feasibility study in patients with ST-segment elevation myocardial infarction. Eur Hear J Cardiovasc Pharmacother. 2015;1:39–45. https://doi.org/10.1093/ehjcvp/pvu021.
Batteux F, Cerles O, Nicco C. Cancer, formes réactives de l’oxygène et neuropathies périphériques chimio-induites. Médecine/Sciences. 2014;30:1078–80. https://doi.org/10.1051/medsci/20143012008.
Karlsson JOG, Andersson RG, Jynge P. Mangafodipir a selective cytoprotectant—with special reference to oxaliplatin and its association to chemotherapy-induced peripheral neuropathy (CIPN). Transl Oncol. 2017;10:641–9. https://doi.org/10.1016/j.tranon.2017.04.012.
Coriat R, Alexandre J, Nicco C, Quinquis L, Benoit E, Chéreau C, et al. Treatment of oxaliplatin-induced peripheral neuropathy by intravenous mangafodipir. J Clin Invest. 2014;124:262–72. https://doi.org/10.1172/JCI68730.
Jaeschke H, Akakpo JY, Umbaugh DS, Ramachandran A. Novel Therapeutic approaches against acetaminophen-induced liver injury and acute liver failure. Toxicol Sci. 2020. https://doi.org/10.1093/toxsci/kfaa002.
Morrison EE, Oatey K, Gallagher B, Grahamslaw J, O’Brien R, Black P, et al. Principal results of a randomised open label exploratory, safety and tolerability study with calmangafodipir in patients treated with a 12 h regimen of N-acetylcysteine for paracetamol overdose (POP trial). EBioMedicine. 2019;46:423–30. https://doi.org/10.1016/j.ebiom.2019.07.013.
Dear JW. Gastrointestinal AEs seen in the POP trial due to SOD mimetic activity of calmangafodipir?—authors’ reply. EBioMedicine. 2019;47:28. https://doi.org/10.1016/j.ebiom.2019.08.039.
Dear J. Randomised open label exploratory, safety and tolerability study with calmangafodipir in patients treated with the 12-h regimen of N-acetylcysteine for paracetamol overdose—the PP100–01 for overdose of paracetamol (POP) trial: study protocol for a randomi. Trials. 2019;20:27. https://doi.org/10.1186/s13063-018-3134-1.
Kelley MR, Wikel JH, Guo C, Pollok KE, Bailey BJ, Wireman R, et al. Identification and characterization of new chemical entities targeting apurinic/apyrimidinic endonuclease 1 for the prevention of chemotherapy-induced peripheral neuropathy. J Pharmacol Exp Ther. 2016;359:300–9. https://doi.org/10.1124/jpet.116.235283.
A study of APX3330 in patients with advanced solid tumors—Full Text View—ClinicalTrials.gov n.d. https://clinicaltrials.gov/ct2/show/NCT03375086?term=apx3330&draw=2&rank=1. Accessed 23 Jan 2020.
Wang W, Xiang P, Chew WS, Torta F, Bandla A, Lopez V, et al. Activation of sphingosine 1-phosphate receptor 2 attenuates chemotherapy-induced neuropathy. J Biol Chem. 2019. https://doi.org/10.1074/jbc.RA119.011699.
Stockstill K, Doyle TM, Yan X, Chen Z, Janes K, Little JW, et al. Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain. J Exp Med. 2018;215:1301–13. https://doi.org/10.1084/jem.20170584.
Huwiler A, Zangemeister-Wittke U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: recent findings and new perspectives. Pharmacol Ther. 2018;185:34–49. https://doi.org/10.1016/j.pharmthera.2017.11.001.
Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391:1263–73. https://doi.org/10.1016/S0140-6736(18)30475-6.
Motyl J, Strosznajder JB. Sphingosine kinase 1/sphingosine-1-phosphate receptors dependent signalling in neurodegenerative diseases. The promising target for neuroprotection in Parkinson’s disease. Pharmacol Rep. 2018;70:1010–4. https://doi.org/10.1016/j.pharep.2018.05.002.
