
Abstract
The mitochondrial network constantly changes and remodels its shape to face the cellular energy demand. In human cells, mitochondrial fusion is regulated by the large, evolutionarily conserved GTPases Mfn1 and Mfn2, which are embedded in the mitochondrial outer membrane, and by OPA1, embedded in the mitochondrial inner membrane. In contrast, the soluble dynamin-related GTPase Drp1 is recruited from the cytosol to mitochondria and is key to mitochondrial fission. A number of new players have been recently involved in Drp1-dependent mitochondrial fission, ranging from large cellular structures such as the ER and the cytoskeleton to the surprising involvement of the endocytic dynamin 2 in the terminal abscission step. Here we review the recent findings that have expanded the mechanistic model for the mitochondrial fission process in human cells and highlight open questions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Discovered by Richard Altmann in the 1840s, mitochondria are thought to derive from the invasion of a pre-eukaryotic cell by an alphaproteobacterium of the Rickettsiales order. The monophyletic origin of mitochondria suggests that this invasion was a unique event and their advent was critical for eukaryotic evolution. This is reflected by the multitude of vital functions mitochondria are responsible for, such as energy production, the biogenesis of essential cellular components, apoptosis, or innate immune signaling. In line with this, mitochondrial dysfunction correlates with a wide variety of pathologies, in particular neurodegenerative disease and cardiac dysfunction [1].
Mitochondria are dynamic organelles
Mitochondria are particularly complex organelles, enveloped by two membranes: the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM), which enclose two compartments, the intermembrane space and the matrix. The matrix harbours the mitochondrial DNA (mtDNA), a highly reduced version of the alphaproteobacterial mitochondrial ancestor’s genome. In mammalian cells, mitochondria are not isolated, bacteria-like organelles, but rather form a dynamic network. Mitochondrial dynamics and the resulting overall morphology of the network are determined by fusion and fission events and by mitochondrial movement, both of which are highly dependent on the cell type and on the functional state of mitochondria.
Mitochondria move along cytoskeletal tracks but the extent of their motility is very variable and both species and cell type dependent [2]. For example, mitochondria appear almost immotile in muscle cells, but motility is crucial for mitochondria distribution in highly polarized cells, such as neurons, where mitochondrial activity is required at the synapse. In this context the kinesin KIF5, along with dyneins and the mitochondrial adaptors Miro and Milton, mediate mitochondria movement along microtubules in higher eukaryotic cells [3], while mitochondria movement mainly relies on actin microfilaments in budding yeast [4].
Mitochondrial fusion and fission often occur simultaneously and in a balanced manner within a cell, and an increase in either activity leads to hyperfused or fragmented mitochondria (Fig. 1). The complex membrane system of mitochondria is a challenge for fusion and fission and requires specialized molecular machineries on the OMM or IMM. It is interesting to note that the key proteins known to date to mediate mitochondrial fusion and fission are all nuclear-encoded, large GTPases, pointing to a common evolutionary origin (Fig. 2a). This may not be surprising, as despite differences in the membrane topology, both mitochondrial fission and fusion require the merging of membranes (Fig. 1a, b, inset), which is achieved through membrane remodeling proteins. Different model organisms including yeast, Drosophila and C.elegans have greatly contributed to our understanding of the molecular mechanisms that underlie mitochondrial fusion and fission. In this review, we will focus on mammalian cells, summarizing mitochondrial fusion and highlighting several recent discoveries that significantly expanded our knowledge on mitochondrial fission.

Mitochondrial fusion and fission. a Mitochondria tethering via homotypic (Mfn1–Mfn1 and Mfn2–Mfn2) and heterotypic (Mfn1–Mfn2) mitofusin interactions promotes OMM fusion, while inner membrane fusion is promoted by OPA1. b Schematic representation of mitochondrial fission. Enlargements of OMM and IMM fusion events that occur during mitochondrial fusion and fission. c Immunofluorescence showing changes in mitochondrial morphology (red) and Drp1 localization (green) in response to different perturbations. Adapted from [182, 190, 202]

Domain structure of Drp1, its modifications and receptors. a The family of mitochondrial dynamics proteins and their bacterial ancestor, the bacterial dynamin-like protein BDLP. Abbreviations: paddle domain (PAD) and pleckstrin homology domain (PH), both responsible for lipid binding; proline-rich domain (PRD), tetratricopeptide (TPR) and coiled–coiled (CC) domains are involved in protein–protein interactions; GTPase effector domain (GED); insert B domain (Ins B); transmembrane domain (TM). b full-length human Drp1 (isoform 1, 736 aa). Numbers indicate the start of a new domain. Posttranslational modifications are colour-coded, and the modified amino acids have been indicated, except for ubiquitination and O-GlcNAcylation (grey), for which sites have not yet been identified. A few commonly used mutants are shown in black: K38A (dominant-negative), G350D (impaired in higher order oligomerization), A395D (natural mutation, impaired in tetramerization and higher order oligomerization). c Drp1 is recruited to the OMM via multiple transmembrane receptors. MiD51 and Mff are able to oligomerize and to interact with each other in the same fission foci. The SAMM50 protein has been shown to interact with the MITOS complex in the IMM
Molecular basis and physiological roles of mitochondrial fusion
During mitochondrial fusion, the outer and inner membranes of two mitochondria fuse in a regulated manner. This permits content exchange and enables cross-complementation of mitochondrial DNA molecules, preventing the accumulation of mutant DNA in a specific mitochondrion [5, 6]. Although content exchange allows the mitochondrial network to adapt to the metabolic demand in a concerted manner (reviewed in [7]), it does not always lead to a completely homogeneous mitochondrial population within a single cell, and the mitochondrial membrane potential can vary among different mitochondria [8]. Mitochondrial fusion appears particularly important during stress and starvation conditions, where it is thought to maximize the efficiency of mitochondrial metabolism through the sharing of metabolites in the matrix (reviewed in [9]). Interestingly, although bacteria are not known to fuse, filamentous bacteria do exist (e.g. syncytial Streptomyces or septated filamentous cyanobacteria). Such filament formation can be induced by mutation in cell division proteins, or upon stress or nutritional changes, conditions potentially also encountered by the alphaproteobacterial mitochondrial ancestor.
In present-day mitochondria fusion is an active process that is mediated by Mitofusin 1 and Mitofusin 2 in human cells (Mfn1 and Mfn2, Fzo1 in yeast [10, 11]). These partially redundant large GTPases are embedded in the OMM, where they promote mitochondrial fusion (Fig. 1a). Mitofusins form hetero-oligomers, promote mitochondrial tethering similar to SNARE proteins and mediate GTP-dependent fusion [12]. Recently, it has been reported that mitofusins fold both in fusion-competent and fusion-incompetent conformations to regulate mitochondrial tethering [13]. An additional mechanism has been recently described for mitochondrial network formation, which would involve mitochondrial tubulation by kinesin KIF5B in concert with mitofusin-mediated fusion [14]. Recently, part of the Mfn1 structure has been solved and found to resemble that of bacterial dynamin-like proteins, uncovering an interesting evolutionary relationship [15, 16]. While Mfn1 has a higher GTPase activity and is ubiquitously expressed [17], Mfn2 displays tissue-specific expression [18] and has been linked to the type 2a subset of Charcot–Marie–Tooth disease, a group of hereditary peripheral neuropathies that can be caused by defects in several of the proteins regulating mitochondrial dynamics [19]. Interestingly, both mitofusins are essential for embryonic development in mice, where they play an important role in the maintenance of mitochondrial DNA (mtDNA) and oxidative phosphorylation [20, 21] and may also have additional protein-specific functions [22]. In addition, Mfn2 plays a role in ER–mitochondria tethering [23]. ER–mitochondria contact sites are crucial for interorganellar communication, including calcium signaling integration and lipid biogenesis [24, 25], and are emerging as an important player in mitochondrial fission (detailed below).
Although to a large extent coordinated, fusion of the OMM and IMM can occur sequentially, as suggested by the isolation of fusion intermediates in vitro and in vivo [26, 27]. This points to the presence of an IMM fusion machinery. A key molecule for IMM fusion is Optic Atrophy 1 (OPA1, Mgm1 in yeast), a three-membrane-pass protein that faces the intermembrane space (Fig. 1a) and is mutated in autosomal dominant optic atrophy [28, 29]. In contrast to Mitofusins, OPA1 does not need to be present on apposing membranes to mediate fusion [30]. In addition, OPA1 participates in the shaping of cristae, IMM invaginations whose remodeling plays an important role in the release of the proapoptotic mitochondrial inner membrane-associated protein cytochrome c during apoptosis [31]. OPA1 is highly regulated, both through differential splicing [32], and through proteolytic processing, which leads to long IMM-anchored OPA1 and short, soluble OPA1. The AAA-protease YME1L and the inner membrane metalloprotease OMA1 are involved in OPA1 processing [33]. Although both long and short OPA1 forms are found in OPA1 supercomplexes, long OPA1 appears to be required for fusion, while short OPA1 may play a role in IMM fission [34]. In mammals, a drop in the mitochondrial membrane potential and other stress factors block fusion and stimulate OPA1 processing by OMA1 [33]. OMA1 activation may, therefore, represent a key point of regulation, as its activation under stress conditions can result in complete conversion of long OPA1 to short OPA1, definitively preventing mitochondrial fusion until new OPA1 is synthesized. Although OPA1 regulation and function have been characterized in detail, further research is needed to uncover mechanistic details of the inner membrane fusion and fission processes, which may involve additional players.
