
Abstract
The endothelium, a tissue that forms a single layer of cells lining various organs and cavities of the body, especially the heart and blood as well as lymphatic vessels, plays a complex role in vascular biology. It contributes to key aspects of vascular homeostasis and is also involved in pathophysiological processes, such as thrombosis, inflammation, and hypertension. Epidemiological data show that high doses of ionizing radiation lead to cardiovascular disease over time. The aim of this review is to summarize the current knowledge on endothelial cell activation and dysfunction after ionizing radiation exposure as a central feature preceding the development of cardiovascular diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For many years after its discovery in the 1800s, the vascular endothelium was believed to be a mere inert, semipermeable barrier between circulating blood and underlying subendothelial tissues. Numerous subsequent studies have led to the current view of the endothelium as a dynamic heterogeneous and distributed organ with essential secretory, synthetic, metabolic, and immunologic functions [1]. In the presence of irritant stimuli, such as dyslipidemia [2, 3], hypertension [4,5,6,7], and pro-inflammatory agents [8,9,10,11], the normal physiological functions of the arterial endothelium are adversely affected [12, 13], starting a chain of molecular changes that leads to atherosclerosis and cardiovascular diseases (CVDs), including coronary artery disease, carotid artery disease, peripheral artery disease, and ischemic stroke [14,15,16].
When cells are exposed to ionizing radiation, they undergo a stress response within less than a microsecond after the hit [17]. This response is initiated by the interaction of ionizing radiation with biological matter, causing damage by interacting directly or indirectly through the formation of reactive oxygen species (ROS) with cellular biomolecules such as DNA, proteins, and lipids. This reaction interferes with all cellular organelles and has the ability to affect their molecular mechanisms. As a result, endothelial activation occurs, causing the quiescent phenotype to switch towards a pro-inflammatory one [18,19,20]. When exposure is prolonged and/or repeated, it can exhaust the protective physiological effect of the endothelium, leading to endothelial dysfunction [21]. This pathological state can thus be seen as a maladaptive response to pathological stimuli and refers to a failure of the endothelium to perform its normal, physiologic functions [22]. As a result, deterioration of the vascular tone, blood hemostasis problems, inflammation, and edema occurs at the site of the affected endothelium [23]. Because the endothelium is a key integrator of vascular risk, pathogenic signals, including ionizing radiation, may converge to produce several pathological conditions [22], atherosclerosis as typified example [24]. Atherosclerosis itself perpetrates vascular damage, resulting in radiation-induced heart disease [25, 26]. According to the current consensus, the term “low dose” is defined as a dose of 0.1 Gy or less throughout this review [27, 28]. The terms “moderate dose” and “high dose” are, respectively, defined in this review as doses between 0.1 Gy and 2 Gy, and equal or higher than 2 Gy. It has been shown that high doses of ionizing radiation-induced cardiovascular diseases in atomic bomb survivors [29] and cancer therapy patients [30].
In the sections below, we summarize current knowledge on the effects of ionizing radiation exposure on the different aspects of endothelial activation and dysfunction. Progress in the knowledge of endothelial pathophysiology has mainly been a consequence of investigations performed with endothelial cells in culture (Table 1) [31], human umbilical vein endothelial cells (HUVECs) being the most commonly used [32]. To standardize experimental conditions, immortalized, well-characterized endothelial cell lines were developed, of which EA.hy926 is the most frequently used [33]. Additional models of endothelial pathophysiology encompass in vitro co-culture and 3D models that mimic in vivo complexity [34,35,36,37,38]. Ex vivo explanted blood vessel segments from animal (commonly used are canine, bovine, porcine, rat and mouse) or human origin are also used, mainly in the field of vascular tone research [39]. Finally, a number of animal models have been used for understanding the mechanisms involved in cardiovascular disease development as they can replicate complex cell–cell and cell–matrix interactions. The most common animal model being the ApoE−/− mice [40, 41]. However, it is difficult to draw general conclusions, because the current literature often describes different endothelial cell models, timepoints, and radiation doses.
Endothelial activation: a pro-inflammatory state
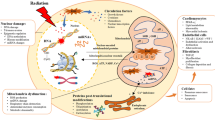
Endothelial cell activation can be defined by the manifestation of a pro-inflammatory phenotype characterized by the expression of chemokines, cytokines, and adhesion molecules that facilitate the recruitment and attachment of circulating leukocytes on the vascular wall [21]. Endothelial cells are typically activated by pro-inflammatory cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-6, released by immune cells upon contact with pathogens [42]. After ionizing radiation exposure, however, endothelial cell activation occurs in a sterile environment without the presence of pathogens, i.e., sterile inflammation (Fig. 1). The prime cause of sterile inflammation following ionizing radiation exposure is activation of the genotoxic stress-induced nuclear factor (NF)-κB pathway, recently reviewed by Hellweg [43]. NF-κB is a heterodimeric transcription factor that is normally sequestered in the cytoplasm as an inactive complex with inhibitor of κB (IκB) [44]. DNA double-strand breaks (DSBs), produced by direct or indirect radiation damage to DNA, act as an initial trigger that results in activation of ataxia telangiectasia mutated protein (ATM). Activated ATM promotes nuclear export of IKK-γ/NF-κB essential modulator (NEMO), a regulatory subunit of the IκB kinase complex that is able to activate NF-κB in the cytoplasm. During this process, a nucleoplasmic signalosome is required for NEMO posttranslational modification and NEMO shuttling to the cytoplasm. While the composition of the nucleoplasmic signalosome is not fully elucidated, p53-induced protein with a death domain (PIDD), receptor interacting protein 1 (RIP1), and poly(ADP-ribose)-polymerase-1 (PARP-1) are known to play supporting roles [43]. In this context, a dose of 8–10 Gy of either γ-rays or X-rays was found to activate the genotoxic stress-induced NF-κB pathway in HUVECs [45, 46].

Radiation-induced sterile inflammation in endothelial cells. Ionizing radiation exposure activates redox-sensitive transcription factor NF-κB via DSB and ATM signaling, induces oxidative stress, and triggers the release of DAMPs. The resulting inflammation leads to the production and secretion of pro-inflammatory cytokines as well as to the expression of a modified repertoire of adhesion molecules by irradiated endothelial cells
Another possible cause of sterile inflammation is oxidative stress, a recognized consequence of endothelial cell exposure to radiation (Fig. 1) [47,48,49,50]. Besides reacting with cellular biomolecules, ROS directly activate redox-sensitive transcription factors nuclear factor (erythroid-derived 2)-like 2 (NRF2), activator protein 1 (AP-1), and NF-κB [44]. AP-1 is a heterodimeric transcription factor composed of members of the Jun, Jun dimerization protein (JDP), FOS, and related activating transcription partner families [51, 52]. Depending on its composition, it plays a role in the expression of several genes involved in cellular differentiation, proliferation, and apoptosis. Examples of AP-1-target genes are transforming growth factor (TGF)α, TGFβ, and IL-2 [51]. Activation of AP-1 during oxidative and inflammatory stimuli is predominantly mediated by mitogen-activated protein kinase (MAPK) signaling [44]. NF-κB is also a redox-regulated transcription factor: inflammatory and/or oxidative stimuli activate a series of upstream kinases, such as MAPKs, IκB kinase, protein kinase C (PKC), and phosphatidylinositide 3-kinases (PI3 K), which then activate NF-κB by phosphorylation-mediated degradation of IκB. Activated NF-κB translocates to the nucleus and induces the expression of a wide array of genes regulating pro-inflammatory mediators TNF-α, IL-8, IL-1, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 [44]. In endothelial cells, NF-κB is involved in the transcriptional regulation of most cytokines and adhesion molecules [53,54,55,56,57].
Another possible cause of endothelial activation is the release of damage-associated molecular patterns (DAMPs) by stressed and dying cells (Fig. 1). Tissue injury emits DAMPs that serve as danger signals to activate danger control (i.e., inflammation for host defense). DAMPs can either be intracellular molecules that signal cell stress and necrosis [high-mobility group box 1 (HMGB1), histones, purine metabolites, uric acid, S100 proteins, heat-shock proteins, and DNA/RNA outside nucleus or mitochondria], matrix constituents that signal extensive matrix remodeling (hyaluronan fragments and glycosaminoglycan fragments) and luminal factors that signal barrier destruction (uromodulin, oxidized low-density lipoprotein). DAMPs activate toll-like receptors, purinergic receptors, and inflammasomes in parenchymal cells and leukocytes. DAMP binding on endothelial cells upregulates pro-inflammatory signaling pathways that lead to NF-κB, MAPK, and interferon regulatory factor 3 (IRF3) signaling [58, 59], resulting in expression of adhesion molecules [intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, and E-selectin] and the release of cytokines [IL-6, IL-8, chemokine C–C motif ligand (CCL) 2, and interferon (IFN) γ] [42, 60,61,62,63]. In this respect, exposure to doses ≥ 2 Gy of X-rays was found to induce a dose-dependent in vitro and in vivo release of HMGB1 [64], known to induce endothelial expression of IL-6, CCL2, ICAM-1, and VCAM-1 [65]. In the murine microvascular endothelial cell line, bEnd.3, irradiation with 10 Gy has been shown to promote HMGB1gene expression [66]. Moreover, NF-κB signaling was found to be upregulated in irradiated arteries of patients treated with radiotherapy, even months or years after radiation exposure [67].
In general, high doses (> 2 Gy) of ionizing radiation induce endothelial activation. Endothelial adhesion molecules ICAM-1 and E-selectin are upregulated in a time- and dose-dependent manner [68,69,70], in part due to NF-κβ activation [71]. Furthermore, the expression of cytokines IL-6 and IL-8 as well as TGF-β was shown to increase after exposure to high doses of ionizing radiation [18, 72, 73] and was further differentially affected by dose quality [74]. For example, in obese ApoE−/− mice, a 14 Gy exposure induced an inflammatory phenotype, accelerating atherosclerotic plaque formation and rupture [75]. In addition, atomic bomb survivors exposed to high doses are more prone to the development of atherosclerosis [29] and demonstrated signs of general inflammation, with increased levels of IL-6 and C-reactive protein (CRP) [76]. Comparatively, the effects of low doses (≤ 2 Gy) of ionizing radiation on endothelial activation are still under debate (also discussed in [77]). On one hand, increased ICAM-1 expression and concomitant leukocyte attachment were detected in in vitro endothelial cell cultures after 0.125–0.5 Gy [78]. In addition, we detected elevated IL-6 and CCL2 levels in human endothelial cells exposed to 0.5 Gy [79]. On the other hand, a decrease in endothelial ICAM-1 and E-selectin expression has been observed after exposure of mice to 0.3 Gy and 1 Gy [69], which caused decreased endothelial adhesiveness to monocytes [69, 80]. This anti-inflammatory effect of low-dose radiation, which was confirmed by others [50, 81,82,83,84,85,86], requires a pre-activation of endothelial cells with pro-inflammatory stimuli TNF-α, IL-1β, or lipopolysaccharide. When these mice were exposed to low amounts of 137Cs delivered in the drinking water, the pro-inflammatory plaque phenotype was diminished [87]. The dampening effect of radiation exposure on endothelial activation has been used for decades for the treatment of benign inflammatory diseases [88, 89]. Today, the use of low-dose radiotherapy for the treatment of chronic inflammatory diseases is rare, due to the debate on possible cancer and non-cancer risks [85].