Zheng Z, Zeng Y-Z, Ren K, Zhu X, Tan Y, Li Y, et al. S1P promotes inflammation-induced tube formation by HLECs via the S1PR1/NF-κB pathway. Int Immunopharmacol. 2019;66:224–35. https://doi.org/10.1016/j.intimp.2018.11.032.
Dorsey SG, Kleckner IR, Barton D, Mustian K, O’Mara A, St. Germain D, et al. The National Cancer Institute Clinical Trials Planning Meeting for Prevention and Treatment of Chemotherapy-Induced Peripheral Neuropathy. J Natl Cancer Inst. 2019;111:531–7. https://doi.org/10.1093/jnci/djz011.
Zhang L, Wang H. FTY720 inhibits the Nrf2/ARE pathway in human glioblastoma cell lines and sensitizes glioblastoma cells to temozolomide. Pharmacol Reports. 2017;69:1186–93. https://doi.org/10.1016/j.pharep.2017.07.003.
Choucry AM, Al-Shorbagy MY, Attia AS, El-Abhar HS. Pharmacological manipulation of Trk, p75NTR, and NGF balance restores memory deficit in global ischemia/reperfusion model in rats. J Mol Neurosci. 2019;68:78–90. https://doi.org/10.1007/s12031-019-01284-1.
Zakharova IO, Sokolova TV, Vlasova YA, Furaev VV, Rychkova MP, Avrova NF. GM1 ganglioside activates ERK1/2 and Akt downstream of Trk tyrosine kinase and protects PC12 cells against hydrogen peroxide toxicity. Neurochem Res. 2014;39:2262–75. https://doi.org/10.1007/s11064-014-1428-6.
Gong G, Yin L, Yuan L, Sui D, Sun Y, Fu H, et al. Ganglioside GM1 protects against high altitude cerebral edema in rats by suppressing the oxidative stress and inflammatory response via the PI3K/AKT-Nrf2 pathway. Mol Immunol. 2018;95:91–8. https://doi.org/10.1016/j.molimm.2018.02.001.
Li Y, North RY, Rhines LD, Tatsui CE, Rao G, Edwards DD, et al. DRG voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J Neurosci. 2018;38:1124–36. https://doi.org/10.1523/JNEUROSCI.0899-17.2017.
Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology. 2014;120:1463–75. https://doi.org/10.1097/ALN.0000000000000176.
Olodanrigan—Novartis—AdisInsight n.d. https://adisinsight.springer.com/drugs/800022957. Accessed 1 Apr 2020.
Disappointment for Australian innovation—Biotech n.d. https://biotechdispatch.com.au/news/disappointment-for-australian-innovation. Accessed 1 Apr 2020.
Sommer C. EAN/PNS: Novel approach in the treatment of neuropathy (Level3) Novel advances in the treatment of pain in neuropathies. n.d.
Search of: ema401—List Results—ClinicalTrials.gov n.d. https://clinicaltrials.gov/ct2/results?cond=&term=ema401&cntry=&state=&city=&dist=. Accessed 23 Jan 2020.
Sisignano M, Baron R, Scholich K, Geisslinger G. Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat Rev Neurol. 2014;10:694–707. https://doi.org/10.1038/nrneurol.2014.211.
Sisignano M, Parnham MJ, Geisslinger G. Drug repurposing for the development of novel analgesics. Trends Pharmacol Sci. 2016;37:172–83. https://doi.org/10.1016/j.tips.2015.11.006.
DiAntonio A. Axon degeneration. Pain. 2019;160:S17–22. https://doi.org/10.1097/j.pain.0000000000001528.
Huppke P, Wegener E, Gilley J, Angeletti C, Kurth I, Drenth JPH, et al. Homozygous NMNAT2 mutation in sisters with polyneuropathy and erythromelalgia. Exp Neurol. 2019;320:112958. https://doi.org/10.1016/j.expneurol.2019.112958.
Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, et al. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.129920.
Zhu J, Chen W, Mi R, Zhou C, Reed N, Höke A. Ethoxyquin prevents chemotherapy-induced neurotoxicity via Hsp90 modulation. Ann Neurol. 2013;74:893–904. https://doi.org/10.1002/ana.24004.
Zhu J, Carozzi VA, Reed N, Mi R, Marmiroli P, Cavaletti G, et al. Ethoxyquin provides neuroprotection against cisplatin-induced neurotoxicity. Sci Rep. 2016;6:28861. https://doi.org/10.1038/srep28861.
Pease-Raissi SE, Pazyra-Murphy MF, Li Y, Wachter F, Fukuda Y, Fenstermacher SJ, et al. Paclitaxel reduces axonal Bclw to initiate IP3R1-dependent axon degeneration. Neuron. 2017;96(373–386):e6. https://doi.org/10.1016/j.neuron.2017.09.034.
Flatters SJL, Dougherty PM, Colvin LA. Clinical and preclinical perspectives on chemotherapy-induced peripheral neuropathy (CIPN): a narrative review. Br J Anaesth. 2017;119:737–49. https://doi.org/10.1093/bja/aex229.
Toyama S, Shimoyama N, Ishida Y, Koyasu T, Szeto HH, Shimoyama M. Characterization of acute and chronic neuropathies induced by oxaliplatin in mice and differential effects of a novel mitochondria-targeted antioxidant on the neuropathies. Anesthesiology. 2014;120:459–73. https://doi.org/10.1097/01.anes.0000435634.34709.65.
Chiu GS, Maj MA, Rizvi S, Dantzer R, Vichaya EG, Laumet G, et al. Pifithrin-μ prevents cisplatin-induced chemobrain by preserving neuronal mitochondrial function. Cancer Res. 2017;77:742–52. https://doi.org/10.1158/0008-5472.CAN-16-1817.
Maj MA, Ma J, Krukowski KN, Kavelaars A, Heijnen CJ. Inhibition of mitochondrial p53 accumulation by PFT-μ prevents cisplatin-induced peripheral neuropathy. Front Mol Neurosci. 2017. https://doi.org/10.3389/fnmol.2017.00108.
Van Helleputte L, Kater M, Cook DP, Eykens C, Rossaert E, Haeck W, et al. Inhibition of histone deacetylase 6 (HDAC6) protects against vincristine-induced peripheral neuropathies and inhibits tumor growth. Neurobiol Dis. 2018;111:59–69. https://doi.org/10.1016/j.nbd.2017.11.011.
Ma J, Kavelaars A, Dougherty PM, Heijnen CJ. Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: targeting the source. Cancer. 2018;124:2289–98. https://doi.org/10.1002/cncr.31248.
Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, et al. Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci. 2012;32:6149–60. https://doi.org/10.1523/JNEUROSCI.6343-11.2012.
Kawata D, Wu Z. Regulatable transgene expression for prevention of chemotherapy-induced peripheral neuropathy. Mol Ther Methods Clin Dev. 2017;6:91–101. https://doi.org/10.1016/j.omtm.2017.06.004.
Al-Mazidi S, Alotaibi M, Nedjadi T, Chaudhary A, Alzoghaibi M, Djouhri L. Blocking of cytokines signalling attenuates evoked and spontaneous neuropathic pain behaviours in the paclitaxel rat model of chemotherapy-induced neuropathy. Eur J Pain. 2018;22:810–21. https://doi.org/10.1002/ejp.1169.
Cerles O, Benoit E, Chéreau C, Chouzenoux S, Morin F, Guillaumot M-A, et al. Niclosamide inhibits oxaliplatin neurotoxicity while improving colorectal cancer therapeutic response. Mol Cancer Ther. 2017;16:300–11. https://doi.org/10.1158/1535-7163.MCT-16-0326.
Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon S-Y, Walters ET, et al. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2013;14:1031–44. https://doi.org/10.1016/j.jpain.2013.03.012.