Mitochondrial fission: division for survival?
Mitochondrial fission divides the organelle into two often not equally sized daughter mitochondria (Fig. 1b). The best-characterized mitochondrial division machinery relies on dynamin-related protein 1 (Drp1/DNML1/DLVP/Dymple, Dnm1 in yeast), which is regulated at multiple levels. Knockout studies in mice showed that deletion of Drp1 is embryonically lethal, indicating that mitochondrial fission is an essential process [35, 36]. Tissue-specific knockout approaches and in vitro studies demonstrated that mitochondrial fission deficiencies result in respiratory defects [37]. At the cellular level, mitochondrial fission is important for organelle distribution during mitosis [38] and for proper distribution of mitochondria to neuronal synapses, where localized energy production supports synaptic functions [36, 39]. Interestingly, organellar and cellular quality control mechanisms have been linked to mitochondrial dynamics. At the organellar level, mitochondrial fission has been implicated in mitophagy, the autophagic process by which defective mitochondria are selectively degraded [40, 41]. Such defective mitochondria are characterized by a loss in the inner membrane potential, and are thereby recognized by the Pink1/Parkin system. Parkin then mediates ubiquitination of several OMM-associated proteins, which are then recognized by different ubiquitin-binding mitophagy receptors, targeting the whole mitochondrion for autophagic degradation (for recent reviews see [42, 43]). Limiting mitochondrial size through fission seems to be a prerequisite for mitochondria engulfment into autophagosomes [40, 44]. The extent to which Drp1 is an active player in mitophagy or not is, however, still debated [40, 43, 45,46,47,48,49].
In addition to mitophagy, mitochondrial fission has been shown to occur during apoptosis and to promote the release of the proapoptotic protein cytochrome c from the intermembrane space [50]. However, later studies indicated that apoptosis can proceed in Drp1-depleted cells [35, 36, 51, 52] and that fission does not necessarily correlate with apoptosis. In the following, we will summarize recent findings in mitochondrial fission, with a particular focus on the role of the cytoskeleton during the fission process.
New insights into Drp1-dependent mitochondrial fission
The dynamin-related protein Drp1
Dynamins are evolutionarily conserved, versatile molecules that are regulated by oligomerization and conformational changes to mediate membrane remodeling. A key protein in mitochondrial fission is Dynamin-related protein 1, which is conserved in opisthokonts (animals and fungi) and plants [53, 54]. Drp1 was first identified by several laboratories through the homology of its GTPase domain with that of endocytic dynamins [55,56,57,58,59,60]. Similar to endocytic dynamins, Drp1 multimerizes to form spirals on the mitochondrial outer membrane and hydrolyzes GTP, which cause a conformational change, enabling spiral compaction and resulting in mitochondrial constriction [61, 62]. Alternative splicing can produce up to eight different Drp1 isoforms, with cell type-specific expression patterns [59, 63, 64]. The best-characterized is isoform 3, which lacks 31 amino acids in the insert b domain [59, 63,64,65,66]. Drp1 has an apparent molecular weight of ~ 80kD and is composed of four domains (Fig. 2b): a GTPase domain (containing an insert A in the neuronal isoform), a middle domain, a variable domain (also called insert B), and a GTPase effector domain (GED), whose C-terminal coiled coil mediates multimerization [67]. The mechanochemical core of the protein comprises the GTPase, middle and GED domains and is sufficient to cause liposome constriction [60]. In contrast to dynamin, Drp1 lacks the pleckstrin homology domain that mediates binding to phosphoinositide-containing membranes, such as the inner leaflet of the plasma membrane (Fig. 2a). This may account for the fact that Drp1 does not localize to clathrin-coated pits [59] and its depletion/mutation does not affect the secretory pathway [68]. However, Drp1 was recently shown to play a role in endocytic vesicle formation in hippocampal neurons, suggesting that recruitment of Drp1 to different cellular structures may be regulated through cell type-specific mechanisms [69].
Role of lipids in mitochondrial fission
For a long time thought to be only structural components of cellular membranes, lipids are now well recognized as active players in membrane remodeling. In addition to serving as recruiting or activating factors for proteins, lipids can affect membrane dynamics by lowering the energy barrier for fusion and fission, two energetically unfavorable processes. Here we briefly summarize the role of phospholipids in Drp1-mediated mitochondrial fission (see [70] for a comprehensive review on mitochondrial lipids).
The signature of mitochondrial membranes is the presence of cardiolipin (CL). CL is an atypical conical lipid and indirect evidence has implicated conical lipids in the generation of membrane curvature, which is required for both membrane fusion and fission [71]. CL is found primarily in the IMM, where it constitutes almost 20% of the total lipid content [72], but is also present in the OMM, in particular at IMM/OMM contact sites [73, 74]. Recent studies have shown that CL binds Drp1 at four lysines located in the variable domain [75]. Additionally, CL appears to recruit Drp1 to the OMM and stimulates the GTPase activity [76, 77]. This stimulation occurs only at concentrations matching those of IMM/OMM contact sites, suggesting that these specialized domains can serve as a platform for Drp1 recruitment and activation. Conversely, a recent report showed that Drp1 regulates CL clustering in liposomes, inducing a CL phase transition, which in turn triggers membrane constriction and fission [78]. An interesting recent report added a new layer of complexity to this picture, showing that mitochondrial phosphatidic acid (PA) can bind Drp1 and inhibit its GTPase activity [79]. PA is formed on the OMM by the action of mitochondrial phospholipase D (mitoPLD), a membrane-tethered enzyme, which utilizes CL as a substrate. The ability of Drp1 to interact with mitoPLD suggests that recruitment of the latter could be a way to balance Drp1 activation.
Drp1-dependent mitochondrial fission as a multistep process
Surprisingly, Drp1, the key player of mitochondrial fission not only targets mitochondria, but also the ER [59, 80, 81] and peroxisomes [82]. This raises the question as to how differential targeting of Drp1 is achieved. Several studies showed that mitochondrial fission is a multistep process, in which specific targeting of Drp1 to the OMM relies on its numerous posttranslational modifications and on its transmembrane receptors in the OMM.
Drp1 regulation by posttranslational modifications
Phosphorylation
Since mitochondria cannot be generated de novo, they have to be segregated into daughter cells during mitosis, which is achieved through extensive mitochondrial fission prior to mitosis. Mitochondrial network fragmentation coincides with phosphorylation of human Drp1 at S616 by Cdk1/cyclin B kinase, but the functional impact of this modification on Drp1 is currently unknown, as its GTPase activity does not seem affected in vitro. Interestingly, as cells exit mitosis, the mitochondrial network is restored via APC/C (anaphase-promoting complex/cyclosome)-mediated degradation of Drp1 [83]. Mitochondrial network fragmentation has been also observed upon phosphorylation of Drp1 S616 by Erk2 in cancer cells, where it appeared to play a role in tumor proliferation [84, 85]. Recently, Drp1 S616 has been found to be phosphorylated also by Cdk5 kinase in neurons, but its effect on Drp1 activity and mitochondrial network morphology is controversial [86, 87].
Drp1 S637 is phosphorylated by cyclic AMP-dependent protein kinase A (PKA) and this modification was found to inhibit the GTPase activity, most likely by preventing intra-molecular interactions between the GED and GTPase domain [88, 89]. This phosphorylation implicated Drp1 as a survival-promoting substrate of PKA, in particular during starvation, where elongation of the mitochondrial network and increased ATP production sustain metabolic needs [90]. Calcineurin was identified as the phosphatase responsible for S637 dephosphorylation [89] and was then shown to mediate calcium-induced, Drp1-dependent mitochondrial fission [91]. Calcineurin-mediated Drp1 dephosphorylation appeared to have a marginal effect on Drp1 oligomerization or its GTPase activity, while strongly regulating the recruitment of Drp1 to the OMM [91, 92]. Both phosphorylation sites are shown in Fig. 2b.
Ubiquitination
Ubiquitin is a 76-amino acid polypeptide, which is covalently attached to lysine residues of target proteins via an enzymatic reaction. Ubiquitin can either regulate protein stability by targeting modified substrates to proteasomal degradation or change their biological function by modulating functional interactions. Drp1 has been shown to be ubiquitinated by the ubiquitin ligase Parkin [93]. This modification targets Drp1 for proteasomal degradation and, therefore, Parkin depletion increased Drp1 levels and mitochondrial fragmentation. The OMM-anchored ubiquitin ligase MARCH5 (also known as MITOL) also plays a role in mitochondrial dynamics; however, it is not clear from the published reports whether MARCH5 specifically regulates fusion or fission [94,95,96]. Several proteins involved in mitochondrial dynamics are reported substrates of MARCH5, including Drp1, Mfn1, Mfn2 and Mid49 [97,98,99,100], possibly explaining the heterogeneity of published results concerning MARCH5 function. A recent report added another layer of complexity by showing that Drp1 is not only a substrate, but also a regulator of MARCH5 activity, along with Mff (Mitochondrial fission factor, a Drp1 receptor on the OMM, see below) [101]. New studies will shed light on the role of MARCH5 activity towards its several identified substrates.