It must be emphasized that endothelial cell activation is a normal part of bodily defense mechanisms. In physiological circumstances, it draws immune cells to sites of infection or tissue injury. The difference between normal physiological and detrimental pathological activation of the endothelium lies in the nature, extent, duration, and combination of pro-inflammatory stimuli. As a consequence of prolonged and/or repeated exposure to a combination of cardiovascular risk factors, the protective effect of endogenous anti-inflammatory systems of endothelial cells can ultimately be depleted, resulting in endothelial dysfunction [21]. An overview of findings supporting endothelial inflammation in different endothelial cell cultures and organs by different radiation qualities and doses is given in Table 2.
Deterioration of the vascular tone
One of the key consequences of endothelial dysfunction is impairment of endothelium-dependent vasodilation due to reduced bioavailability of vasodilators, particularly nitric oxide (NO), and/or to elevated levels of endothelium-derived contracting factors, i.e., endothelins, prostaglandin, and thromboxane [23, 90,91,92,93]. The role of NO and its reactive intermediates in the endothelial radiation response largely remains an open question [94]. What is known is that, after exposure of endothelial cells to ionizing radiation, NO is rapidly deactivated by superoxide radicals, resulting in the formation of vasotoxic peroxynitrites [95, 96] (Fig. 2). Irradiation-induced oxidative stress also causes endothelial NO synthase (eNOS) uncoupling due to inadequate availability of its redox-sensitive cofactor tetrahydrobiopterin, resulting in eNOS-dependent production of superoxide and diminished release of NO [97]. From 1 to 4 days after irradiation, doses of 6 Gy and higher were found to promote eNOS expression and activity, leading to NO production and NO-induced angiogenesis with a concomitant increase in tumor blood flow [98, 99]. eNOS activation after endothelial irradiation depends on components of the DNA damage response pathway, namely, ATM and heat-shock protein 90, which phosphorylate Ser1179 of eNOS, leading to enhanced eNOS activity [100]. However, most of endothelial DNA damage signaling ceases within 24 h after irradiation [79], explaining why irradiation acutely but not chronically enhances NO availability. At later timepoints, endothelium-dependent vasodilation is compromised. Timing also depends on the dose and on the nature of the irradiated endothelial bed. For example, reduced endothelium-dependent vasodilation was found in rabbit carotid arterial rings 20 h after irradiation with 8 Gy and 16 Gy [95], in rabbit ear arteries 1 week after irradiation with 10 Gy, 20 Gy, and 45 Gy [101, 102], in rabbit aorta 9 days after whole-body irradiation with 1 Gy, 2 Gy, and 4 Gy [103] and in rat aorta 6 months after irradiation with 15 Gy [104]. In humans, endothelium-dependent vasodilation was found to be impaired both in vitro and in vivo in carotid arteries 4–6 weeks after neck irradiation (total pre-operative dose of radiation averaged 47.9 Gy ± 2.8 Gy) [105]. In addition, impaired endothelium-dependent vasodilation of axillary arteries was reported in breast cancer radiotherapy patients more than 3 year after radiotherapy (no average dose assigned) [106].

Irradiation-induced deterioration of the vascular tone. Ionizing radiation exposure induces oxidative stress and DNA damage in endothelial cells (left), leading to decreased NO levels and altered production and/or secretion of vasoactive compounds resulting in an initial vasodilation followed by vasoconstriction. In addition, VSMC irradiation induces oxidative stress and DNA damage, resulting in an initial reduction of cellular viability and proliferation as well as vasodilation (right). In the long run, oxidative stress results in Ca2+ release from intracellular stores and increased VSMC proliferation, supporting vasoconstriction
NO is not the sole vasoactive substance produced and released by the endothelium. The production of prostacyclin, a potent endothelium-derived vasodilator, is also affected by radiation exposure. Basal prostacyclin release was found to be unaffected in irradiated HUVECs at doses up to 25 Gy [107]. However, when endothelial cells were stimulated with exogenous arachidonic acid, a precursor of endothelial prostacyclin, prostacyclin levels decreased 15 min after irradiation [108], increased within 1 day after irradiation [109,110,111,112,113] and then decreased again thereafter in a radiation dose-dependent way [107, 111, 114, 115]. The short-term stimulatory effect of radiation on prostacyclin production is believed to be caused by oxidative stress [116, 117] and cell damage [111]. Endothelium-dependent hyperpolarization-related signaling was unaffected after endothelial irradiation, thereby serving as a reserve defense mechanism for vasorelaxation [103, 118]. Conversely, levels of vasoconstrictor endothelin-1 were increased after in vitro [119, 120] and in vivo [121, 122] radiation exposure with doses ranging from 0.2 to 20 Gy. In addition, the endothelial production and release of vasoconstrictor angiotensin II by endothelial cells, in bovine pulmonary arterial endothelial cells and HUVECs [123, 124] and in pulmonary endothelial cells collected from irradiated rats [125, 126] increased dose- and time-dependently starting 24 h after exposure to 5–30 Gy. Overall, one can conclude that endothelial irradiation induces initial vasodilation during the first couple of days after irradiation, followed by chronic vasoconstriction with compromised endothelium-dependent vasodilation.
Besides affecting the endothelial layer of blood vessels, ionizing radiation can also directly affect vascular smooth muscle cells (VSMCs; Fig. 2). In culture in the absence of endothelial cells, VSMCs underwent decreased proliferation after a 1.25–20 Gy exposure [127,128,129], with a reduction of viable cells only 15 days after exposure [128, 129]. Surviving VSMCs demonstrated reduced contractibility [129], but maintained a contractile phenotype after exposure to 10–20 Gy [130]. In contrast, when VSMCs were co-cultured with endothelial cells and both were irradiated together with 2–10 Gy, VSMCs changed from a normal contractile to a fibrogenic phenotype [73] associated with the pathogenesis of atherosclerosis [131]. Fibrosis was induced by TGFβ released by irradiated endothelial cells, resulting in small mothers against decapentaplegic (SMAD) signaling in VSMCs [73]. Exposure to 6 Gy also mediated increased myofilament Ca2+ sensitivity in isolated rat thoracic aortic VSMCs 9 and 30 days after exposure [132, 133]. Furthermore, oxidative stress has been shown to induce vasoconstriction by promoting Ca2+ release from VSMC intracellular stores [134] and by upregulating VSMC proliferation by either their secretion of cyclophilin A [135] or by the binding of oxidative stress products hydroperoxyoctadecadienoic acids and 4-hydroxy-2-nonenal to VSMCs [136, 137]. An overview of findings supporting deterioration of vascular tone by different radiation qualities and doses is given in Table 3.
Procoagulatory and prothrombotic phenotype
In addition to altered vascular tone, vascular damage shifts the homeostatic balance towards a procoagulant and prothrombotic endothelial cell phenotype [138]. Because prostacyclin and NO are the main anticoagulatory agents secreted by endothelial cells [139], their decreased production after radiation exposure results in platelet aggregation and blood clot formation (Fig. 3). However, molecular mechanisms responsible for loss of endothelial thromboresistance are more complex. An irradiated endothelium indeed increases the synthesis of von Willebrand factor (vWF) [140,141,142,143,144] and platelet-activating factor [145] while reducing thrombomodulin [68, 146, 147] and prostacyclin production [108, 117, 148], as well as its fibrinolytic activity [149,150,151]. These changes promote platelet adhesion and aggregation and the development of platelet–fibrin thrombi [152,153,154,155]. Cytokines produced during endothelial activation (e.g., IL-6 and CCL2) further affect hemostasis by inducing the expression of tissue factor, tissue plasminogen activator, and vWF [156,157,158]. In this context, irradiation with 14 Gy was shown to induce atherosclerotic plaques with an inflammatory phenotype prone to hemorrhage in ApoE−/− obese mice [75], which may accelerate atherosclerosis [159]. An overview of findings supporting the procoagulatory and prothrombotic effect on endothelial cells by different radiation qualities and doses is given in Table 4.

Irradiation-induced procoagulatory and prothrombotic state in endothelial cells. Endothelial irradiation results in a decreased production of anticoagulants prostacyclin and NO, resulting in a procoagulatory state. In addition, endothelial cell activation and general vascular damage result in elevated secretion of prothrombotic proteins (e.g., vWF) and a reduced fibrinolytic activity producing a prothrombotic state
Endothelial cell retraction and death
Besides edema formation in surrounding tissues caused by endothelial inflammation and tissue injury [160, 161], exposure to radiation doses as low as 2 Gy can induce a transient and rapid decrease in the integrity of in vitro human endothelial barriers through cell detachment and loss of platelet endothelial cell adhesion molecule (PECAM)-1 [162, 163] (Fig. 4). Rapid loss of endothelial monolayer integrity depends on cytoskeletal reorganization due to actin stress fiber formation and redistribution of vascular endothelial (VE)-cadherin junctions, resulting in endothelial retraction [164,165,166,167]. At higher doses, a more direct cause of increased vascular permeability is of course endothelial cell death [168, 169]. Sensitivity of endothelial cells to cell-reproductive death after ionizing radiation can be assessed by clonogenic assays, the method of choice in such situation [170]. Radiosensitivity varies between endothelial cells from different vascular beds, with HUVECs being the most sensitive and HHSEC being the most radioresistant among the tested ones [171]. In addition, sensitivity to cell-reproductive death depends on radiation quality, with the relative biological effectiveness of α-particles estimated at 5.5 and 4.6 for 10% survival of A549 cells and EA.hy926 cells, respectively [172]. Doses as low as 0.1 Gy can reduce the surviving fraction of EA.hy926 cells [172, 173]. Doses higher than 5 Gy induce endothelial cell apoptosis by the production of ceramide [174, 175]: irradiation activates stress-activated c-Jun N-terminal kinases (JNKs), resulting in the conversion of sphingomyelin to ceramide by neutral sphingomyelinase and the subsequent activation of caspase-3 [176, 177]. In addition, endothelial apoptosis at doses higher than 5 Gy can also be induced by persistent DNA damage, resulting in p53 accumulation and activation of the caspase pathway [178, 179]. Mechanisms behind endothelial cytotoxicity of lower doses are less known. For example, apoptotic EA.hy926 cell death was not increased after exposure to 0.2 Gy, but well after exposure to 5 Gy [180]. In another study, TNF-α-activated endothelial cells were shown to have a discontinuous induction of apoptosis, with a relative maximum at 0.3 Gy and 3 Gy and a relative minimum at 0.5 Gy [82]. In addition, our group observed a dose-dependent increase in endothelial cell apoptosis from 0.5 Gy in HUVECs and from 0.1 Gy in EA.hy926 cells [173]. In vivo, compromised barrier function is involved in the pathogenesis of vascular failure, including atherosclerosis [23, 181, 182]. An overview of findings supporting the induction of endothelial cell retraction and cell death by different radiation qualities and doses is given in Table 5.

Irradiation-induced retraction and death of endothelial cells. Ionizing radiation exposure is able to decrease PECAM-1 expression, redistribute VE-cadherin, and produce actin stress fibers leading to endothelial retraction. Depending on the radiation dose, radiation quality, and inherent radiation sensitivity, ionizing radiation can activate the caspase pathway by ceramide formation and persistent p53 signaling, causing endothelial cell death. As a consequence of endothelial retraction and cell death, the physiological endothelial barrier is compromised
Mitochondrial dysfunction
Recent years have seen increasing interest for radiation-induced mitochondrial dysfunction as a cause of endothelial dysfunction in the context of cardiovascular disease [183,184,185,186,187]. In most mammalian cells, mitochondria are primarily considered as the major suppliers of cellular energy in the form of ATP produced by oxidative phosphorylation (OXPHOS) [186]. However, mitochondria are only present in modest number in endothelial cells [185, 188] and produce a low proportion of the total amount of cellular energy [189,190,191]. Thus, endothelial mitochondria are more likely to primarily serve as important signaling organelles [192]. While mitochondria are linked to endothelial function (reviewed in [185]) and endothelial mitochondria are known to play a role in vascular diseases (reviewed in [186]), data on the effect of ionizing radiation on endothelial mitochondria in general are scarce. It was shown that in vitro endothelial cells exposed to 5–20 Gy of γ-rays lose their mitochondrial membrane potential and that mitochondrial ROS production increased 24–72 h after exposure [193]. Furthermore, murine cardiac microvascular endothelial cells irradiated with 8 and 16 Gy X-rays acquired protein expression profiles associated with mitochondrial dysfunction [194]. In light of the caveats in current knowledge, sections below will focus on three main mitochondrial functions hypothesized to be disturbed in endothelial cells after exposure to ionizing radiation: Ca2+ regulation, control of cell death, and oxidative stress signaling.