Laura B, Elisabetta B, Adelchi RP, Roberto R, Loredana C, Andrea A, et al. CXCR1/2 pathways in paclitaxel-induced neuropathic pain. Oncotarget. 2017. https://doi.org/10.18632/oncotarget.15533.
Moepps B. CXCR1 and CXCR2 and ligands. Compend Inflamm Dis. 2016. https://doi.org/10.1007/978-3-7643-8550-7_223.
Brandolini L, Castelli V, Aramini A, Giorgio C, Bianchini G, Russo R, et al. DF2726A, a new IL-8 signalling inhibitor, is able to counteract chemotherapy-induced neuropathic pain. Sci Rep. 2019;9:11729. https://doi.org/10.1038/s41598-019-48231-z.
Kwiatkowski K, Mika J. The importance of chemokines in neuropathic pain development and opioid analgesic potency. Pharmacol Rep. 2018;70:821–30. https://doi.org/10.1016/j.pharep.2018.01.006.
Montague K, Malcangio M. The therapeutic potential of monocyte/macrophage manipulation in the treatment of chemotherapy-induced painful neuropathy. Front Mol Neurosci. 2017. https://doi.org/10.3389/fnmol.2017.00397.
Qin B, Luo N, Li Y, Gong D, Zheng J, Tan X, et al. Protective effect of gastrodin on peripheral neuropathy induced by anti-tumor treatment with vincristine in rat models. Drug Chem Toxicol. 2018. https://doi.org/10.1080/01480545.2018.1547739.
Illias AM, Gist AC, Zhang H, Kosturakis AK, Dougherty PM. Chemokine CCL2 and its receptor CCR2 in the dorsal root ganglion contribute to oxaliplatin-induced mechanical hypersensitivity. Pain. 2018;159:1308–16. https://doi.org/10.1097/j.pain.0000000000001212.
Tsubota M, Fukuda R, Hayashi Y, Miyazaki T, Ueda S, Yamashita R, et al. Role of non-macrophage cell-derived HMGB1 in oxaliplatin-induced peripheral neuropathy and its prevention by the thrombin/thrombomodulin system in rodents: negative impact of anticoagulants. J Neuroinflammation. 2019;16:199. https://doi.org/10.1186/s12974-019-1581-6.
Sandborn WJ, Bhandari BR, Randall C, Younes ZH, Romanczyk T, Xin Y, et al. Andecaliximab [anti-matrix metalloproteinase-9] induction therapy for ulcerative colitis: a randomised, double-blind, placebo-controlled, phase 2/3 study in patients with moderate to severe disease. J Crohn’s Colitis. 2018;12:1021–9. https://doi.org/10.1093/ecco-jcc/jjy049.
Schreiber S, Siegel CA, Friedenberg KA, Younes ZH, Seidler U, Bhandari BR, et al. A phase 2, randomized, placebo-controlled study evaluating matrix metalloproteinase-9 inhibitor, andecaliximab, in patients with moderately to severely active Crohn’s disease. J Crohn’s Colitis. 2018;12:1014–20. https://doi.org/10.1093/ecco-jcc/jjy070.
Shah MA, Starodub A, Sharma S, Berlin J, Patel M, Wainberg ZA, et al. Andecaliximab/GS-5745 alone and combined with mFOLFOX6 in advanced gastric and gastroesophageal junction adenocarcinoma: results from a phase I study. Clin Cancer Res. 2018;24:3829–37. https://doi.org/10.1158/1078-0432.CCR-17-2469.
Kumar V, Soni P, Garg M, Kamholz S, Chandra AB. Emerging therapies in the management of advanced-stage gastric cancer. Front Pharmacol. 2018;9:404. https://doi.org/10.3389/fphar.2018.00404.
Tonello R, Lee SH, Berta T. Monoclonal antibody targeting the matrix metalloproteinase 9 prevents and reverses paclitaxel-induced peripheral neuropathy in mice. J Pain. 2019;20:515–27. https://doi.org/10.1016/j.jpain.2018.11.003.