Sumoylation
Small ubiquitin-like modifier (SUMO) is a ubiquitin-related protein, which can be covalently linked to a target protein. SUMOylation regulates protein conformation (and hence activity), localization and interaction with cellular partners [102]. The SUMO-conjugating enzyme Ubc9 and SUMO itself were identified as Drp1 interacting partners by yeast two-hybrid analysis and SUMO partially colocalized with Drp1 at fission sites [103, 104]. Later work identified MAPL (mitochondrial-anchored protein ligase) as the mitochondrial-anchored E3 SUMO ligase able to directly SUMOylate Drp1 in the variable domain [105, 106], Fig. 2b. Interestingly, four lysines are differentially spliced into three of the Drp1 isoforms, raising the possibility that SUMOylation might also account for functional differences among Drp1 isoforms [106]: SUMOylation led to increased Drp1 stabilization on the OMM and increased mitochondrial fission [104]. Drp1 SUMOylation is reversed by the SUMO protease SENP5 [107], in particular during mitosis, where Drp1 deSUMOylation correlated with increased binding and release of Drp1 to/from the OMM, thereby stimulating fission [108]. Phosphorylation and deSUMOyation thus appear to converge in activating Drp1 during mitosis.
S-nitrosylation
Nitric oxide (NO) is not only a neurotransmitter of the central nervous system: in addition to its signaling role, NO can be covalently linked to thiol groups of target proteins, such as cysteine residues, in a process called S-nitrosylation. S-nitrosylation can affect protein function, stability or subcellular location [109]. Drp1 was found to be S-nitrosylated on C644 (Fig. 2b) upon NO overproduction due to pathological conditions, such as accumulation of β-amyloid aggregates, key mediators of Alzheimer disease [110, 111]. This correlated with rapid Drp1-dependent mitochondrial fragmentation, followed by energy production failure and cell death [110]. However, these findings were subsequently questioned by Bossy et al. [112], who failed to detect a change in Drp1 oligomerization and GTPase activity upon S-nitrosylation. Additionally, OPA1 was also found to be S-nitrosylated. Therefore, at present S-nitrosylation of Drp1 may not be uniquely responsible for NO-induced mitochondrial fission.
O-GlcNAcylation
O-GlcNAcylation refers to the reversible but covalent attachment of N-acetyl-glucosamine to serine or threonine residues of a target protein. O-GlcNAcylation of Drp1 was shown to increase Drp1 GTP-binding activity, Drp1 translocation to the OMM and mitochondrial fission in cardiomyocytes [113]. O-GlcNAcylation of Drp1 was accompanied by decreased levels of Ser637 phosphorylation; further investigations are thus required to discriminate whether the effects observed upon O-GlcNAcylation are mediated by decreased Ser637 phosphorylation or whether O-GlcNAcylation directly affects Drp1 function. Recently, decreased O-GlcNAcylation levels were shown to result in an increase in Drp1-dependent mitochondrial fission, along with a decrease in the mitochondrial membrane potential and mitochondrial content [114]. An interesting aspect is that O-GlcNAcylation directly links mitochondrial morphology with the metabolic state of the cell, as the latter regulates cellular levels of the O-GlcNAc donor UDP-GlcNAc [115].
Drp1 recruitment through receptors on the OMM: Mff, MiD49/51, and Fis1
Although being the master regulator of mitochondrial fission, more than 95% of Drp1 is cytosolic [68]. Hence, Drp1 recruitment to mitochondria is an early and key step in mitochondrial division, which is stimulated by the presence of four single-pass transmembrane Drp1 receptors that are anchored to the OMM: mitochondrial fission factor (Mff), the mitochondrial dynamics proteins 49 and 51 (MiD49 and MiD51) and Fis1.
Fis1
Fis1 was the first identified receptor for Drp1 [116, 117]. In yeast, Fis1 appeared to regulate mitochondrial morphology by recruiting the yeast Drp1 homologue Dnm1 to the OMM in concert with the partially redundant adaptor proteins Mdv1 and Caf4 [118, 119]. However, no Mdv1 and Caf4 homologs have been found in mammals, suggesting that the machineries for mitochondrial fission have diverged during evolution. In agreement with this hypothesis, to date there is mixed evidence about the role of Fis1 in mammals. Some reports have involved Fis1 in Drp1-dependent mitochondrial fission [120,121,122], and shown it binds Drp1 [120, 123]. These data were not reproduced in other studies [121, 124, 125]; furthermore, Fis1 knockout cells displayed very mild fission defects, raising questions about the precise role of Fis1 in Drp1 recruitment and mitochondrial fission [92, 123].
Fis1 was recently proposed to play a specific role in stress-induced mitochondrial fission [126, 127]. Indeed, induction of mitophagy caused Fis1 to form a complex with several ER proteins and Mff-recruited Drp1. Fis1 overexpression has in turn been shown to trigger mitophagy [40, 90, 128]. Fis1 deletion induces accumulation of the autophagy marker LC3 at mitochondria during stress-induced, Parkin-mediated mitophagy [127, 129] and Fis1 knockout cells show severely impaired mitophagy [128]. Thus, the role of Fis1 in mitochondrial dynamics may be restricted to specific physiological conditions such as apoptosis and mitophagy.
Mff
Mff is a 30-kDa C-tail-anchored membrane protein, which is considered a prime Drp1 receptor at both mitochondria and peroxisomes, as for both organelles its overexpression stimulates fission and Drp1 recruitment, while its ablation induces elongation [123, 130]. Unlike Fis1, Mff accumulates in discrete foci, where it recruits dimeric or higher order complexes of Drp1 [131]. Interestingly, the formation of Mff clusters is lost in Drp1 knockout cells, suggesting that Drp1 binding in turn influences Mff oligomerization [132]. Mff might promote Drp1 recruitment to facilitate severing of mitochondria specifically at ER-mitochondria contact sites, as suggested by imaging experiments and by mutagenesis data on Drp1 [133, 134]. The regulation of Drp1 by Mff is complicated by the presence of several Mff isoforms [130], which were recently shown to have a differential effect on the Drp1 isoforms [77]. Additionally, Mff also appears to increase the Drp1 GTPase activity and may thereby promote Drp1 spiral compaction and consequently the severing of mitochondria [131, 135]. Interestingly, Mff appears to be restricted to metazoans, highlighting that the overall conserved mitochondrial fission machinery can accommodate additional proteins, likely providing new ways to regulate fission.
MiD49/MiD51
Evolutionarily, mitochondrial division (MiD) proteins appeared after Mff, since they are only present in chordates, possibly reflecting a higher specialization in the mitochondrial division machinery. MiDs were unambiguously identified as Drp1 receptors through immunoprecipitation and yeast two-hybrid assays [124]. As in the case of Mff [123], the interaction between Drp1 and MiDs also appears to be transient, requiring chemical crosslinking for its detection [124]. MiDs bear an N-terminal transmembrane domain that anchors them to the OMM, and are not present on peroxisomes. The structures of the MiD49- and MiD51-soluble domains have been recently solved, revealing a similar nucleotidyltransferase domain [136,137,138]. Despite this similarity, MiD49 and MiD51 differ in their nucleotide-binding capacity, as MiD49 lacks the critical residues required for nucleotide binding. Interestingly, a MiD51 mutant deficient in nucleotide binding was still capable to recruit Drp1 to mitochondria, but appeared unable to promote mitochondrial fission [137].
Cooperation or specialization?
Why does Drp1 need several receptors to localize to mitochondria? Although the receptors may play independent and partially redundant functions in Drp1 recruitment [92, 125, 132], recent work showed that Mff and MiDs colocalize with the ER at mitochondrial fission sites, suggesting functional cooperation of the receptors [139]. Given that Mff preferentially interacts with dimeric Drp1 and stimulates Drp1 GTPase activity [65, 131], while MiD51 inhibits it [125, 140], a plausible speculation might be that Mff first recruits dimeric Drp1 at mitochondria and MiDs would then promote Drp1 self-assembly, while inhibiting its GTPase activity. Finally, Mff would stimulate the GTPase activity of the assembled Drp1 oligomer to promote fission. The canonical Drp1 receptors Mff and MiD49/51 may thus cooperate under specific conditions or in specific cell types. The picture has been recently further complicated by additional OMM proteins shown to also promote Drp1 recruitment, such as SAMM50, a component of the SAM complex, which inserts beta barrels in the OMM [141]. SAMM50 has been shown to interact with Drp1 and induce Drp1-dependent mitochondrial fission through an unknown mechanism [142]. Interestingly, SAMM50 could coordinate outer and inner membrane fission, as it also interacts with VDAC, a component of OMM/IMM contact sites and with the MINOS/MITOS complex in the IMM.