Normal cytosolic Ca2+ concentrations are maintained approximately 10,000 times lower than extracellular Ca2+ concentrations by plasma membrane and endoplasmic reticulum Ca2+ ATPases. Because these transport proteins require ATP for Ca2+ transport, mitochondria are indirectly involved in this form of Ca2+ regulation [195, 196]. In addition, mitochondria can also directly sequester Ca2+ and, thereby, regulate intracellular concentrations by their inner membrane uniporter rapid mode of Ca2+ uptake into heart mitochondria (RaM), which is driven by the proton electrochemical potential. Conversely, mitochondria release Ca2+ via the 2Na+/Ca2+- and 2H+/Ca2+-exchanger. Increased mitochondrial Ca2+ activates dehydrogenase enzymes in mitochondria and increases ATP synthase activity, leading to increased NADH and ATP production [197]. Sparse evidence exists that altered mitochondrial calcium contributes to endothelial dysfunction in cardiovascular diseases. For example, in diabetes, high glucose levels were shown to elevate mitochondrial Ca2+ levels in human endothelial cells, thereby increasing mitochondrial free radical production [198]. Furthermore, mitochondrial Ca2+ regulates the intensity of TNF-α-induced inflammation in mouse lung microvascular endothelium [199]. In addition, flow-induced dilation of human coronary arterioles was found to be mediated by Ca2+ influx via the transient receptor potential vanilloid type 4 (TRPV4) channel that is in closely apposition with endothelial mitochondria, resulting in mitochondrial ROS release in coronary artery endothelial cells [200]. Finally, the mitochondrial Ca2+ uniporter can potentiate endothelial cell migration [201], and its levels are markedly decreased in endothelial cells derived from CVD patients [202]. While it is known that mitochondrial Ca2+ signaling is affected by ionizing radiation (reviewed in [203, 204]), there is a lack of experimental studies on the role of mitochondrial Ca2+ in the irradiated endothelium.
Importantly, mitochondria are also central executioners of apoptosis. In normal state, anti-apoptotic proteins of the B-cell lymphoma (Bcl)-2 family located on the outer mitochondrial membrane inhibit pro-apoptotic effector proteins Bcl-2-associated protein X (BAX) and Bcl-2 homologous antagonist killer (BAK) [205]. In response to cytotoxic stress, Bcl-2 homology 3 (BH3)-only proteins inhibit Bcl-2 proteins, resulting in BAX and BAK activation. BAX and BAK form oligomers that permeabilize the mitochondrial outer membrane, mediating the release of cytochrome c into the cytosol [206, 207]. Cytosolic cytochrome c promotes the activation of caspase 9 by apoptotic protease activating factor 1 (APAF1), which in turn activates effector caspases that induce cell death [208]. Dysregulation of these vital functions can promote endothelial inflammation, apoptosis, and senescence, which are all linked to the development and progression of atherosclerosis [169, 185,186,187, 209].
As a byproduct of OXPHOS, a small amount of O2 undergoes monoelectronic reduction mainly at complexes I and III of the mitochondrial electron transport chain, resulting in the generation of O ·−2 [210]. As a result, mitochondria are a main generation site of ROS within cells [210]. Formed O ·−2 is converted to H2O2 by SOD2 inside mitochondria, which is able to activate the redox-sensitive transcription factors NRF2, AP-1, and NF-κB [44]. Because mitochondria are an important cellular source of ROS [169, 211, 212], they are closely related to oxidative stress signaling. At relatively low levels, mitochondria-derived ROS are signaling molecules that support normal or compensatory cellular functions involved in hypoxic adaptation, immunity, cellular differentiation, and longevity [213]. However, excessive mitochondrial ROS levels can cause oxidative stress [186]. This is problematic, because mitochondria themselves are critical targets of ROS [214], leading to irreversible damage to mitochondrial DNA, membrane lipids, and proteins [215].
In an effort to combat oxidative challenge, ROS and RNS activate cellular transcription factors, NRF2, NF-κB, and AP-1, resulting in increased expression of ROS-detoxifying enzymes catalase, SOD, glutathione peroxidase (GPx), glutathione S-transferases (GST), and heme oxygenase-1 (HO-1) [216,217,218]. NRF2 is believed to be the main regulator of cellular resistance to pro-oxidants. Because NRF2 controls basal and induced expression of an array of antioxidant response element-dependent genes, including HO-1, SOD2, and GPx [219, 220], it is not surprising that this factor is induced after radiation exposure in both normal and cancerous cells (0.05–8 Gy) [217, 221, 222]. NRF2 also confers cellular radioresistance [223,224,225,226] by mediation of DNA repair, by regulating genes from the homologous recombination DNA repair pathway, and oxidative defense in both normal and cancerous cells [222, 227]. In addition, NRF2 upregulation has been implicated in oxidative stress-induced endothelial dysfunction [228]. Because NRF2 mediates gene expression resulting in both high NADPH production and the production, regeneration and utilization of GSH, thioredoxin, and peroxiredoxin, upregulation of NRF2 leads to increased levels of these antioxidants after irradiation (0.25–20 Gy) in lymphocytes and glioma cells [229, 230]. Of note, elevated levels of several mammalian peroxiredoxin isoforms have been evidenced after a 10 Gy radiation exposure of mouse testis and liver, further enhancing cellular defense mechanisms [231,232,233,234]. Both cumulative and acute radiation exposure can disrupt the cellular redox balance. However, oxidative stress only prevails when pro-oxidant levels eventually overwhelm cellular antioxidant systems, an event marked by enzyme inactivation, a low GSH/glutathione disulfide ratio, and a decreased pool of low molecular weight antioxidants. The consequence of such redox imbalance is manifested by modifications of nucleic acids, lipids, proteins, and other biomolecules [216, 235].
If radiation doses are high enough to overwhelm cellular antioxidant responses, oxidative stress can induce mitochondrial dysfunction by ROS-induced ROS release [236]. As a consequence, radiation-induced oxidative stress that normally disappears within seconds after exposure [214] can lead to the initiation of a self-amplifying cycle, giving rise to long-term ROS production [237] and concomitant mitochondrial dysfunction [169]. During this process, mitochondrial DNA seems to be particularly sensitive to oxidative damage because of its limited DNA repair capacity, lack of protective histones, a high exon to intron ratio and its close proximity to the electron transport chain [238]. In agreement, a range of studies demonstrated changes in mitochondrial function and number after exposure of cells or tissues to high doses of ionizing radiation [239,240,241,242]. Doses of 5–20 Gy of γ-radiation were found to induce a dose-dependent increase in ROS levels with a decrease in mitochondrial activity [193].
Furthermore, 15 Gy of X-rays induced persistent oxidative stress in endothelial cells, linked to mitochondrial dysfunction and premature senescence [243]. Effects of low radiation doses have been less studied on mitochondrial dysfunction in endothelial cells. Doses of 1.5 Gy, 4 Gy, and 10 Gy were found to influence mitochondrial membrane potential in HUVECs 2 days after exposure. While the mitochondrial potential reverted back to control level by days 5 and 6 in 1.5 and 4 Gy irradiated cells, respectively, 10 Gy resulted in persistently decreased mitochondrial activity [244]. In another example, the respiratory capacity of cardiac mitochondria was significantly reduced 40 weeks after local heart irradiation in ApoE−/− mice with a single X-ray dose of 2 Gy [245]. In addition, 0.1 Gy and 0.5 Gy were found to reduce mitochondrial signaling in murine hippocampus and cortex [246]. An overview of findings supporting the induction of mitochondrial dysfunction in endothelial cells by different radiation qualities and doses is given in Table 6.
Premature endothelial senescence
Aging of the vascular system predisposes the cardiovascular system to the development of diseases, even in the absence of other risk factors [247]. On a cellular level, vascular aging corresponds to endothelial cell senescence [248, 249], a phenomenon that refers to irreversible arrest of endothelial cell renewal. At a molecular level, senescence is induced and maintained by p53 and p16-Rb pathways that inhibit cell-cycle progression [250]. Both pathways are activated either during attrition of telomeres, referred to as replicative senescence [251, 252], or during stress situations independently of telomere shortening, referred to as stress-induced premature senescence [253]. For instance, limited availability of nutrients and growth factors, chromatin perturbations, improper cell contacts, and oxidative stress prematurely induces senescence via cell stress [254]. Oxidative stress is of special importance, because it induces and accelerates senescence at multiple molecular levels: accelerated telomere shortening [254], induction of DNA damage leading to p53 activation [255], NO scavenging decreasing its bioavailability [256], and mitochondrial dysfunction [257].
Endothelial senescence is currently emerging as a contributor to the pathogenesis of atherosclerosis by increasing ROS production, decreasing NO availability, and increasing the production of pro-inflammatory molecules IL-6, IL-1, IL-8, CCL2, and ICAM-1 [257,258,259]. All these molecules indeed contribute to the development and progression of atherosclerosis [209, 257], as indirectly evidenced by the presence of senescent endothelial cells in human atherosclerotic plaques [258]. These observations have to be taken with caution: identification of senescent endothelial cells suggests an association with atherosclerosis, but cannot be used to evidence a causal relationship between endothelial senescence and development and/or progression of atherosclerosis. Considering that ionizing radiation induces oxidative stress [254, 260], DSBs [261], and telomere shortening [260], it is not surprising that it constitutes a stressor that can evoke premature senescence in cells. Several in vitro studies demonstrated that high dose (4–50 Gy) [45, 262,263,264,265,266,267] and medium dose (0.5 Gy) [79] radiation exposure as well as chronic radiation exposure to low doses [268,269,270] induces premature endothelial cell senescence. An overview of findings supporting the induction of premature senescence in endothelial cells by different radiation qualities and doses is given in Table 7.
Conclusions
In current times, it has become a common practice to use ionizing radiation during justified diagnostic and therapeutic medical procedures. High doses of ionizing radiation lead to cardiac dysfunction over time [29, 30]. However, precise biological and molecular mechanisms are still elusive. In this review, we substantiated the hypothesis that ionizing radiation exposure can induce endothelial activation and dysfunction (Fig. 5). Mechanisms of action predominantly involve induction of a pro-inflammatory state by genotoxic stress, oxidative stress and DAMP release, deterioration of the vascular tone by compromising endothelium-dependent vasodilatation, induction of a procoagulatory and prothrombotic endothelial phenotype by perturbing hemostasis, inducing endothelial cell retraction and death leading to loss of vessel integrity, and induction of mitochondrial dysfunction and premature senescence. Together with other vascular risk factors (e.g., dyslipidemia and hypertension), ionizing radiation can initiate and/or trigger the progression of several pathological conditions, such as atherosclerosis and subsequent radiation-induced heart disease. While these effects are well documented at high doses, there is to date little knowledge on the effect at low doses of ionizing radiation. Furthermore, little is known about the effects of chronic radiation exposure, the exposure to different kinds of radiation qualities (in particular particle radiation), the evolution of endothelial dysfunction in time, and the role of endothelial heterogeneity. Future studies are required to advance the understanding of the mechanisms that lead to endothelial dysfunction, as they may relate to the induction and/or progression of cardiovascular diseases following exposure to ionizing radiation. Further understanding of radiation-induced endothelial dysfunction could lead to advances in the development of countermeasures, such as antioxidant therapy [271, 272], for cardiovascular disease in radiation-exposed individuals.