Robinson CR, Zhang H, Dougherty PM. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience. 2014;274:308–17. https://doi.org/10.1016/j.neuroscience.2014.05.051.
Sekiguchi F, Domoto R, Nakashima K, Yamasoba D, Yamanishi H, Tsubota M, et al. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: evidence for a neuroimmune crosstalk. Neuropharmacology. 2018;141:201–13. https://doi.org/10.1016/j.neuropharm.2018.08.040.
Hu L-Y, Zhou Y, Cui W-Q, Hu X-M, Du L-X, Mi W-L, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) dependent microglial activation promotes cisplatin-induced peripheral neuropathy in mice. Brain Behav Immun. 2018;68:132–45. https://doi.org/10.1016/j.bbi.2017.10.011.
Pachman DR, Dockter T, Zekan PJ, Fruth B, Ruddy KJ, Ta LE, et al. A pilot study of minocycline for the prevention of paclitaxel-associated neuropathy: ACCRU study RU221408I. Support Care Cancer. 2017;25:3407–16. https://doi.org/10.1007/s00520-017-3760-2.
Robinson CR, Dougherty PM. Spinal astrocyte gap junction and glutamate transporter expression contributes to a rat model of bortezomib-induced peripheral neuropathy. Neuroscience. 2015;285:1–10. https://doi.org/10.1016/j.neuroscience.2014.11.009.
Cerles O, Gonçalves TC, Chouzenoux S, Benoit E, Schmitt A, Bennett Saidu NE, et al. Preventive action of benztropine on platinum-induced peripheral neuropathies and tumor growth. Acta Neuropathol Commun. 2019;7:9. https://doi.org/10.1186/s40478-019-0657-y.
Kyte SL, Toma W, Bagdas D, Meade JA, Schurman LD, Lichtman AH, et al. Nicotine prevents and reverses paclitaxel-induced mechanical allodynia in a mouse model of CIPN. J Pharmacol Exp Ther. 2018;364:110–9. https://doi.org/10.1124/jpet.117.243972.
Toma W, Kyte SL, Bagdas D, Jackson A, Meade JA, Rahman F, et al. The α7 nicotinic receptor silent agonist R-47 prevents and reverses paclitaxel-induced peripheral neuropathy in mice without tolerance or altering nicotine reward and withdrawal. Exp Neurol. 2019;320:113010. https://doi.org/10.1016/j.expneurol.2019.113010.
Sałat K, Jakubowska A, Kulig K. Cebranopadol: a first-in-class potent analgesic agent with agonistic activity at nociceptin/orphanin FQ and opioid receptors. Expert Opin Investig Drugs. 2015;24:837–44. https://doi.org/10.1517/13543784.2015.1036985.
Sałat K, Furgała A, Sałat R. Evaluation of cebranopadol, a dually acting nociceptin/orphanin FQ and opioid receptor agonist in mouse models of acute, tonic, and chemotherapy-induced neuropathic pain. Inflammopharmacology. 2018;26:361–74. https://doi.org/10.1007/s10787-017-0405-5.
Chłoń-Rzepa G, Ślusarczyk M, Jankowska A, Gawalska A, Bucki A, Kołaczkowski M, et al. Novel amide derivatives of 1,3-dimethyl-2,6-dioxopurin-7-yl-alkylcarboxylic acids as multifunctional TRPA1 antagonists and PDE4/7 inhibitors: a new approach for the treatment of pain. Eur J Med Chem. 2018;158:517–33. https://doi.org/10.1016/j.ejmech.2018.09.021.
Resta F, Micheli L, Laurino A, Spinelli V, Mello T, Sartiani L, et al. Selective HCN1 block as a strategy to control oxaliplatin-induced neuropathy. Neuropharmacology. 2018;131:403–13. https://doi.org/10.1016/j.neuropharm.2018.01.014.