Determining the site of fission along a mitochondrion
Initial constriction occurs at ER–mitochondrial contact sites
ER–mitochondria contact sites were described already in the 1960s [143] and are visible by EM, but the molecular nature of the proteinaceous tether between the two organelles is still largely obscure. In mammalian cells, Mfn2 is thought to tether the ER and mitochondria [23, 144] and ER–mitochondrial contacts were also found to contain a complex of the IP3 receptor, the chaperone Grp75 and the voltage-gated anion channel VDAC [145]. Additional proteins found at contact sites include Rab32 [146], mammalian target of rapamycin complex 2 (mTORC2, [147], PACS2 [148]), and the recently identified Syntaxin 17, an evolutionarily conserved SNARE protein, a possibly ancestral tethering molecule [149]. ER–mitochondria contact sites can span up to 20% of the mitochondrial surface [150] and were mainly thought to serve as a platform for lipid exchange and Ca2+ signaling [24, 25]. In a spearheading work, Friedman and colleagues showed that ER–mitochondria contact sites also play a role in mitochondrial division: 88% of the mitochondrial fission events were found to be marked by ER tubules, which wrapped around mitochondria, and appeared to promote fission by constricting mitochondria in a process termed ER-associated mitochondrial division (ERMD) [133]. Intriguingly, the ER seems to play an analogous role in the fission of endosomes [151]. ER-marked mitochondrial constrictions show a diameter comparable to that of Drp1 helices and may, therefore, physically facilitate Drp1 recruitment or be actively sensed by Drp1. The ability of dynamin and dynamin-like proteins to detect membrane deformations [152], polymerize, and induce local membrane rearrangements seems to be ancestral, as a bacterial dynamin-like protein has recently been shown to accumulate in foci of antibiotic or phage-induced membrane deformations to protect membrane integrity [153]. The ER could also induce mitochondrial fission by promoting lipid insertion at contact sites or by clustering Mff and MiD49/51 [133, 139], which is expected to promote Drp1 recruitment. In addition, proteins present at ER-induced preconstrictions may support local Drp1 activation, as shown for MiD51 [154] and Syntaxin 17, which has been recently shown to specifically bind GTP-loaded Drp1 [149].
ER-associated mitochondrial division preferentially occurs next to the nucleoid
Mitochondria are the only organelles of animal cells that harbour their own circular genome (mtDNA), which associates with proteins that pack it into a nucleoid. As for the nuclear genome, the distribution of mtDNA molecules to daughter mitochondria is a crucial event. Early studies noticed that nucleoids are often localize at the tips of mitochondria [155, 156] or next to Drp1-marked constrictions [157], suggesting a link to mitochondrial fission. Ban-Ishihara then found that mitochondrial fission occurs in the vicinity of a nucleoid in 70% of all cases, and nucleoids cluster in Drp1-deficient cells [158]. This confirmed previous work showing that mitochondrial fission prevents nucleoid clustering and mtDNA loss in cellulo, later also shown in vivo [159, 160]. Interestingly, the nucleoid position correlated with ER–mitochondria contact sites in 85% of cases, raising the unsolved question as to how the nucleoid position is sensed by the ER across the two mitochondrial membranes and vice versa. Not only do nucleoids seem to communicate their position to the ER across the IMM and OMM, but recent data showed that the number of ERMD events increases up to threefold in presence of replicating nucleoids [161], highlighting the importance of the functional state of the nucleoid. mtDNA replication was found to occur upstream of Drp1 recruitment and mitochondrial division, and blocking nucleoid replication had been previously shown to cause Drp1 and Mff redistribution [158]. Whether ERMD at replicating nucleoids equally distributes the newly synthesized mtDNA into daughter mitochondria appears controversial [158, 161].
The role of the cytoskeleton in Drp1-dependent fission
Actin and actin-binding proteins
The first hint that actin may be involved in mitochondrial dynamics came from an early study in which actin depolymerization by cytochalasin D was shown to slow down mitochondrial fusion in mammalian cells [162]. In a pioneering work, De Vos and colleagues subsequently showed that actin depolymerization by cytochalasin D or latrunculin A impairs CCCP-induced mitochondrial fission by preventing Drp1 recruitment to mitochondria ([163], see Table 1 for an overview of drugs commonly used to manipulate mitochondrial dynamics). Stabilization of actin filaments through Wiskott–Aldrich Syndrome protein (WASP) overexpression was shown to increase mitochondrial length in Drosophila neurons, impairing Drp1 recruitment and eventually leading to neurodegeneration [164]. In search for factors mediating the actin-dependent localization of Drp1 to mitochondria, Myosin II was identified as a crucial player, a finding that the authors confirmed in mammalian cells.
The mechanism by which actin polymerization at mitochondria would regulate Drp1 recruitment has been recently investigated in more detail. Ji et al. showed that actin polymerization on mitochondria supports Drp1 maturation by incorporation of small Drp1 oligomers already associated with mitochondria. Interestingly, also MiD-containing complexes have been shown to coalesce and mature, raising the possibility that MiD foci maturation might also be dependent on actin dynamics [139]. Actin polymerization on mitochondria was found to precede the appearance of large Drp1 oligomers at fission sites upon ionomycin-induced mitochondrial fission [135]. In vitro studies showed that actin directly binds Drp1 in a GTP-dependent manner, stimulating its GTPase activity, which is further increased by the soluble domain of Mff [135, 165].
The identification of the ER as a major player in regulating mitochondrial pre-constriction upstream of Drp1 has set the stage for the discovery of the role played by the ER-localized form of inverted formin 2 (INF2) [166]. Together with the Arp2/3 complex and tandem monomer-binding proteins (TMBPs), formins constitute one of the three actin nucleator families characterized to date (reviewed in [167]). Interestingly, INF2 mutations have been associated with a subset of Charcot–Marie–Tooth disease [168], a disease in which several other genes regulating mitochondrial morphology have also been implicated, such as Mfn2, GDAP1 or DNML2 (https://ghr.nlm.nih.gov/condition/charcot-marie-tooth-disease). INF2 is a differentially spliced protein, which promotes the polymerization of linear actin filaments [169]. Depletion of the ER-targeted form of INF2 was found to increase the mitochondrial length and reduce Drp1 localization at mitochondria. Recently, Manor et al. identified Spire1C as a new player in INF2-dependent mitochondrial fission [170]. Spire1C, a splicing isoform of Spire1, localizes to the OMM and belongs to the TMBP actin nucleator proteins. TMBPs bind the pointed end of the actin filament, leaving the fast-growing barbed end free to associate with formins that stimulate further growth [171]. Thus, the discovery of Spire1C may explain why INF2 induced actin polymerization specifically at ER-mitochondria contact sites despite displaying an even ER membrane staining [169]. In addition, pull-down assays revealed a direct interaction between the soluble domains of Spire1C and INF2. Importantly, inhibition of the Spire1C–INF2 interaction decreases ER–mitochondria overlaps and mitochondrial fission [170]. Spire1C overexpression induces actin polymerization on mitochondria and a significant increase in fission, while its downregulation had the opposite effect, inducing mitochondrial elongation. Further work is needed to uncover the signals that regulate the interaction between Spire1C and INF2.
How does INF2-induced actin polymerization provide the force required for mitochondrial (pre)constriction? Higgs and collaborators recently identified Myosin II as a downstream effector in INF2-dependent mitochondrial division [172]. Myosin II was found at mitochondrial fission sites in an actin- and INF2-dependent manner and its depletion or chemical inhibition decreased mitochondrial fission rates and mitochondria-associated Drp1 levels [135, 172]. These findings support a model in which the INF2-mediated polymerization of antiparallel actin bundles at the ER-mitochondria contact sites leads to recruitment of Myosin II, whose activation by an as yet unidentified signal would promote actin filament pulling, resulting in mitochondrial constriction (Fig. 3a). This process has been termed mitokinesis, in analogy to cytokinesis and to the term mitochondriokinesis introduced by Kuroiwa [173]. Of note, Myosin II has already been described to regulate membrane fission in other cellular pathways such as cytokinesis [174], phagocytosis [175] and trans-Golgi fission [176].

The cytoskeleton contributes to Drp1-dependent mitochondrial fission. a Linear actin polymerized by INF2 and Spire1C at the ER–mitochondria contact sites contributes to mitochondrial constriction with the aid of Myosin II, promoting Drp1 recruitment and mitochondrial fission. b Arp2/3 complex-mediated polymerization of branched actin on the OMM regulates Drp1 dynamics and mitochondrial fission. c Septins interact with Drp1 to regulate Drp1-mediated mitochondrial fission
Recent work identified transient actin structures on the OMM that are not limited to ER–mitochondria contact sites [135] (see also Fig. 3b). Interestingly, impairment of Drp1-dependent fission was associated with an accumulation of F-actin on the OMM, suggesting that actin disassembly requires Drp1 activity. The authors identified Arp2/3-dependent actin polymerization as an additional actin polymerization mode that participates to mitochondrial fission [177]. The Arp2/3 complex [178] differs from formins and TMBPs in that it creates branched actin structures. Intriguingly, depletion of Arp2/3, cofilin and cortactin impaired FCCP-induced fission, while resulting in the accumulation of Drp1 oligomers on highly elongated mitochondria. These oligomers were hypothesized to represent non-functional Drp1 fission complexes, similar to what has been observed upon MiD49/51 overexpression, which leads to mitochondrial elongation with a concomitant accumulation of actin and non-functional Drp1 on the OMM [124].