Ionizing radiation can induce both endothelial cell activation and dysfunction. The resulting vasoconstrictive, pro-inflammatory, procoagulatory, prothrombotic, and prohypertrophic environment can initiate and/or trigger the progression of several pathological cardiovascular conditions, together with other vascular risk factors (e.g., dyslipidemia and hypertension)
References
Fishman AP (1982) Endothelium: a distributed organ of diverse capabilities. Ann N Y Acad Sci 401:1–8
Creager MA, Cooke JP, Mendelsohn ME, Gallagher SJ, Coleman SM, Loscalzo J, Dzau VJ (1990) Impaired vasodilation of forearm resistance vessels in hypercholesterolemic humans. J Clin Invest 86(1):228–234. https://doi.org/10.1172/JCI114688
Masumoto A, Hirooka Y, Hironaga K, Eshima K, Setoguchi S, Egashira K, Takeshita A (2001) Effect of pravastatin on endothelial function in patients with coronary artery disease (cholesterol-independent effect of pravastatin). Am J Cardiol 88(11):1291–1294
Bragulat E, de la Sierra A, Antonio MT, Coca A (2001) Endothelial dysfunction in salt-sensitive essential hypertension. Hypertension 37(2 Pt 2):444–448
Yoshida M, Imaizumi T, Ando S, Hirooka Y, Harada S, Takeshita A (1991) Impaired forearm vasodilatation by acetylcholine in patients with hypertension. Heart Vessels 6(4):218–223
Gilligan DM, Guetta V, Panza JA, Garcia CE, Quyyumi AA, Cannon RO 3rd (1994) Selective loss of microvascular endothelial function in human hypercholesterolemia. Circulation 90(1):35–41
Panza JA, Quyyumi AA, Brush JE Jr, Epstein SE (1990) Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med 323(1):22–27. https://doi.org/10.1056/NEJM199007053230105
Bergholm R, Leirisalo-Repo M, Vehkavaara S, Makimattila S, Taskinen MR, Yki-Jarvinen H (2002) Impaired responsiveness to NO in newly diagnosed patients with rheumatoid arthritis. Arterioscler Thromb Vasc Biol 22(10):1637–1641
Datta D, Ferrell WR, Sturrock RD, Jadhav ST, Sattar N (2007) Inflammatory suppression rapidly attenuates microvascular dysfunction in rheumatoid arthritis. Atherosclerosis 192(2):391–395. https://doi.org/10.1016/j.atherosclerosis.2006.05.034
Piper MK, Raza K, Nuttall SL, Stevens R, Toescu V, Heaton S, Gardner-Medwin J, Hiller L, Martin U, Townend J, Bacon PA, Gordon C (2007) Impaired endothelial function in systemic lupus erythematosus. Lupus 16(2):84–88. https://doi.org/10.1177/0961203306074842
Sari I, Okan T, Akar S, Cece H, Altay C, Secil M, Birlik M, Onen F, Akkoc N (2006) Impaired endothelial function in patients with ankylosing spondylitis. Rheumatology (Oxford) 45(3):283–286. https://doi.org/10.1093/rheumatology/kei145
Deanfield J, Donald A, Ferri C, Giannattasio C, Halcox J, Halligan S, Lerman A, Mancia G, Oliver JJ, Pessina AC, Rizzoni D, Rossi GP, Salvetti A, Schiffrin EL, Taddei S, Webb DJ, Working Group on E, Endothelial Factors of the European Society of H (2005) Endothelial function and dysfunction. Part I: Methodological issues for assessment in the different vascular beds: a statement by the Working Group on Endothelin and Endothelial Factors of the European Society of Hypertension. J Hypertens 23(1):7–17
Widmer RJ, Lerman A (2014) Endothelial dysfunction and cardiovascular disease. Glob Cardiol Sci Pract 3:291–308. https://doi.org/10.5339/gcsp.2014.43
Davignon J, Ganz P (2004) Role of endothelial dysfunction in atherosclerosis. Circulation 109(23 Suppl 1):27–32. https://doi.org/10.1161/01.cir.0000131515.03336.f8
Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr, Lerman A (2000) Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 101(9):948–954
Boron WF, Boulpaep EL (2017) Medical physiology, 3rd edn. Elsevier, Philadelphia
Azzam EI, Jay-Gerin JP, Pain D (2012) Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett 327(1–2):48–60. https://doi.org/10.1016/j.canlet.2011.12.012
Baselet B, Belmans N, Coninx E, Lowe D, Janssen A, Michaux A, Tabury K, Raj K, Quintens R, Benotmane MA, Baatout S, Sonveaux P, Aerts A (2017) Functional gene analysis reveals cell cycle changes and inflammation in endothelial cells irradiated with a single X-ray dose. Front Pharmacol 8:213. https://doi.org/10.3389/fphar.2017.00213
Meeren AV, Bertho JM, Vandamme M, Gaugler MH (1997) Ionizing radiation enhances IL-6 and IL-8 production by human endothelial cells. Mediators Inflamm 6(3):185–193. https://doi.org/10.1080/09629359791677
Jaillet C, Morelle W, Slomianny MC, Paget V, Tarlet G, Buard V, Selbonne S, Caffin F, Rannou E, Martinez P, Francois A, Foulquier F, Allain F, Milliat F, Guipaud O (2017) Radiation-induced changes in the glycome of endothelial cells with functional consequences. Sci Rep 7(1):5290. https://doi.org/10.1038/s41598-017-05563-y
Deanfield JE, Halcox JP, Rabelink TJ (2007) Endothelial function and dysfunction: testing and clinical relevance. Circulation 115(10):1285–1295. https://doi.org/10.1161/CIRCULATIONAHA.106.652859
Steyers CM 3rd, Miller FJ Jr (2014) Endothelial dysfunction in chronic inflammatory diseases. Int J Mol Sci 15(7):11324–11349. https://doi.org/10.3390/ijms150711324
Bonetti PO, Lerman LO, Lerman A (2003) Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol 23(2):168–175
Michiels C (2003) Endothelial cell functions. J Cell Physiol 196(3):430–443. https://doi.org/10.1002/jcp.10333
Darby SC, Cutter DJ, Boerma M, Constine LS, Fajardo LF, Kodama K, Mabuchi K, Marks LB, Mettler FA, Pierce LJ, Trott KR, Yeh ET, Shore RE (2010) Radiation-related heart disease: current knowledge and future prospects. Int J Radiat Oncol Biol Phys 76(3):656–665. https://doi.org/10.1016/j.ijrobp.2009.09.064
Yusuf SW, Venkatesulu BP, Mahadevan LS, Krishnan S (2017) Radiation-induced cardiovascular disease: a clinical perspective. Front Cardiovasc Med 4:66. https://doi.org/10.3389/fcvm.2017.00066
ICRP (2007) The 2007 Recommendations of the International Commission on Radiological Protection. ICRP Publication 103. Ann ICRP 37(2–4)
(2006) Committee to Assess Health Risks from Exposure to Low Levels of Ionizing Radiation. National Research Council. Health Risks from Exposure to Low Levels of Ionizing Radiation: BEIR VII—Phase 2. National Academy Press, Washington, DC, pp 1–406
Shimizu Y, Kodama K, Nishi N, Kasagi F, Suyama A, Soda M, Grant EJ, Sugiyama H, Sakata R, Moriwaki H, Hayashi M, Konda M, Shore RE (2010) Radiation exposure and circulatory disease risk: Hiroshima and Nagasaki atomic bomb survivor data, 1950–2003. BMJ 340:b5349. https://doi.org/10.1136/bmj.b5349
Darby SC, Ewertz M, McGale P, Bennet AM, Blom-Goldman U, Bronnum D, Correa C, Cutter D, Gagliardi G, Gigante B, Jensen MB, Nisbet A, Peto R, Rahimi K, Taylor C, Hall P (2013) Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 368(11):987–998. https://doi.org/10.1056/NEJMoa1209825
Onat D, Brillon D, Colombo PC, Schmidt AM (2011) Human vascular endothelial cells: a model system for studying vascular inflammation in diabetes and atherosclerosis. Curr Diab Rep 11(3):193–202. https://doi.org/10.1007/s11892-011-0182-2
Bouis D, Hospers GA, Meijer C, Molema G, Mulder NH (2001) Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis 4(2):91–102
Edgell CJ, McDonald CC, Graham JB (1983) Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA 80(12):3734–3737
Bishop ET, Bell GT, Bloor S, Broom IJ, Hendry NF, Wheatley DN (1999) An in vitro model of angiogenesis: basic features. Angiogenesis 3(4):335–344
Donovan D, Brown NJ, Bishop ET, Lewis CE (2001) Comparison of three in vitro human ‘angiogenesis’ assays with capillaries formed in vivo. Angiogenesis 4(2):113–121
Wallace CS, Truskey GA (2010) Direct-contact co-culture between smooth muscle and endothelial cells inhibits TNF-alpha-mediated endothelial cell activation. Am J Physiol Heart Circ Physiol 299(2):H338–H346. https://doi.org/10.1152/ajpheart.01029.2009
Rainger GE, Nash GB (2001) Cellular pathology of atherosclerosis: smooth muscle cells prime cocultured endothelial cells for enhanced leukocyte adhesion. Circ Res 88(6):615–622
Acheva A, Aerts A, Baatout S, Rombouts C, Salomaa S, Manda K, Hildebrandt G, Kämäräinen M (2014) Human 3-D tissue models in radiation biology—current status and future perspectives. Int J Radiat Res 12(2):81–98
Rezvan A, Ni CW, Alberts-Grill N, Jo H (2011) Animal, in vitro, and ex vivo models of flow-dependent atherosclerosis: role of oxidative stress. Antioxid Redox Signal 15(5):1433–1448. https://doi.org/10.1089/ars.2010.3365
Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T (2007) Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol 27(8):1706–1721. https://doi.org/10.1161/ATVBAHA.107.142570
Ohashi R, Mu H, Yao Q, Chen C (2004) Cellular and molecular mechanisms of atherosclerosis with mouse models. Trends Cardiovasc Med 14(5):187–190. https://doi.org/10.1016/j.tcm.2004.04.002
Liao JK (2013) Linking endothelial dysfunction with endothelial cell activation. J Clin Invest 123(2):540–541. https://doi.org/10.1172/JCI66843
Hellweg CE (2015) The nuclear factor kappaB pathway: a link to the immune system in the radiation response. Cancer Lett 368(2):275–289. https://doi.org/10.1016/j.canlet.2015.02.019
Surh YJ, Kundu JK, Na HK, Lee JS (2005) Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr 135(12 Suppl):2993S–3001S
Dong X, Tong F, Qian C, Zhang R, Dong J, Wu G, Hu Y (2015) NEMO modulates radiation-induced endothelial senescence of human umbilical veins through NF-kappaB signal pathway. Radiat Res 183(1):82–93. https://doi.org/10.1667/RR13682.1
Hallahan D, Clark ET, Kuchibhotla J, Gewertz BL, Collins T (1995) E-selectin gene induction by ionizing radiation is independent of cytokine induction. Biochem Biophys Res Commun 217(3):784–795. https://doi.org/10.1006/bbrc.1995.2841
Hall EJ, Giaccia AJ (2012) Radiobiology for the radiologist, 7th edn. Lippincott Williams & Wilkins, Philadelphia
Collins-Underwood JR, Zhao W, Sharpe JG, Robbins ME (2008) NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radic Biol Med 45(6):929–938. https://doi.org/10.1016/j.freeradbiomed.2008.06.024
Choi SH, Kim M, Lee HJ, Kim EH, Kim CH, Lee YJ (2016) Effects of NOX1 on fibroblastic changes of endothelial cells in radiationinduced pulmonary fibrosis. Mol Med Rep 13(5):4135–4142. https://doi.org/10.3892/mmr.2016.5090