Furgała A, Fijałkowski Ł, Nowaczyk A, Sałat R, Sałat K. Time-shifted co-administration of sub-analgesic doses of ambroxol and pregabalin attenuates oxaliplatin-induced cold allodynia in mice. Biomed Pharmacother. 2018;106:930–40. https://doi.org/10.1016/j.biopha.2018.07.039.
Al-Massri KF, Ahmed LA, El-Abhar HS. Mesenchymal stem cells in chemotherapy-induced peripheral neuropathy: a new challenging approach that requires further investigations. J Tissue Eng Regen Med. 2019. https://doi.org/10.1002/term.2972.
Zhong S, Zhou Z, Liang Y, Cheng X, Li Y, Teng W, et al. Targeting strategies for chemotherapy-induced peripheral neuropathy: does gut microbiota play a role? Crit Rev Microbiol. 2019;45:369–93. https://doi.org/10.1080/1040841X.2019.1608905.
Defaye M, Gervason S, Altier C, Berthon J-Y, Ardid D, Filaire E, et al. Microbiota: a novel regulator of pain. J Neural Transm. 2019. https://doi.org/10.1007/s00702-019-02083-z.
Pratt C, Campbell MD. The effect of bifidobacterium on reducing symptomatic abdominal pain in patients with irritable bowel syndrome: a systematic review. Probiotics Antimicrob Proteins. 2019. https://doi.org/10.1007/s12602-019-09609-7.
Santucci NR, Saps M, van Tilburg MA. New advances in the treatment of paediatric functional abdominal pain disorders. Lancet Gastroenterol Hepatol. 2019. https://doi.org/10.1016/S2468-1253(19)30256-0.
Castelli V, Palumbo P, D’Angelo M, Moorthy NK, Antonosante A, Catanesi M, et al. Probiotic DSF counteracts chemotherapy induced neuropathic pain. Oncotarget. 2018. https://doi.org/10.18632/oncotarget.25524.
Toume K, Hou Z, Yu H, Kato M, Maesaka M, Bai Y, et al. Search of anti-allodynic compounds from Plantaginis Semen, a crude drug ingredient of Kampo formula “Goshajinkigan”. J Nat Med. 2019;73:761–8. https://doi.org/10.1007/s11418-019-01327-2.
Zhang Q-Y, Wang F-X, Jia K-K, Kong L-D. Natural product interventions for chemotherapy and radiotherapy-induced side effects. Front Pharmacol. 2018;9:1253. https://doi.org/10.3389/fphar.2018.01253.
Wu B-Y, Liu C-T, Su Y-L, Chen S-Y, Chen Y-H, Tsai M-Y. A review of complementary therapies with medicinal plants for chemotherapy-induced peripheral neuropathy. Complement Ther Med. 2019;42:226–32. https://doi.org/10.1016/j.ctim.2018.11.022.
Liu Y, May BH, Zhang AL, Guo X, Lu C, Xue CC, et al. Integrative herbal medicine for chemotherapy-induced peripheral neuropathy and hand-foot syndrome in colorectal cancer: a systematic review and meta-analysis. Integr Cancer Ther. 2019;18:153473541881783. https://doi.org/10.1177/1534735418817833.
Hoshino N, Ganeko R, Hida K, Sakai Y. Goshajinkigan for reducing chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Int J Clin Oncol. 2018;23:434–42. https://doi.org/10.1007/s10147-017-1229-4.
Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nat Rev Dis Prim. 2017;3:17002. https://doi.org/10.1038/nrdp.2017.2.
Acknowledgements
The data of the author’s own research presented in this paper were obtained with financial support of the National Science Centre, Poland [Grant Number UMO-2015/17/B/NZ7/02937].
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sałat, K. Chemotherapy-induced peripheral neuropathy—part 2: focus on the prevention of oxaliplatin-induced neurotoxicity. Pharmacol. Rep 72, 508–527 (2020). https://doi.org/10.1007/s43440-020-00106-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-020-00106-1