Given that the Arp2/3 complex is involved in actin polymerization at several subcellular locations [167], an interesting open question is how differential regulation is achieved. Recent data on different models showed that the composition of the Arp2/3 complex is variable; while Arp2 and Arp3 are invariant and essential core subunits, the other subunits (ArpC1–ArpC5) play a regulatory role, and their substitution or posttranslational modification appears to determine localization and activity of the complex (reviewed in [179]). Future work will elucidate the exact composition of the Arp2/3 complex acting in mitochondrial fission, and its associated molecules, including the nucleation promoting factors that activate it.
A detailed description of the F-actin dynamics on the entire mitochondrial network in cultured HeLa cells showed that a wave of transient actin polymerization forms on a subset of mitochondria, rapidly cycling through the entire mitochondrial network and locally coinciding with fission [180]. These cyclic waves of actin-induced mitochondrial fission were proposed to regulate mitochondrial morphology at the level of the whole mitochondrial network. Actin appeared to assemble preferentially on elongated mitochondria, promoting their fragmentation, then promptly disassembled to allow refusion. Drug inhibition experiments showed that this process depends on Arp2/3 and formins, confirming the involvement of both actin polymerizing factors in actin-driven mitochondrial fission. Indeed, INF2/Spire1C- and Arp2/3-mediated actin polymerization may be to some extent functionally redundant, allowing Arp2/3-mediated actin polymerization to take over in cells that express low levels of INF2, such as epithelial or immune cells [181]. This would implicate that according to the tissue, different types of actin structures (linear versus branched) can fulfil a similar function in mitochondrial division. We currently lack a clear view of the actin filament structure during mitochondrial fission, a comprehensive list of the associated components, as well as the mechanisms that stimulate it beyond uncouplers [163, 177] and increases in intracellular calcium [135, 182].
Targeted actin polymerization participates in membrane fission in a number of different processes, such as endocytosis [183] and endosome fission [184], where it might act in conjunction with the ER [151]. An intriguing parallel can also be drawn with the intracellular bacterium Listeria monocytogenes, which initiates actin polymerization at the bacterial surface through the surface protein ActA [185]. This bacterial-induced local actin polymerization has been shown to compensate for a fission defect caused by deletion of a bacterial murein hydrolase [186] and also to regulate the timing of the division cycle of wild-type Listeria inside cells [187]. Whether the mechanisms by which actin stimulates this plethora of fission processes are the same or not remains an open question.
Septins
In contrast to actin, septins have been implicated in mitochondrial fission only very recently. Septins are a family of GTP-binding proteins discovered more than 40 years ago as regulators of cytokinesis in yeast [188, 189]. In humans there are 13 septin-coding genes, with many of them being expressed in a tissue-specific pattern. Now fully accepted as components of the cytoskeleton, septins can heterooligomerize into non-polar filaments, which can then assemble into higher order structures such as rings, bundles and gauzes. At present septins have been involved in diverse cellular processes such as cytokinesis, ciliogenesis, axon guidance and endocytosis and are thought to restrict protein diffusion at membranous structures such as the cell cortex, the yeast bud neck, and the ER, as well as restricting bacterial actin-based motility [188, 189].
A recent study highlighted a new role for septins as regulators of Drp1-dependent mitochondrial fission [190] (Fig. 3c). Sept2 was found to interact directly with Drp1, and depletion of Sept2 or Sept7 increased mitochondrial length. Sept2 depletion also delayed FCCP-induced mitochondrial fission and looping. About 30% of the mitochondrial constrictions appeared to be positive for Sept2, indicating that it might transiently associate with fission sites or be involved in a subset of fission events, as it has been proposed for INF2 and Myosin II [191]. What is the functional role of the Sept2–Drp1 interaction? Imaging coupled to mitochondrial fractionation experiments revealed that Sept2 is important for efficient recruitment of Drp1 to mitochondria, as Sept2-depleted cells displayed less Drp1 on mitochondria, similar to INF2 and Myosin II depletion [166, 172] (Fig. 2c). Septins might facilitate Drp1 recruitment through multiple mechanisms: first, septins might directly promote mitochondrial pre-constriction and Drp1 retention. Second, it is tempting to speculate that septins function as a scaffold at mitochondria and promote functional interactions of Drp1 with other proteins involved in mitochondrial fission, similar to the function that septins have already been shown to fulfil in other contexts, such as cytokinesis [192]. For example, septins might promote posttranslational modifications of Drp1 that allow its retention onto mitochondria. Multiple septins have indeed been shown to interact with the SUMOylation machinery by yeast two-hybrid screening and could, therefore, act as a scaffold to promote Drp1 SUMOylation. Another way septins might promote Drp1 function is by affecting its oligomerization status on mitochondria, similar to actin [135]. In this context, Ji et al. noticed cytosolic Drp1 oligomers, which may be associated with cytoskeletal structures. In agreement with this, we observed septin-associated oligomeric Drp1 structures in the cytosol (our unpublished observation), while Strack et al. found microtubule association of a specific Drp1 isoform, Drp1-x01 [66]. Different cytoskeletal structures may hence act as a cytosolic reservoir for delivering Drp1 oligomers to mitochondria.
Dynamin performs the terminal abscission step
In vitro studies have shown that Drp1 is able to induce liposome tubulation and branching [60], but no fission has been observed in contrast to what has been observed with the endocytic Dynamin 1, which tubulates and severs liposomes in a GTP-dependent fashion [193]. Lee et al. have recently solved this conundrum by showing that the ubiquitous endocytic Dynamin 2 (Dyn2) terminates mitochondrial abscission downstream of Drp1 [194] (Fig. 4). Dyn2 required both its GTPase activity, as well as its PH and polyproline lipid-binding domains to mediate mitochondrial fission, although the structural arrangement of these domains with respect to the OMM remains unclear. Strikingly, Dyn2 mutations have been found associated with Charcot–Marie–Tooth type 2 disease and a recent case report showed that the Dyn2 R369W mutation induced multiple mtDNA deletions [195], reinforcing the link between mitochondrial dynamics and mtDNA maintenance. Interestingly, peroxisome fission has also been shown to require a cooperation between the mitochondrial dynamin Dnm1p and the endocytic dynamin Vps1p in yeast [196]. In mammalian cells, the ER, Arp2/3, the actomyosin system and Dyn2 [184] (Fig. 4) are all implicated in both endosomal and mitochondrial fission, raising the question as to how specific targeting is achieved [151, 184, 196, 197].

Mitochondrial fission is a multistep process. a The site of fission on mitochondria is first marked by an ER tubule which constricts mitochondria by wrapping around them. b Drp1 is recruited to the OMM with the aid of receptors, actin and septins, oligomerizes and initiates mitochondrial constriction. c Dynamin is recruited through an unknown mechanism and d drives mitochondrial abscission upon GTP hydrolysis
From an evolutionary perspective, it is interesting to note that an implication for endocytic dynamins in mitochondrial fission has been also shown in eukaryotes as distinct as the red algae Cyanidioschyzon merolae and the parasite Trypanosoma brucei [198, 199]. Consistent with the partnership between Dyn2 and Drp1 uncovered by Lee et al., a recent study proposed that the evolutionary ancestor of endocytic dynamins and Drp1 mediated both mitochondrial and vesicle abscission [53].
In contrast to Drp1, Dyn2 seems to associate only very briefly with mitochondrial division sites and segregates asymmetrically, suggesting important differences in the assembly and disassembly kinetics of Drp1 and Dyn2 [194]. Like endocytic dynamins, Drp1 disassembly requires GTP hydrolysis [60] and in addition possibly calcium [194], but although advances have been made by structure–function studies [200, 201], relatively little is known concerning the regulation of this essential step of the fission cycle.
Drp1-independent fission
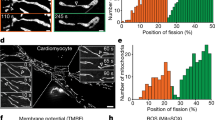
The canonical mitochondrial fission process occurs through Drp1 (Fig. 3); however, accumulating evidence suggests the presence of Drp1-independent fission mechanisms. For instance, during L. monocytogenes infection of epithelial cells, the mitochondrial network undergoes a dramatic fragmentation with concomitant impairment of the respiration capacity [182]. Surprisingly, Drp1 oligomers are absent from fragmented mitochondria (Fig. 1c). Analysis of this phenomenon in Drp1-depleted cells as well as in Drp1KO MEF revealed that L. monocytogenes-induced mitochondrial fragmentation is Drp1 independent [202]. Another example is provided by the abscission of mitochondria-derived vesicles (MDVs) [203, 204]. MDVs are induced by oxidative stress in a Parkin and Pink1-dependent manner, bud off mitochondria in a Drp1-independent manner, and then deliver their content to the lysosome to allow the selective removal of damaged proteins from the OMM [204].
Recently, mitophagy was reported to also occur in the absence of Drp1 [48]. Through live cell imaging experiments, the authors showed that upon induction of mitophagy, a mitochondrial bud emerges and is progressively isolated from the parental mitochondrion to finally detach by a still unidentified mechanism, which does not rely on Drp1. It is interesting to note that while Drp1 is required for embryonic development, it appears to be dispensable for the viability of in vitro-cultured cells [35]. This further suggests the existence of Drp1-independent mitochondrial fission mechanisms with a basal activity, which can be boosted under specific conditions such as oxidative stress, mitophagy induction or Listeria infection.