Large M, Reichert S, Hehlgans S, Fournier C, Rodel C, Rodel F (2014) A non-linear detection of phospho-histone H2AX in EA.hy926 endothelial cells following low-dose X-irradiation is modulated by reactive oxygen species. Radiat Oncol 9:80. https://doi.org/10.1186/1748-717x-9-80
Angel P, Karin M (1991) The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1072(2–3):129–157
Wisdom R (1999) AP-1: one switch for many signals. Exp Cell Res 253(1):180–185. https://doi.org/10.1006/excr.1999.4685
Gaboury JP, Anderson DC, Kubes P (1994) Molecular mechanisms involved in superoxide-induced leukocyte-endothelial cell interactions in vivo. Am J Physiol 266(2 Pt 2):H637–H642
Rahman A, Bando M, Kefer J, Anwar KN, Malik AB (1999) Protein kinase C-activated oxidant generation in endothelial cells signals intercellular adhesion molecule-1 gene transcription. Mol Pharmacol 55(3):575–583
Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM (1993) Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest 92(4):1866–1874. https://doi.org/10.1172/JCI116778
Awad EM, Khan SY, Sokolikova B, Brunner PM, Olcaydu D, Wojta J, Breuss JM, Uhrin P (2013) Cold induces reactive oxygen species production and activation of the NF-kappa B response in endothelial cells and inflammation in vivo. J Thromb Haemost 11(9):1716–1726. https://doi.org/10.1111/jth.12357
Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M (2000) Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 20(10):2175–2183
Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF (2003) Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 101(7):2652–2660. https://doi.org/10.1182/blood-2002-05-1300
Land WG (2015) The role of damage-associated molecular patterns (DAMPs) in human diseases: part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ Med J 15(2):e157–e170
Jirik FR, Podor TJ, Hirano T, Kishimoto T, Loskutoff DJ, Carson DA, Lotz M (1989) Bacterial lipopolysaccharide and inflammatory mediators augment IL-6 secretion by human endothelial cells. J Immunol 142(1):144–147
Goebeler M, Gillitzer R, Kilian K, Utzel K, Brocker EB, Rapp UR, Ludwig S (2001) Multiple signaling pathways regulate NF-kappaB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood 97(1):46–55
Yoshida A, Yoshida S, Khalil AK, Ishibashi T, Inomata H (1998) Role of NF-kappaB-mediated interleukin-8 expression in intraocular neovascularization. Invest Ophthalmol Vis Sci 39(7):1097–1106
Kim I, Moon SO, Kim SH, Kim HJ, Koh YS, Koh GY (2001) Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem 276(10):7614–7620. https://doi.org/10.1074/jbc.M009705200
Wang L, He L, Bao G, He X, Fan S, Wang H (2016) Ionizing radiation induces HMGB1 cytoplasmic translocation and extracellular release. Guo Ji Fang She Yi Xue He Yi Xue Za Zhi 40(2):91–99
Sun W, Jiao Y, Cui B, Gao X, Xia Y, Zhao Y (2013) Immune complexes activate human endothelium involving the cell-signaling HMGB1-RAGE axis in the pathogenesis of lupus vasculitis. Lab Invest 93(6):626–638. https://doi.org/10.1038/labinvest.2013.61
Zhou H, Jin C, Cui L, Xing H, Liu J, Liao W, Liao H, Yu Y (2018) HMGB1 contributes to the irradiation-induced endothelial barrier injury through receptor for advanced glycation endproducts (RAGE). J Cell Physiol 233(9):6714–6721. https://doi.org/10.1002/jcp.26341
Halle M, Gabrielsen A, Paulsson-Berne G, Gahm C, Agardh HE, Farnebo F, Tornvall P (2010) Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J Am Coll Cardiol 55(12):1227–1236. https://doi.org/10.1016/j.jacc.2009.10.047
Schultz-Hector S, Trott KR (2007) Radiation-induced cardiovascular diseases: is the epidemiologic evidence compatible with the radiobiologic data? Int J Radiat Oncol Biol Phys 67(1):10–18. https://doi.org/10.1016/j.ijrobp.2006.08.071
Hildebrandt G, Maggiorella L, Rodel F, Rodel V, Willis D, Trott KR (2002) Mononuclear cell adhesion and cell adhesion molecule liberation after X-irradiation of activated endothelial cells in vitro. Int J Radiat Biol 78(4):315–325. https://doi.org/10.1080/09553000110106027
Hallahan DE, Virudachalam S (1997) Intercellular adhesion molecule 1 knockout abrogates radiation induced pulmonary inflammation. Proc Natl Acad Sci USA 94(12):6432–6437
Hallahan DE, Virudachalam S, Kuchibhotla J (1998) Nuclear factor kappaB dominant negative genetic constructs inhibit X-ray induction of cell adhesion molecules in the vascular endothelium. Cancer Res 58(23):5484–5488
Van Der Meeren A, Squiban C, Gourmelon P, Lafont H, Gaugler MH (1999) Differential regulation by IL-4 and IL-10 of radiation-induced IL-6 and IL-8 production and ICAM-1 expression by human endothelial cells. Cytokine 11(11):831–838. https://doi.org/10.1006/cyto.1999.0497
Milliat F, Francois A, Isoir M, Deutsch E, Tamarat R, Tarlet G, Atfi A, Validire P, Bourhis J, Sabourin JC, Benderitter M (2006) Influence of endothelial cells on vascular smooth muscle cells phenotype after irradiation: implication in radiation-induced vascular damages. Am J Pathol 169(4):1484–1495. https://doi.org/10.2353/ajpath.2006.060116
Baselet B, Azimzadeh O, Erbeldinger N, Bakshi MV, Dettmering T, Janssen A, Ktitareva S, Lowe DJ, Michaux A, Quintens R, Raj K, Durante M, Fournier C, Benotmane MA, Baatout S, Sonveaux P, Tapio S, Aerts A (2017) Differential impact of single-dose fe ion and X-ray irradiation on endothelial cell transcriptomic and proteomic responses. Front Pharmacol 8:570. https://doi.org/10.3389/fphar.2017.00570
Stewart FA, Heeneman S, Te Poele J, Kruse J, Russell NS, Gijbels M, Daemen M (2006) Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE−/− mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am J Pathol 168(2):649–658. https://doi.org/10.2353/ajpath.2006.050409
Hayashi T, Morishita Y, Khattree R, Misumi M, Sasaki K, Hayashi I, Yoshida K, Kajimura J, Kyoizumi S, Imai K, Kusunoki Y, Nakachi K (2012) Evaluation of systemic markers of inflammation in atomic-bomb survivors with special reference to radiation and age effects. FASEB J 26(11):4765–4773. https://doi.org/10.1096/fj.12-215228
Baselet B, Rombouts C, Benotmane AM, Baatout S, Aerts A (2016) Cardiovascular diseases related to ionizing radiation: the risk of low-dose exposure (Review). Int J Mol Med 38(6):1623–1641. https://doi.org/10.3892/ijmm.2016.2777
Cervelli T, Panetta D, Navarra T, Andreassi MG, Basta G, Galli A, Salvadori PA, Picano E, Del Turco S (2014) Effects of single and fractionated low-dose irradiation on vascular endothelial cells. Atherosclerosis 235(2):510–518. https://doi.org/10.1016/j.atherosclerosis.2014.05.932
Baselet B, Belmans N, Coninx E, Lowe D, Janssen A, Michaux A, Tabury K, Raj K, Quintens R, Benotmane MA, Baatout S, Sonveaux P, Aerts A (2017) Functional gene analysis reveals cell cycle changes and inflammation in endothelial cells irradiated with a single X-ray dose. Front Pharmacol 12:12. https://doi.org/10.3389/fphar.2017.00213
Kern PM, Keilholz L, Forster C, Hallmann R, Herrmann M, Seegenschmiedt MH (2000) Low-dose radiotherapy selectively reduces adhesion of peripheral blood mononuclear cells to endothelium in vitro. Radiother Oncol 54(3):273–282
Large M, Hehlgans S, Reichert S, Gaipl US, Fournier C, Rodel C, Weiss C, Rodel F (2015) Study of the anti-inflammatory effects of low-dose radiation: the contribution of biphasic regulation of the antioxidative system in endothelial cells. Strahlenther Onkol 191(9):742–749. https://doi.org/10.1007/s00066-015-0848-9
Rodel F, Frey B, Capalbo G, Gaipl U, Keilholz L, Voll R, Hildebrandt G, Rodel C (2010) Discontinuous induction of X-linked inhibitor of apoptosis in EA.hy.926 endothelial cells is linked to NF-kappaB activation and mediates the anti-inflammatory properties of low-dose ionising-radiation. Radiother Oncol 97(2):346–351. https://doi.org/10.1016/j.radonc.20
Rodel F, Hofmann D, Auer J, Keilholz L, Rollinghoff M, Sauer R, Beuscher HU (2008) The anti-inflammatory effect of low-dose radiation therapy involves a diminished CCL20 chemokine expression and granulocyte/endothelial cell adhesion. Strahlenther Onkol 184(1):41–47. https://doi.org/10.1007/s00066-008-1776-8
Rodel F, Kamprad F, Sauer R, Hildebrandt G (2002) Functional and molecular aspects of anti-inflammatory effects of low-dose radiotherapy. Strahlenther Onkol 178(1):1–9
Rodel F, Keilholz L, Herrmann M, Sauer R, Hildebrandt G (2007) Radiobiological mechanisms in inflammatory diseases of low-dose radiation therapy. Int J Radiat Biol 83(6):357–366. https://doi.org/10.1080/09553000701317358
Rodel F, Schaller U, Schultze-Mosgau S, Beuscher HU, Keilholz L, Herrmann M, Voll R, Sauer R, Hildebrandt G (2004) The induction of TGF-beta(1) and NF-kappaB parallels a biphasic time course of leukocyte/endothelial cell adhesion following low-dose X-irradiation. Strahlenther Onkol 180(4):194–200. https://doi.org/10.1007/s00066-004-1237-y
Le Gallic C, Phalente Y, Manens L, Dublineau I, Benderitter M, Gueguen Y, Lehoux S, Ebrahimian TG (2015) Chronic internal exposure to low dose 137Cs induces positive impact on the stability of atherosclerotic plaques by reducing inflammation in ApoE−/− Mice. PLoS One 10(6):e0128539. https://doi.org/10.1371/journal.pone.0128539
Trott KR, Kamprad F (1999) Radiobiological mechanisms of anti-inflammatory radiotherapy. Radiother Oncol 51(3):197–203
Seegenschmiedt MH, Katalinic A, Makoski HB, Haase W, Gademann G, Hassenstein E (1999) Radiotherapy of benign diseases: a pattern of care study in Germany. Strahlenther Onkol 175(11):541–547
Lerman A, Burnett JC Jr (1992) Intact and altered endothelium in regulation of vasomotion. Circulation 86(6 Suppl):III12–III19
Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C (2012) The role of nitric oxide on endothelial function. Curr Vasc Pharmacol 10(1):4–18
Mombouli JV, Vanhoutte PM (1999) Endothelial dysfunction: from physiology to therapy. J Mol Cell Cardiol 31(1):61–74. https://doi.org/10.1006/jmcc.1998.0844
Vanhoutte PM, Shimokawa H, Feletou M, Tang EH (2017) Endothelial dysfunction and vascular disease—a 30th anniversary update. Acta Physiol (Oxf) 219(1):22–96. https://doi.org/10.1111/apha.12646
Gisone P, Dubner D, Del Perez Rosario M, Michelin S, Puntarulo S (2004) The role of nitric oxide in the radiation-induced effects in the developing brain. Vivo 18(3):281–292
Hong CW, Kim YM, Pyo H, Lee JH, Kim S, Lee S, Noh JM (2013) Involvement of inducible nitric oxide synthase in radiation-induced vascular endothelial damage. J Radiat Res 54(6):1036–1042. https://doi.org/10.1093/jrr/rrt066
Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271(5 Pt 1):C1424–C1437. https://doi.org/10.1152/ajpcell.1996.271.5.C1424
Pathak R, Cheema AK, Boca SM, Krager KJ, Hauer-Jensen M, Aykin-Burns N (2015) Modulation of radiation response by the tetrahydrobiopterin pathway. Antioxidants (Basel) 4(1):68–81. https://doi.org/10.3390/antiox4010068
Sonveaux P, Brouet A, Havaux X, Gregoire V, Dessy C, Balligand JL, Feron O (2003) Irradiation-induced angiogenesis through the up-regulation of the nitric oxide pathway: implications for tumor radiotherapy. Cancer Res 63(5):1012–1019
Sonveaux P, Dessy C, Brouet A, Jordan BF, Gregoire V, Gallez B, Balligand JL, Feron O (2002) Modulation of the tumor vasculature functionality by ionizing radiation accounts for tumor radiosensitization and promotes gene delivery. FASEB J 16(14):1979–1981. https://doi.org/10.1096/fj.02-0487fje
Nagane M, Yasui H, Sakai Y, Yamamori T, Niwa K, Hattori Y, Kondo T, Inanami O (2015) Activation of eNOS in endothelial cells exposed to ionizing radiation involves components of the DNA damage response pathway. Biochem Biophys Res Commun 456(1):541–546. https://doi.org/10.1016/j.bbrc.2014.12.002
Qi F, Sugihara T, Hattori Y, Yamamoto Y, Kanno M, Abe K (1998) Functional and morphological damage of endothelium in rabbit ear artery following irradiation with cobalt60. Br J Pharmacol 123(4):653–660. https://doi.org/10.1038/sj.bjp.0701654
Maynard KI, Stewart-Lee AL, Milner P, Burnstock G (1992) X-irradiation attenuates relaxant responses in the rabbit ear artery. Br J Pharmacol 105(1):126–128
Soloviev AI, Tishkin SM, Parshikov AV, Ivanova IV, Goncharov EV, Gurney AM (2003) Mechanisms of endothelial dysfunction after ionized radiation: selective impairment of the nitric oxide component of endothelium-dependent vasodilation. Br J Pharmacol 138(5):837–844. https://doi.org/10.1038/sj.bjp.0705079
Menendez JC, Casanova D, Amado JA, Salas E, Garcia-Unzueta MT, Fernandez F, de la Lastra LP, Berrazueta JR (1998) Effects of radiation on endothelial function. Int J Radiat Oncol Biol Phys 41(4):905–913
Sugihara T, Hattori Y, Yamamoto Y, Qi F, Ichikawa R, Sato A, Liu MY, Abe K, Kanno M (1999) Preferential impairment of nitric oxide-mediated endothelium-dependent relaxation in human cervical arteries after irradiation. Circulation 100(6):635–641
Beckman JA, Thakore A, Kalinowski BH, Harris JR, Creager MA (2001) Radiation therapy impairs endothelium-dependent vasodilation in humans. J Am Coll Cardiol 37(3):761–765
Verheij M, Koomen GC, van Mourik JA, Dewit L (1994) Radiation reduces cyclooxygenase activity in cultured human endothelial cells at low doses. Prostaglandins 48(6):351–366
Hosoi Y, Yamamoto M, Ono T, Sakamoto K (1993) Prostacyclin production in cultured endothelial cells is highly sensitive to low doses of ionizing radiation. Int J Radiat Biol 63(5):631–638
Friedman M, Saunders DS, Madden MC, Chaney EL, Kwock L (1986) The effects of ionizing radiation on the pulmonary endothelial cell uptake of alpha-aminoisobutyric acid and synthesis of prostacyclin. Radiat Res 106(2):171–181
Rubin DB, Drab EA, Ts’ao CH, Gardner D, Ward WF (1985) Prostacyclin synthesis in irradiated endothelial cells cultured from bovine aorta. J Appl Physiol 58(2):592–597
Eldor A, Vlodavsky I, HyAm E, Atzmon R, Fuks Z (1983) The effect of radiation on prostacyclin (PGI2) production by cultured endothelial cells. Prostaglandins 25(2):263–279
Hahn GL, Menconi MJ, Cahill M, Polgar P (1983) The influence of gamma radiation on arachidonic acid release and prostacyclin synthesis. Prostaglandins 25(6):783–791
Sinzinger H, Firbas W, Cromwell M (1982) Radiation induced alterations in rabbit aortic prostacyclin formation. Prostaglandins 24(3):323–329. https://doi.org/10.1016/0090-6980(82)90159-9
Allen JB, Sagerman RH, Stuart MJ (1981) Irradiation decreases vascular prostacyclin formation with no concomitant effect on platelet thromboxane production. Lancet 2(8257):1193–1196
Sinzinger H, Cromwell M, Firbas W (1984) Long-lasting depression of rabbit aortic prostacyclin formation by single-dose irradiation. Radiat Res 97(3):533–536
Estrada-Garcia L, Carrera-Rotllan J, Puig-Parellada P (2002) Effects of oxidative stress and antioxidant treatments on eicosanoid synthesis and lipid peroxidation in long term human umbilical vein endothelial cells culture. Prostaglandins Other Lipid Mediat 67(1):13–25
Eldor A, Vlodavsky I, Riklis E, Fuks Z (1987) Recovery of prostacyclin capacity of irradiated endothelial cells and the protective effect of vitamin C. Prostaglandins 34(2):241–255
Ivanova IV, Kislova OV, Soloviev AI (2012) Endothelium-derived hyperpolarizing factor as a reserve defense mechanism of the vascular control under ionizing radiation impact 3(2):161–173. https://doi.org/10.1615/IntJPhysPathophys.v3.i2.60
Lanza V, Fadda P, Iannone C, Negri R (2007) Low-dose ionizing radiation stimulates transcription and production of endothelin by human vein endothelial cells. Radiat Res 168(2):193–198. https://doi.org/10.1667/RR0780.1
Lanza V, Pretazzoli V, Olivieri G, Pascarella G, Panconesi A, Negri R (2005) Transcriptional response of human umbilical vein endothelial cells to low doses of ionizing radiation. J Radiat Res 46(2):265–276
Fardid R, Najafi M, Salajegheh A, Kazemi E, Rezaeyan A (2017) Radiation-induced non-targeted effect in vivo: evaluation of cyclooygenase-2 and endothelin-1 gene expression in rat heart tissues. J Cancer Res Ther 13(1):51–55. https://doi.org/10.4103/0973-1482.203601
Merlin SL, Brock GB, Begin LR, Hiou Tim FF, Macramalla AN, Seyam RM, Shenouda G, Dion SB (2001) New insights into the role of endothelin-1 in radiation-associated impotence. Int J Impot Res 13(2):104–109. https://doi.org/10.1038/sj.ijir.3900652
Papapetropoulos A, Burch SE, Topouzis S, Catravas JD (1993) Radiation-induced alterations in angiotensin converting enzyme activity in cultured bovine pulmonary arterial endothelial cell monolayers. Toxicol Appl Pharmacol 120(1):96–105. https://doi.org/10.1006/taap.1993.1091
Wei J, Xu H, Liu Y, Li B, Zhou F (2017) Effect of captopril on radiation-induced TGF-beta1 secretion in EA.Hy926 human umbilical vein endothelial cells. Oncotarget 8(13):20842–20850. https://doi.org/10.18632/oncotarget.15356
Ward WF, Kim YT, Molteni A, Solliday NH (1988) Radiation-induced pulmonary endothelial dysfunction in rats: modification by an inhibitor of angiotensin converting enzyme. Int J Radiat Oncol Biol Phys 15(1):135–140
Cao S, Wu R (2012) Expression of angiotensin II and aldosterone in radiation-induced lung injury. Cancer Biol Med 9(4):254–260. https://doi.org/10.7497/j.issn.2095-3941.2012.04.006
Heckenkamp J, Leszczynski D, Schiereck J, Kung J, LaMuraglia GM (1999) Different effects of photodynamic therapy and gamma-irradiation on vascular smooth muscle cells and matrix: implications for inhibiting restenosis. Arterioscler Thromb Vasc Biol 19(9):2154–2161
Gajdusek CM, Tian H, London S, Zhou D, Rasey J, Mayberg MR (1996) Gamma radiation effect on vascular smooth muscle cells in culture. Int J Radiat Oncol Biol Phys 36(4):821–828
Keller PF, Verin V, Ziegler T, Mermillod B, Popowski Y, Delafontaine P (2001) Gamma-irradiation markedly inhibits the hydrated collagen gel contradiction by arterial smooth muscle cells. J Investig Med 49(3):258–264. https://doi.org/10.2310/6650.2001.33970
Heckenkamp J, Nigri GR, Waterman PR, Overhaus M, Kossodo SC, Lamuraglia GM (2004) Gamma-irradiation modulates vascular smooth muscle cell and extracellular matrix function: implications for neointimal development. J Vasc Surg 39(5):1097–1103. https://doi.org/10.1016/j.jvs.2003.12.021
Rudijanto A (2007) The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones 39(2):86–93
Soloviev A, Tishkin S, Ivanova I, Zelensky S, Dosenko V, Kyrychenko S, Moreland RS (2009) Functional and molecular consequences of ionizing irradiation on large conductance Ca2+-activated K+ channels in rat aortic smooth muscle cells. Life Sci 84(5–6):164–171. https://doi.org/10.1016/j.lfs.2008.11.015
Soloviev AI, Tishkin SM, Zelensky SN, Ivanova IV, Kizub IV, Pavlova AA, Moreland RS (2005) Ionizing radiation alters myofilament calcium sensitivity in vascular smooth muscle: potential role of protein kinase C. Am J Physiol Regul Integr Comp Physiol 289(3):R755–R762. https://doi.org/10.1152/ajpregu.00748.2004
Roveri A, Coassin M, Maiorino M, Zamburlini A, van Amsterdam FT, Ratti E, Ursini F (1992) Effect of hydrogen peroxide on calcium homeostasis in smooth muscle cells. Arch Biochem Biophys 297(2):265–270
Satoh K, Nigro P, Berk BC (2010) Oxidative stress and vascular smooth muscle cell growth: a mechanistic linkage by cyclophilin A. Antioxid Redox Signal 12(5):675–682. https://doi.org/10.1089/ars.2009.2875
Ruef J, Rao GN, Li F, Bode C, Patterson C, Bhatnagar A, Runge MS (1998) Induction of rat aortic smooth muscle cell growth by the lipid peroxidation product 4-hydroxy-2-nonenal. Circulation 97(11):1071–1078
Rao GN, Alexander RW, Runge MS (1995) Linoleic acid and its metabolites, hydroperoxyoctadecadienoic acids, stimulate c-Fos, c-Jun, and c-Myc mRNA expression, mitogen-activated protein kinase activation, and growth in rat aortic smooth muscle cells. J Clin Invest 96(2):842–847. https://doi.org/10.1172/JCI118130
Pearson JD (1999) Endothelial cell function and thrombosis. Baillieres Best Pract Res Clin Haematol 12(3):329–341
Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM (1998) Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 91(10):3527–3561
van Kleef E, Verheij M, te Poele H, Oussoren Y, Dewit L, Stewart F (2000) In vitro and in vivo expression of endothelial von Willebrand factor and leukocyte accumulation after fractionated irradiation. Radiat Res 154(4):375–381