Open questions
Although the appearance of the mitochondrial fission machinery has been postulated to precede that of fusion in evolution [173], no conserved machinery has been identified that would mediate fission of the inner mitochondrial membrane. A bacterial-derived system which induces fission from the matrix through an FtsZ homolog was detected across multiple eukaryotic species, suggesting that it was an ancestral mode of division [54], but little is known in mammalian cells. Because mammals lack a recognizable homolog of the bacterial fission protein FtsZ [53, 54] and the appearance of the organelle-dedicated dynamin Drp1 through gene duplication was found to coincide with the loss of FtsZ [53], Drp1 is currently thought to simultaneously sever both the OMM and the IMM. In agreement with this hypothesis, activation of Drp1 results in a fragmented mitochondrial network. But matrix constrictions have been observed in the absence of functional Drp1 in mammalian cells, suggesting the presence of an inner membrane fission machinery [205] which would respond to uncouplers or other stimuli that induce fission by acting directly on mitochondria. A few candidates for IMM fission have been proposed, including the inner membrane proteins TMEM11 [206] and MTGM/Romo1 [207], intermembrane space proteins as MTP18 [208] and the short, soluble form of OPA1 [209]. While overexpression of these candidates induced mitochondrial fission, their depletion did not always yield the hyperfusion phenotype that would be intuitively expected. We anticipate that the model for mitochondrial fission will be further refined by integrating the characterization of new players with the advent of new imaging techniques that allow high-resolution imaging of mitochondria with limited photodamage [135, 210].
References
Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148:1145–1159
Kuznetsov AV, Hermann M, Saks V, Hengster P, Margreiter R (2009) The cell-type specificity of mitochondrial dynamics. Int J Biochem Cell Biol 41:1928–1939
Barnhart EL (2016) Mechanics of mitochondrial motility in neurons. Curr Opin Cell Biol 38:90–99
Peraza-Reyes L, Crider DG, Pon LA (2010) Mitochondrial manoeuvres: latest insights and hypotheses on mitochondrial partitioning during mitosis in Saccharomyces cerevisiae. BioEssays 32:1040–1049
Chen H, Chomyn A, Chan DC (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280:26185–26192
Yang WY (2015) Optogenetic probing of mitochondrial damage responses. Ann N Y Acad Sci 1350:48–51
Wai T, Langer T (2016) Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab 27:105–117
Collins TJ, Berridge MJ, Lipp P, Bootman MD (2002) Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J 21:1616–1627
Silva Ramos E, Larsson NG, Mourier A (2016) Bioenergetic roles of mitochondrial fusion. Biochem Biophys Acta 1857:1277–1283
Santel A, Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114:867–874
Rojo M, Legros F, Chateau D, Lombes A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115:1663–1674
Hoppins S, Nunnari J (2009) The molecular mechanism of mitochondrial fusion. Biochem Biophys Acta 1793:20–26
Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW II (2016) Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540:74–79
Wang C, Du W, Su QP, Zhu M, Feng P, Li Y, Zhou Y, Mi N, Zhu Y, Jiang D, Zhang S, Zhang Z, Sun Y, Yu L (2015) Dynamic tubulation of mitochondria drives mitochondrial network formation. Cell Res 25:1108–1120
Qi Y, Yan L, Yu C, Guo X, Zhou X, Hu X, Huang X, Rao Z, Lou Z, Hu J (2016) Structures of human mitofusin 1 provide insight into mitochondrial tethering. J Cell Biol 215:621–629
Schrepfer E, Scorrano L (2016) Mitofusins, from Mitochondria to Metabolism. Mol Cell 61:683–694
Ishihara N, Eura Y, Mihara K (2004) Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117:6535–6546
Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnaune-Pelloquin L, Davezac N, Mils V, Miquel MC, Rojo M, Belenguer P (2016) Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Disease 90:3–19
Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36:449–451
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200
Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141:280–289
Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101:15927–15932
Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernandez-Alvarez MI, Zorzano A, De Stefani D, Dorn GW 2nd, Scorrano L (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA 113:11249–11254
Giacomello M, Pellegrini L (2016) The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ 23:1417–1427
Phillips MJ, Voeltz GK (2016) Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17:69–82
Meeusen S, McCaffery JM, Nunnari J (2004) Mitochondrial fusion intermediates revealed in vitro. Science 305:1747–1752
Malka F, Guillery O, Cifuentes-Diaz C, Guillou E, Belenguer P, Lombes A, Rojo M (2005) Separate fusion of outer and inner mitochondrial membranes. EMBO Rep 6:853–859
Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26:211–215
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26:207–210
Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20:3525–3532
Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126:177–189
Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, Lenaers G, Belenguer P, Hamel CP (2001) Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet 109:584–591
MacVicar T, Langer T (2016) OPA1 processing in cell death and disease—the long and short of it. J Cell Sci 129:2297–2306
Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T (2015) Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350:aad0116
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11:958–966
Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H (2009) The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186:805–816
Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H (2012) Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol 197:535–551
Horbay R, Bilyy R (2016) Mitochondrial dynamics during cell cycling. Apoptosis 21:1327–1335
Schwarz TL (2013) Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446
Narendra D, Tanaka A, Suen DF, Youle RJ (2009) Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 5:706–708
Wei H, Liu L, Chen Q (2015) Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochem Biophys Acta 1853:2784–2790
Anding AL, Baehrecke EH (2017) Cleaning house: selective autophagy of organelles. Dev Cell 41:10–22
Buhlman L, Damiano M, Bertolin G, Ferrando-Miguel R, Lombes A, Brice A, Corti O (2014) Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochem Biophys Acta 1843:2012–2026
Rambold AS, Lippincott-Schwartz J (2011) Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 10:4032–4038
Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33:2798–2813
Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21:273–285
Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, Saigusa T, Yoshimori T, Sakai Y, Mihara K, Kanki T (2016) Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol 215:649–665
Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1:515–525
Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC (2006) Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol 26:7397–7408
Estaquier J, Arnoult D (2007) Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 14:1086–1094
Purkanti R, Thattai M (2015) Ancient dynamin segments capture early stages of host-mitochondrial integration. Proc Natl Acad Sci USA 112:2800–2805
Leger MM, Petru M, Zarsky V, Eme L, Vlcek C, Harding T, Lang BF, Elias M, Dolezal P, Roger AJ (2015) An ancestral bacterial division system is widespread in eukaryotic mitochondria. Proc Natl Acad Sci USA 112:10239–10246
Shin HW, Shinotsuka C, Torii S, Murakami K, Nakayama K (1997) Identification and subcellular localization of a novel mammalian dynamin-related protein homologous to yeast Vps1p and Dnm1p. J Biochem 122:525–530
Imoto M, Tachibana I, Urrutia R (1998) Identification and functional characterization of a novel human protein highly related to the yeast dynamin-like GTPase Vps1p. J Cell Sci 111(Pt 10):1341–1349
Kamimoto T, Nagai Y, Onogi H, Muro Y, Wakabayashi T, Hagiwara M (1998) Dymple, a novel dynamin-like high molecular weight GTPase lacking a proline-rich carboxyl-terminal domain in mammalian cells. J Biol Chem 273:1044–1051
Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143:351–358
Yoon Y, Pitts KR, Dahan S, McNiven MA (1998) A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J Cell Biol 140:779–793
Yoon Y, Pitts KR, McNiven MA (2001) Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell 12:2894–2905
Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE (2011) Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol 18:20–26
Francy CA, Alvarez FJ, Zhou L, Ramachandran R, Mears JA (2015) The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. J Biol Chem 290:11692–11703
Howng SL, Sy WD, Cheng TS, Lieu AS, Wang C, Tzou WS, Cho CL, Hong YR (2004) Genomic organization, alternative splicing, and promoter analysis of human dynamin-like protein gene. Biochem Biophys Res Commun 314:766–772
Uo T, Dworzak J, Kinoshita C, Inman DM, Kinoshita Y, Horner PJ, Morrison RS (2009) Drp1 levels constitutively regulate mitochondrial dynamics and cell survival in cortical neurons. Exp Neurol 218:274–285
Macdonald PJ, Francy CA, Stepanyants N, Lehman L, Baglio A, Mears JA, Qi X, Ramachandran R (2016) Distinct splice variants of dynamin-related protein 1 differentially utilize mitochondrial fission factor as an effector of cooperative GTPase activity. J Biol Chem 291:493–507
Strack S, Wilson TJ, Cribbs JT (2013) Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J Cell Biol 201:1037–1051
Frohlich C, Grabiger S, Schwefel D, Faelber K, Rosenbaum E, Mears J, Rocks O, Daumke O (2013) Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J 32:1280–1292
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
Li H, Alavian KN, Lazrove E, Mehta N, Jones A, Zhang P, Licznerski P, Graham M, Uo T, Guo J, Rahner C, Duman RS, Morrison RS, Jonas EA (2013) A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat Cell Biol 15:773–785
Frohman MA (2015) Role of mitochondrial lipids in guiding fission and fusion. J Mol Med 93:263–269
Kooijman EE, Chupin V, de Kruijff B, Burger KN (2003) Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic 4:162–174
Tatsuta T, Langer T (2017) Intramitochondrial phospholipid trafficking. Biochem Biophys Acta 1862:81–89
Ardail D, Lerme F, Louisot P (1990) Further characterization of mitochondrial contact sites: effect of short-chain alcohols on membrane fluidity and activity. Biochem Biophys Res Commun 173:878–885