Boerma M, Kruse JJ, van Loenen M, Klein HR, Bart CI, Zurcher C, Wondergem J (2004) Increased deposition of von Willebrand factor in the rat heart after local ionizing irradiation. Strahlenther Onkol 180(2):109–116. https://doi.org/10.1007/s00066-004-1138-0
Stewart FA, Te Poele JA, Van der Wal AF, Oussoren YG, Van Kleef EM, Kuin A, Verheij M, Dewit LG (2001) Radiation nephropathy—the link between functional damage and vascular mediated inflammatory and thrombotic changes. Acta Oncol 40(8):952–957
Sporn LA, Rubin P, Marder VJ, Wagner DD (1984) Irradiation induces release of von Willebrand protein from endothelial cells in culture. Blood 64(2):567–570
Jahroudi N, Ardekani AM, Greenberger JS (1996) Ionizing irradiation increases transcription of the von Willebrand factor gene in endothelial cells. Blood 88(10):3801–3814
McManus LM, Ostrom KK, Lear C, Luce EB, Gander DL, Pinckard RN, Redding SW (1993) Radiation-induced increased platelet-activating factor activity in mixed saliva. Lab Invest 68(1):118–124
Wang J, Zheng H, Ou X, Fink LM, Hauer-Jensen M (2002) Deficiency of microvascular thrombomodulin and up-regulation of protease-activated receptor-1 in irradiated rat intestine: possible link between endothelial dysfunction and chronic radiation fibrosis. Am J Pathol 160(6):2063–2072. https://doi.org/10.1016/S0002-9440(10)61156-X
Zhou Q, Zhao Y, Li P, Bai X, Ruan C (1992) Thrombomodulin as a marker of radiation-induced endothelial cell injury. Radiat Res 131(3):285–289
Leigh PJ, Cramp WA, MacDermot J (1984) Identification of the prostacyclin receptor by radiation inactivation. J Biol Chem 259(20):12431–12436
Henderson BW, Bicher HI, Johnson RJ (1983) Loss of vascular fibrinolytic activity following irradiation of the liver–an aspect of late radiation damage. Radiat Res 95(3):646–652
Svanberg L, Astedt B, Kullander S (1976) On radiation-decreased fibrinolytic activity of vessel walls. Acta Obstet Gynecol Scand 55(1):49–51
Ts’ao CH, Ward WF, Port CD (1983) Radiation injury in rat lung. III. Plasminogen activator and fibrinolytic inhibitor activities. Radiat Res 96(2):301–308
Wang HF, Li XD, Chen YM, Yuan LB, Foye WO (1991) Radiation-protective and platelet aggregation inhibitory effects of five traditional Chinese drugs and acetylsalicylic acid following high-dose gamma-irradiation. J Ethnopharmacol 34(2–3):215–219
Schneider MD (1977) Functional aspects of blood platelets in irradiated burros. Am J Vet Res 38(2):209–216
Bicher HI, D’Agostino L, Doss LL, Kaufman N, Amigone J (1977) Prevention of ionizing radiation-induced liver microcirculation changes by the use of flow improvers. Adv Exp Med Biol 94:383–389
Kennedy AR, Maity A, Sanzari JK (2016) A review of radiation-induced coagulopathy and new findings to support potential prevention strategies and treatments. Radiat Res 186(2):121–140. https://doi.org/10.1667/RR14406.1
Kerr R, Stirling D, Ludlam CA (2001) Interleukin 6 and haemostasis. Br J Haematol 115(1):3–12
K-i Inoue, Takano H, Yanagisawa R, Shimada A, Yoshikawa T (2006) Role of interleukin-6 in coagulatory and hemostatic disturbance during inflammation. Ensho Saisei 26(1):40–43. https://doi.org/10.2492/jsir.26.40
Neumann FJ, Ott I, Marx N, Luther T, Kenngott S, Gawaz M, Kotzsch M, Schomig A (1997) Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb Vasc Biol 17(12):3399–3405
Hoving S, Heeneman S, Gijbels MJ, Te Poele JA, Visser N, Cleutjens J, Russell NS, Daemen MJ, Stewart FA (2012) Irradiation induces different inflammatory and thrombotic responses in carotid arteries of wildtype C57BL/6J and atherosclerosis-prone ApoE(−/−) mice. Radiother Oncol 105(3):365–370. https://doi.org/10.1016/j.radonc.2012.11.001
Wiig H (2011) Pathophysiology of tissue fluid accumulation in inflammation. J Physiol 589(Pt 12):2945–2953. https://doi.org/10.1113/jphysiol.2011.206136
Scallan J, Huxley V, Korthuis R (2010) Pathophysiology of Edema formation. Capillary fluid exchange: regulation, functions, and pathology. Morgan & Claypool Life Sciences, San Rafael
Sharma P, Templin T, Grabham P (2013) Short term effects of gamma radiation on endothelial barrier function: uncoupling of PECAM-1. Microvasc Res 86:11–20. https://doi.org/10.1016/j.mvr.2012.11.007
Templin T, Sharma P, Guida P, Grabham P (2016) Short-term effects of low-LET radiation on the endothelial barrier: uncoupling of PECAM-1 and the production of endothelial microparticles. Radiat Res 186(6):602–613. https://doi.org/10.1667/RR14510.1
Gabrys D, Greco O, Patel G, Prise KM, Tozer GM, Kanthou C (2007) Radiation effects on the cytoskeleton of endothelial cells and endothelial monolayer permeability. Int J Radiat Oncol Biol Phys 69(5):1553–1562. https://doi.org/10.1016/j.ijrobp.2007.08.039
Waters CM, Taylor JM, Molteni A, Ward WF (1996) Dose-response effects of radiation on the permeability of endothelial cells in culture. Radiat Res 146(3):321–328
Kantak SS, Diglio CA, Onoda JM (1993) Low dose radiation-induced endothelial cell retraction. Int J Radiat Biol 64(3):319–328
Onoda JM, Kantak SS, Diglio CA (1999) Radiation induced endothelial cell retraction in vitro: correlation with acute pulmonary edema. Pathol Oncol Res 5(1):49–55
Asai K, Kudej RK, Shen YT, Yang GP, Takagi G, Kudej AB, Geng YJ, Sato N, Nazareno JB, Vatner DE, Natividad F, Bishop SP, Vatner SF (2000) Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler Thromb Vasc Biol 20(6):1493–1499
Yu E, Mercer J, Bennett M (2012) Mitochondria in vascular disease. Cardiovasc Res 95(2):173–182. https://doi.org/10.1093/cvr/cvs111
Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C (2006) Clonogenic assay of cells in vitro. Nat Protoc 1(5):2315–2319. https://doi.org/10.1038/nprot.2006.339
Park MT, Oh ET, Song MJ, Lee H, Park HJ (2012) Radio-sensitivities and angiogenic signaling pathways of irradiated normal endothelial cells derived from diverse human organs. J Radiat Res 53(4):570–580. https://doi.org/10.1093/jrr/rrs011
Riquier H, Wera AC, Heuskin AC, Feron O, Lucas S, Michiels C (2013) Comparison of X-ray and alpha particle effects on a human cancer and endothelial cells: survival curves and gene expression profiles. Radiother Oncol 106(3):397–403. https://doi.org/10.1016/j.radonc.2013.02.017
Rombouts C, Aerts A, Beck M, De Vos WH, Van Oostveldt P, Benotmane MA, Baatout S (2013) Differential response to acute low dose radiation in primary and immortalized endothelial cells. Int J Radiat Biol 89(10):841–850. https://doi.org/10.3109/09553002.2013.806831
Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, Kolesnick RN (1994) Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med 180(2):525–535
Kolesnick R, Fuks Z (2003) Radiation and ceramide-induced apoptosis. Oncogene 22(37):5897–5906. https://doi.org/10.1038/sj.onc.1206702
Yabu T, Shiba H, Shibasaki Y, Nakanishi T, Imamura S, Touhata K, Yamashita M (2015) Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ 22(2):258–273. https://doi.org/10.1038/cdd.2014.128
Haimovitz-Friedman A, Kolesnick RN, Fuks Z (1997) Ceramide signaling in apoptosis. Br Med Bull 53(3):539–553
Dikomey E, Dahm-Daphi J, Brammer I, Martensen R, Kaina B (1998) Correlation between cellular radiosensitivity and non-repaired double-strand breaks studied in nine mammalian cell lines. Int J Radiat Biol 73(3):269–278
Langley RE, Bump EA, Quartuccio SG, Medeiros D, Braunhut SJ (1997) Radiation-induced apoptosis in microvascular endothelial cells. Br J Cancer 75(5):666–672
Pluder F, Barjaktarovic Z, Azimzadeh O, Mortl S, Kramer A, Steininger S, Sarioglu H, Leszczynski D, Nylund R, Hakanen A, Sriharshan A, Atkinson MJ, Tapio S (2011) Low-dose irradiation causes rapid alterations to the proteome of the human endothelial cell line EA.hy926. Radiat Environ Biophys 50(1):155–166. https://doi.org/10.1007/s00411-010-0342-9
Hirase T, Node K (2012) Endothelial dysfunction as a cellular mechanism for vascular failure. Am J Physiol Heart Circ Physiol 302(3):H499–H505. https://doi.org/10.1152/ajpheart.00325.2011
Chistiakov DA, Orekhov AN, Bobryshev YV (2015) Endothelial barrier and its abnormalities in cardiovascular disease. Front Physiol 6:365. https://doi.org/10.3389/fphys.2015.00365
Davidson SM, Duchen MR (2007) Endothelial mitochondria: contributing to vascular function and disease. Circ Res 100(8):1128–1141. https://doi.org/10.1161/01.RES.0000261970.18328.1d
Groschner LN, Waldeck-Weiermair M, Malli R, Graier WF (2012) Endothelial mitochondria—less respiration, more integration. Pflugers Arch 464(1):63–76. https://doi.org/10.1007/s00424-012-1085-z
Kluge MA, Fetterman JL, Vita JA (2013) Mitochondria and endothelial function. Circ Res 112(8):1171–1188. https://doi.org/10.1161/CIRCRESAHA.111.300233
Tang X, Luo YX, Chen HZ, Liu DP (2014) Mitochondria, endothelial cell function, and vascular diseases. Front Physiol 5:175. https://doi.org/10.3389/fphys.2014.00175
Szewczyk A, Jarmuszkiewicz W, Koziel A, Sobieraj I, Nobik W, Lukasiak A, Skup A, Bednarczyk P, Drabarek B, Dymkowska D, Wrzosek A, Zablocki K (2015) Mitochondrial mechanisms of endothelial dysfunction. Pharmacol Rep 67(4):704–710. https://doi.org/10.1016/j.pharep.2015.04.009
Blouin A, Bolender RP, Weibel ER (1977) Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol 72(2):441–455
Culic O, Gruwel ML, Schrader J (1997) Energy turnover of vascular endothelial cells. Am J Physiol 273(1 Pt 1):C205–C213
De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154(3):651–663. https://doi.org/10.1016/j.cell.2013.06.037
Krutzfeldt A, Spahr R, Mertens S, Siegmund B, Piper HM (1990) Metabolism of exogenous substrates by coronary endothelial cells in culture. J Mol Cell Cardiol 22(12):1393–1404
Quintero M, Colombo SL, Godfrey A, Moncada S (2006) Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci USA 103(14):5379–5384. https://doi.org/10.1073/pnas.0601026103
Hu S, Gao Y, Zhou H, Kong F, Xiao F, Zhou P, Chen Y (2017) New insight into mitochondrial changes in vascular endothelial cells irradiated by gamma ray. Int J Radiat Biol 93(5):470–476. https://doi.org/10.1080/09553002.2017.1286048