Schlattner U, Tokarska-Schlattner M, Rousseau D, Boissan M, Mannella C, Epand R, Lacombe ML (2014) Mitochondrial cardiolipin/phospholipid trafficking: the role of membrane contact site complexes and lipid transfer proteins. Chem Phys Lipid 179:32–41
Bustillo-Zabalbeitia I, Montessuit S, Raemy E, Basanez G, Terrones O, Martinou JC (2014) Specific interaction with cardiolipin triggers functional activation of dynamin-related protein 1. PLoS One 9:e102738
Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P, Martinou JC (2010) Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142:889–901
Macdonald PJ, Stepanyants N, Mehrotra N, Mears JA, Qi X, Sesaki H, Ramachandran R (2014) A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol Biol Cell 25:1905–1915
Stepanyants N, Macdonald PJ, Francy CA, Mears JA, Qi X, Ramachandran R (2015) Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell 26:3104–3116
Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, Frohman MA, Ramachandran R, Iijima M, Sesaki H (2016) Coincident phosphatidic acid interaction restrains Drp1 in mitochondrial division. Mol Cell 63:1034–1043
Pitts KR, Yoon Y, Krueger EW, McNiven MA (1999) The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol Biol Cell 10:4403–4417
Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4:815–826
Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M (2003) Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem 278:8597–8605
Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, Yang CS, Tang W, An J, Ilkayeva OR, Rathmell JC, Newgard CB, Kornbluth S (2011) Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell 22:1207–1216
Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 57:537–551
Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, Chipuk JE (2015) Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell 57:521–536
Cho B, Cho HM, Kim HJ, Jeong J, Park SK, Hwang EM, Park JY, Kim WR, Kim H, Sun W (2014) CDK5-dependent inhibitory phosphorylation of Drp1 during neuronal maturation. Exp Mol Med 46:e105
Jahani-Asl A, Huang E, Irrcher I, Rashidian J, Ishihara N, Lagace DC, Slack RS, Park DS (2015) CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum Mol Genet 24:4573–4583
Chang CR, Manlandro CM, Arnoult D, Stadler J, Posey AE, Hill RB, Blackstone C (2010) A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J Biol Chem 285:32494–32503
Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8:939–944
Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13:589–598
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105:15803–15808
Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24:659–667
Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q (2011) Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286:11649–11658
Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura-Hoshino M, Sada K, Hotta H, Yamamura H, Inatome R, Yanagi S (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J 25:3618–3626
Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S (2006) MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep 7:1019–1022
Karbowski M, Neutzner A, Youle RJ (2007) The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol 178:71–84
Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H (2010) Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci 123:619–626
Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, Inatome R, Yanagi S (2013) MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell 51:20–34
Park YY, Nguyen OT, Kang H, Cho H (2014) MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Disease 5:e1172
Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C, Karbowski M (2016) Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell 27:349–359
Cherok E, Xu S, Li S, Das S, Meltzer WA, Zalzman M, Wang C, Karbowski M (2016) Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol Biol Cell 28(3):396–410
Geiss-Friedlander R, Melchior F (2007) Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 8:947–956
Harder Z, Zunino R, McBride H (2004) Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14:340–345
Wasiak S, Zunino R, McBride HM (2007) Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177:439–450
Braschi E, Zunino R, McBride HM (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10:748–754
Figueroa-Romero C, Iniguez-Lluhi JA, Stadler J, Chang CR, Arnoult D, Keller PJ, Hong Y, Blackstone C, Feldman EL (2009) SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J 23:3917–3927
Zunino R, Schauss A, Rippstein P, Andrade-Navarro M, McBride HM (2007) The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci 120:1178–1188
Zunino R, Braschi E, Xu L, McBride HM (2009) Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem 284:17783–17795
Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H (2013) Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem 288:26473–26479
Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E (2006) Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 25:3900–3911
Yuan H, Gerencser AA, Liot G, Lipton SA, Ellisman M, Perkins GA, Bossy-Wetzel E (2007) Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ 14:462–471
Bossy B, Petrilli A, Klinglmayr E, Chen J, Lutz-Meindl U, Knott AB, Masliah E, Schwarzenbacher R, Bossy-Wetzel E (2010) S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer’s disease. J Alzheimer’s Disease 20(Suppl 2):S513–S526
Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates JR 3rd, Hoshijima M, Dillmann W (2012) Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287:30024–30034
Sacoman JL, Dagda RY, Burnham-Marusich AR, Dagda RK, Berninsone PM (2017) Mitochondrial O-GlcNAc transferase (mOGT) regulates mitochondrial structure, function and survival in HeLa cells. J Biol Chem 292:4499–4518
Marshall S, Bacote V, Traxinger RR (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem 266:4706–4712
Fekkes P, Shepard KA, Yaffe MP (2000) Gag3p, an outer membrane protein required for fission of mitochondrial tubules. J Cell Biol 151:333–340
Mozdy AD, McCaffery JM, Shaw JM (2000) Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol 151:367–380
Tieu Q, Nunnari J (2000) Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol 151:353–366
Griffin EE, Graumann J, Chan DC (2005) The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol 170:237–248
Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23:5409–5420
James DI, Parone PA, Mattenberger Y, Martinou JC (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278:36373–36379
Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT (2004) Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci 117:1201–1210
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191:1141–1158
Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT (2011) MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12:565–573
Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT (2016) Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci 129:2170–2181
Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S (2011) Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 30:556–568
Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JT, Wang C, Cho JH, Hattori N, Youle RJ, van der Bliek AM (2014) Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell 25:145–159
Rojansky R, Cha MY, Chan DC (2016) Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife 5
Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ (2014) Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3:e01612
Gandre-Babbe S, van der Bliek AM (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19:2402–2412
Clinton RW, Francy CA, Ramachandran R, Qi X, Mears JA (2016) Dynamin-related protein 1 oligomerization in solution impairs functional interactions with membrane-anchored mitochondrial fission factor. J Biol Chem 291:478–492
Otera H, Miyata N, Kuge O, Mihara K (2016) Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol 212:531–544
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334:358–362
Strack S, Cribbs JT (2012) Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J Biol Chem 287:10990–11001
Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN (2015) Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 4:e11553
Loson OC, Meng S, Ngo H, Liu R, Kaiser JT, Chan DC (2015) Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein Sci 24:386–394
Loson OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, Chan DC (2014) The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 22:367–377
Richter V, Singh AP, Kvansakul M, Ryan MT, Osellame LD (2015) Splitting up the powerhouse: structural insights into the mechanism of mitochondrial fission. Cell Mol Life Sci 72:3695–3707
Elgass KD, Smith EA, LeGros MA, Larabell CA, Ryan MT (2015) Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J Cell Sci 128:2795–2804
Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT (2013) Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem 288:27584–27593
Schmidt O, Pfanner N, Meisinger C (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11:655–667
Liu S, Gao Y, Zhang C, Li H, Pan S, Wang X, Du S, Deng Z, Wang L, Song Z, Chen S (2016) SAMM50 affects mitochondrial morphology through the association of Drp1 in mammalian cells. FEBS Lett 590:1313–1323
Copeland DE, Dalton AJ (1959) An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol 5:393–396
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R (2006) Mitochondrial dynamics and Ca2+ signaling. Biochem Biophys Acta 1763:442–449
Bui M, Gilady SY, Fitzsimmons RE, Benson MD, Lynes EM, Gesson K, Alto NM, Strack S, Scott JD, Simmen T (2010) Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J Biol Chem 285:31590–31602
Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN (2013) Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 110:12526–12534
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24:717–729
Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, Kudo Y, Baba M, Baba N, Cheng J, Fujimoto T, Ishihara N, Ortiz-Sandoval C, Barlow LD, Raturi A, Dohmae N, Wakana Y, Inoue H, Tani K, Dacks JB, Simmen T, Tagaya M (2015) A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell 32:304–317
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766
Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK (2014) ER contact sites define the position and timing of endosome fission. Cell 159:1027–1041
Roux A, Koster G, Lenz M, Sorre B, Manneville JB, Nassoy P, Bassereau P (2010) Membrane curvature controls dynamin polymerization. Proc Natl Acad Sci USA 107:4141–4146
Sawant P, Eissenberger K, Karier L, Mascher T, Bramkamp M (2016) A dynamin-like protein involved in bacterial cell membrane surveillance under environmental stress. Environ Microbiol 18:2705–2720
Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM (2013) Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci USA 110:E1342–E1351
Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C (2001) Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet 28:223–231
Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA (2002) Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion 1:425–435
Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN (2003) Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell 14:1583–1596
Ban-Ishihara R, Ishihara T, Sasaki N, Mihara K, Ishihara N (2013) Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc Natl Acad Sci USA 110:11863–11868
Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, Barja F, Martinou JC (2008) Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One 3:e3257
Ishihara T, Ban-Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, Aoki H, Katada S, Nakada K, Nomura M, Mizushima N, Mihara K, Ishihara N (2015) Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol 35:211–223
Lewis SC, Uchiyama LF, Nunnari J (2016) ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353:aaf5549
Mattenberger Y, James DI, Martinou JC (2003) Fusion of mitochondria in mammalian cells is dependent on the mitochondrial inner membrane potential and independent of microtubules or actin. FEBS Lett 538:53–59
De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP (2005) Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr Biol 15:678–683
DuBoff B, Gotz J, Feany MB (2012) Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75:618–632
Hatch AL, Ji WK, Merrill RA, Strack S, Higgs HN (2016) Actin filaments as dynamic reservoirs for Drp1 recruitment. Mol Biol Cell 27:3109–3121
Korobova F, Ramabhadran V, Higgs HN (2013) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339:464–467
Skau CT, Waterman CM (2015) Specification of Architecture and Function of Actin Structures by Actin Nucleation Factors. Ann Rev Biophys 44:285–310
Boyer O, Nevo F, Plaisier E, Funalot B, Gribouval O, Benoit G, Huynh Cong E, Arrondel C, Tete MJ, Montjean R, Richard L, Karras A, Pouteil-Noble C, Balafrej L, Bonnardeaux A, Canaud G, Charasse C, Dantal J, Deschenes G, Deteix P, Dubourg O, Petiot P, Pouthier D, Leguern E, Guiochon-Mantel A, Broutin I, Gubler MC, Saunier S, Ronco P, Vallat JM, Alonso MA, Antignac C, Mollet G (2011) INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med 365:2377–2388
Chhabra ES, Ramabhadran V, Gerber SA, Higgs HN (2009) INF2 is an endoplasmic reticulum-associated formin protein. J Cell Sci 122:1430–1440
Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, Spudich J, Lippincott-Schwartz J (2015) A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 4:e08828
Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK (2005) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433:488–494
Korobova F, Gauvin TJ, Higgs HN (2014) A role for myosin II in mammalian mitochondrial fission. Curr Biol 24:409–414
Kuroiwa T, Nishida K, Yoshida Y, Fujiwara T, Mori T, Kuroiwa H, Misumi O (2006) Structure, function and evolution of the mitochondrial division apparatus. Biochem Biophys Acta 1763:510–521
Yumura S (2001) Myosin II dynamics and cortical flow during contractile ring formation in Dictyostelium cells. J Cell Biol 154:137–146
Araki N, Hatae T, Furukawa A, Swanson JA (2003) Phosphoinositide-3-kinase-independent contractile activities associated with Fc gamma-receptor-mediated phagocytosis and macropinocytosis in macrophages. J Cell Sci 116:247–257
Miserey-Lenkei S, Chalancon G, Bardin S, Formstecher E, Goud B, Echard A (2010) Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex. Nat Cell Biol 12:645–654
Li S, Xu S, Roelofs BA, Boyman L, Lederer WJ, Sesaki H, Karbowski M (2015) Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J Cell Biol 208:109–123
Welch MD, Iwamatsu A, Mitchison TJ (1997) Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385:265–269
Pizarro-Cerda J, Charbit A, Enninga J, Lafont F, Cossart P (2016) Manipulation of host membranes by the bacterial pathogens Listeria, Francisella, Shigella and Yersinia. Semin Cell Dev Biol 60:155–167
Moore AS, Wong YC, Simpson CL, Holzbaur EL (2016) Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat Commun 7:12886
Ramabhadran V, Korobova F, Rahme GJ, Higgs HN (2011) Splice variant-specific cellular function of the formin INF2 in maintenance of Golgi architecture. Mol Biol Cell 22:4822–4833
Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P (2011) Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci USA 108:3612–3617
Grassart A, Cheng AT, Hong SH, Zhang F, Zenzer N, Feng Y, Briner DM, Davis GD, Malkov D, Drubin DG (2014) Actin and dynamin2 dynamics and interplay during clathrin-mediated endocytosis. J Cell Biol 205:721–735
Gautreau A, Oguievetskaia K, Ungermann C (2014) Function and regulation of the endosomal fusion and fission machineries. Cold Spring Harb perspect Biol 6
Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P (1992) L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531
Alonzo F 3rd, McMullen PD, Freitag NE (2011) Actin polymerization drives septation of Listeria monocytogenes namA hydrolase mutants, demonstrating host correction of a bacterial defect. Infect Immun 79:1458–1470
Siegrist MS, Aditham AK, Espaillat A, Cameron TA, Whiteside SA, Cava F, Portnoy DA, Bertozzi CR (2015) Host actin polymerization tunes the cell division cycle of an intracellular pathogen. Cell Rep 11:499–507
Fung KY, Dai L, Trimble WS (2014) Cell and molecular biology of septins. Int Rev Cell Mol Biol 310:289–339
Mostowy S, Cossart P (2012) Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol 13:183–194
Pagliuso A, Tham TN, Stevens JK, Lagache T, Persson R, Salles A, Olivo-Marin JC, Oddos S, Spang A, Cossart P, Stavru F (2016) A role for septin 2 in Drp1-mediated mitochondrial fission. EMBO Rep 17:858–873
Hatch AL, Gurel PS, Higgs HN (2014) Novel roles for actin in mitochondrial fission. J Cell Sci 127:4549–4560
Joo E, Surka MC, Trimble WS (2007) Mammalian SEPT2 is required for scaffolding nonmuscle myosin II and its kinases. Dev Cell 13:677–690
Sweitzer SM, Hinshaw JE (1998) Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell 93:1021–1029
Lee JE, Westrate LM, Wu H, Page C, Voeltz GK (2016) Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540:139–143
Gal A, Inczedy-Farkas G, Pal E, Remenyi V, Bereznai B, Geller L, Szelid Z, Merkely B, Molnar MJ (2015) The coexistence of dynamin 2 mutation and multiple mitochondrial DNA (mtDNA) deletions in the background of severe cardiomyopathy and centronuclear myopathy. Clin Neuropathol 34:89–95
van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL (1993) Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol 122:553–563
Gormal RS, Nguyen TH, Martin S, Papadopulos A, Meunier FA (2015) An acto-myosin II constricting ring initiates the fission of activity-dependent bulk endosomes in neurosecretory cells. J Neurosci 35:1380–1389
Nishida K, Takahara M, Miyagishima SY, Kuroiwa H, Matsuzaki M, Kuroiwa T (2003) Dynamic recruitment of dynamin for final mitochondrial severance in a primitive red alga. Proc Natl Acad Sci USA 100:2146–2151
Chanez AL, Hehl AB, Engstler M, Schneider A (2006) Ablation of the single dynamin of T. brucei blocks mitochondrial fission and endocytosis and leads to a precise cytokinesis arrest. J Cell Sci 119:2968–2974
Mattila JP, Shnyrova AV, Sundborger AC, Hortelano ER, Fuhrmans M, Neumann S, Muller M, Hinshaw JE, Schmid SL, Frolov VA (2015) A hemi-fission intermediate links two mechanistically distinct stages of membrane fission. Nature 524:109–113
Cahill TJ, Leo V, Kelly M, Stockenhuber A, Kennedy NW, Bao L, Cereghetti G, Harper AR, Czibik G, Lao C, Bellahcene M, Steeples V, Ghaffari S, Yavari A, Mayer A, Poulton J, Ferguson DJ, Scorrano L, Hettiarachchi NT, Peers C, Boyle J, Hill RB, Simmons A, Watkins H, Dear TN, Ashrafian H (2015) Resistance of dynamin-related protein 1 oligomers to disassembly impairs mitophagy. Resulting in myocardial inflammation and heart failure. J Biol Chem 290:25907–25919
Stavru F, Palmer AE, Wang C, Youle RJ, Cossart P (2013) Atypical mitochondrial fission upon bacterial infection. Proc Natl Acad Sci USA 110:16003–16008
Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM (2012) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22:135–141
McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33:282–295
Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, Deutsch M, Zurgil N, Reynolds N, Shirihai OS (2006) Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am J Physiol Cell Physiol 291:C176–C184
Rival T, Macchi M, Arnaune-Pelloquin L, Poidevin M, Maillet F, Richard F, Fatmi A, Belenguer P, Royet J (2011) Inner-membrane proteins PMI/TMEM11 regulate mitochondrial morphogenesis independently of the DRP1/MFN fission/fusion pathways. EMBO Rep 12:223–230
Zhao J, Liu T, Jin SB, Tomilin N, Castro J, Shupliakov O, Lendahl U, Nister M (2009) The novel conserved mitochondrial inner-membrane protein MTGM regulates mitochondrial morphology and cell proliferation. J Cell Sci 122:2252–2262
Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A (2005) The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci 118:3049–3059
Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204:919–929
Lo CY, Chen S, Creed SJ, Kang M, Zhao N, Tang BZ, Elgass KD (2016) Novel super-resolution capable mitochondrial probe, MitoRed AIE, enables assessment of real-time molecular mitochondrial dynamics. Sci Rep 6:30855
Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M (2008) CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182:573–585
Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM (2017) The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex i inhibitor that modulates reactive oxygen species. Dev Cell 40(583–594):e6
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204
Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J 28:1589–1600
Acknowledgements
We apologize to those colleagues whose work we could not cite due to space constraints. Work in the Cossart laboratory is funded by Institut Pasteur, Inserm (U604), ERC (Grant 670823–BacCellEpi).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pagliuso, A., Cossart, P. & Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 75, 355–374 (2018). https://doi.org/10.1007/s00018-017-2603-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2603-0