Azimzadeh O, Sievert W, Sarioglu H, Merl-Pham J, Yentrapalli R, Bakshi MV, Janik D, Ueffing M, Atkinson MJ, Multhoff G, Tapio S (2015) Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long-term radiation-induced vascular dysfunction. J Proteome Res 14(2):1203–1219. https://doi.org/10.1021/pr501141b
Gunter KK, Gunter TE (1994) Transport of calcium by mitochondria. J Bioenerg Biomembr 26(5):471–485
Gunter TE, Pfeiffer DR (1990) Mechanisms by which mitochondria transport calcium. Am J Physiol 258(5 Pt 1):C755–C786
Griffiths EJ, Rutter GA (2009) Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 1787(11):1324–1333. https://doi.org/10.1016/j.bbabio.2009.01.019
Paltauf-Doburzynska J, Malli R, Graier WF (2004) Hyperglycemic conditions affect shape and Ca2 + homeostasis of mitochondria in endothelial cells. J Cardiovasc Pharmacol 44(4):423–436
Rowlands DJ, Islam MN, Das SR, Huertas A, Quadri SK, Horiuchi K, Inamdar N, Emin MT, Lindert J, Ten VS, Bhattacharya S, Bhattacharya J (2011) Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J Clin Invest 121(5):1986–1999. https://doi.org/10.1172/JCI43839
Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD, Zhang DX (2012) Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circul Physiol 302(3):H634–H642. https://doi.org/10.1152/ajpheart.00717.2011
Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, Gandhirajan RK, Vagnozzi RJ, Ferrer LM, Sreekrishnanilayam K, Natarajaseenivasan K, Vallem S, Force T, Choi ET, Cheung JY, Madesh M (2013) MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep 5(6):1576–1588. https://doi.org/10.1016/j.celrep.2013.11.026
Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M (2012) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151(3):630–644. https://doi.org/10.1016/j.cell.2012.10.011
Kam WW, Banati RB (2013) Effects of ionizing radiation on mitochondria. Free Radic Biol Med 65:607–619. https://doi.org/10.1016/j.freeradbiomed.2013.07.024
Decrock E, Hoorelbeke D, Ramadan R, Delvaeye T, De Bock M, Wang N, Krysko DV, Baatout S, Bultynck G, Aerts A, Vinken M, Leybaert L (1864) (2017) Calcium, oxidative stress and connexin channels, a harmonious orchestra directing the response to radiotherapy treatment? Biochim Biophys Acta 6:1099–1120. https://doi.org/10.1016/j.bbamcr.2017.02.007
Brunelle JK, Letai A (2009) Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 122(Pt 4):437–441. https://doi.org/10.1242/jcs.031682
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90(3):405–413
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD (1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275(5303):1132–1136
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91(4):479–489
Wang Y, Boerma M, Zhou D (2016) Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res 186(2):153–161. https://doi.org/10.1667/RR14445.1
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417(1):1–13. https://doi.org/10.1042/BJ20081386
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120(4):483–495. https://doi.org/10.1016/j.cell.2005.02.001
Little MP, Tawn EJ, Tzoulaki I, Wakeford R, Hildebrandt G, Paris F, Tapio S, Elliott P (2008) A systematic review of epidemiological associations between low and moderate doses of ionizing radiation and late cardiovascular effects, and their possible mechanisms. Radiat Res 169(1):99–109. https://doi.org/10.1667/RR1070.1
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48(2):158–167. https://doi.org/10.1016/j.molcel.2012.09.025
Riley PA (1994) Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol 65(1):27–33
Kowaltowski AJ, Vercesi AE (1999) Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med 26(3–4):463–471
Mikkelsen RB, Wardman P (2003) Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 22(37):5734–5754. https://doi.org/10.1038/sj.onc.1206663
McDonald JT, Kim K, Norris AJ, Vlashi E, Phillips TM, Lagadec C, Della Donna L, Ratikan J, Szelag H, Hlatky L, McBride WH (2010) Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res 70(21):8886–8895. https://doi.org/10.1158/0008-5472.CAN-10-0171
Patwardhan RS, Checker R, Sharma D, Sandur SK, Sainis KB (2013) Involvement of ERK-Nrf-2 signaling in ionizing radiation induced cell death in normal and tumor cells. PLoS One 8(6):e65929. https://doi.org/10.1371/journal.pone.0065929
Kensler TW, Wakabayashi N, Biswal S (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47:89–116. https://doi.org/10.1146/annurev.pharmtox.46.120604.141046
Kang KW, Lee SJ, Kim SG (2005) Molecular mechanism of nrf2 activation by oxidative stress. Antioxid Redox Signal 7(11–12):1664–1673. https://doi.org/10.1089/ars.2005.7.1664
Chen N, Wu L, Yuan H, Wang J (2015) ROS/autophagy/Nrf2 pathway mediated low-dose radiation induced radio-resistance in human lung adenocarcinoma A549 cell. Int J Biol Sci 11(7):833–844. https://doi.org/10.7150/ijbs.10564
Jayakumar S, Pal D, Sandur SK (2015) Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat Res 779:33–45. https://doi.org/10.1016/j.mrfmmm.2015.06.007
Khan NM, Sandur SK, Checker R, Sharma D, Poduval TB, Sainis KB (2011) Pro-oxidants ameliorate radiation-induced apoptosis through activation of the calcium-ERK1/2-Nrf2 pathway. Free Radic Biol Med 51(1):115–128. https://doi.org/10.1016/j.freeradbiomed.2011.03.037
Sekhar KR, Freeman ML (2015) Nrf2 promotes survival following exposure to ionizing radiation. Free Radic Biol Med 88(Pt B):268–274. https://doi.org/10.1016/j.freeradbiomed.2015.04.035
Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S (2010) Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid Redox Signal 13(11):1627–1637. https://doi.org/10.1089/ars.2010.3219
Zhao Q, Mao A, Yan J, Sun C, Di C, Zhou X, Li H, Guo R, Zhang H (2016) Downregulation of Nrf2 promotes radiation-induced apoptosis through Nrf2 mediated Notch signaling in non-small cell lung cancer cells. Int J Oncol 48(2):765–773. https://doi.org/10.3892/ijo.2015.3301
Anuranjani Bala M (2014) Concerted action of Nrf2-ARE pathway, MRN complex, HMGB1 and inflammatory cytokines—implication in modification of radiation damage. Redox Biol 2:832–846. https://doi.org/10.1016/j.redox.2014.02.008
Chen B, Lu Y, Chen Y, Cheng J (2015) The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol 225(3):R83–R99. https://doi.org/10.1530/JOE-14-0662
Hoshi Y, Tanooka H, Miyazaki K, Wakasugi H (1997) Induction of thioredoxin in human lymphocytes with low-dose ionizing radiation. Biochim Biophys Acta 1359(1):65–70
Chen WC, McBride WH, Iwamoto KS, Barber CL, Wang CC, Oh YT, Liao YP, Hong JH, de Vellis J, Shau H (2002) Induction of radioprotective peroxiredoxin-I by ionizing irradiation. J Neurosci Res 70(6):794–798. https://doi.org/10.1002/jnr.10435
An JH, Kim J, Seong J (2004) Redox signaling by ionizing radiation in mouse liver. Ann N Y Acad Sci 1030:86–94. https://doi.org/10.1196/annals.1329.011
Gao MC, Jia XD, Wu QF, Cheng Y, Chen FR, Zhang J (2011) Silencing Prx1 and/or Prx5 sensitizes human esophageal cancer cells to ionizing radiation and increases apoptosis via intracellular ROS accumulation. Acta Pharmacol Sin 32(4):528–536. https://doi.org/10.1038/aps.2010.235
Lee K, Park JS, Kim YJ, Soo Lee YS, Sook Hwang TS, Kim DJ, Park EM, Park YM (2002) Differential expression of Prx I and II in mouse testis and their up-regulation by radiation. Biochem Biophys Res Commun 296(2):337–342
Woolston CM, Storr SJ, Ellis IO, Morgan DA, Martin SG (2011) Expression of thioredoxin system and related peroxiredoxin proteins is associated with clinical outcome in radiotherapy treated early stage breast cancer. Radiother Oncol 100(2):308–313. https://doi.org/10.1016/j.radonc.2011.05.029
Reisz JA, Bansal N, Qian J, Zhao W, Furdui CM (2014) Effects of ionizing radiation on biological molecules—mechanisms of damage and emerging methods of detection. Antioxid Redox Signal 21(2):260–292. https://doi.org/10.1089/ars.2013.5489
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94(3):909–950. https://doi.org/10.1152/physrev.00026.2013
Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, Inanami O (2012) Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med 53(2):260–270. https://doi.org/10.1016/j.freeradbiomed.2012.04.033
Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 94(2):514–519
Prithivirajsingh S, Story MD, Bergh SA, Geara FB, Ang KK, Ismail SM, Stevens CW, Buchholz TA, Brock WA (2004) Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett 571(1–3):227–232. https://doi.org/10.1016/j.febslet.2004.06.078
Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB (2001) Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res 61(10):3894–3901
Wang L, Kuwahara Y, Li L, Baba T, Shin RW, Ohkubo Y, Ono K, Fukumoto M (2007) Analysis of common deletion (CD) and a novel deletion of mitochondrial DNA induced by ionizing radiation. Int J Radiat Biol 83(7):433–442. https://doi.org/10.1080/09553000701370878
Schilling-Toth B, Sandor N, Kis E, Kadhim M, Safrany G, Hegyesi H (2011) Analysis of the common deletions in the mitochondrial DNA is a sensitive biomarker detecting direct and non-targeted cellular effects of low dose ionizing radiation. Mutat Res 716(1–2):33–39. https://doi.org/10.1016/j.mrfmmm.2011.07.018
Lafargue A, Degorre C, Corre I, Alves-Guerra MC, Gaugler MH, Vallette F, Pecqueur C, Paris F (2017) Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic Biol Med 108:750–759. https://doi.org/10.1016/j.freeradbiomed.2017.04.019
Helm A, Lee R, Durante M, Ritter S (2016) The influence of C-ions and X-rays on human umbilical vein endothelial cells. Front Oncol 6:5. https://doi.org/10.3389/fonc.2016.00005
Barjaktarovic Z, Shyla A, Azimzadeh O, Schulz S, Haagen J, Dorr W, Sarioglu H, Atkinson MJ, Zischka H, Tapio S (2013) Ionising radiation induces persistent alterations in the cardiac mitochondrial function of C57BL/6 mice 40 weeks after local heart exposure. Radiother Oncol 106(3):404–410. https://doi.org/10.1016/j.radonc.2013.01.017
Kempf SJ, Moertl S, Sepe S, von Toerne C, Hauck SM, Atkinson MJ, Mastroberardino PG, Tapio S (2015) Low-dose ionizing radiation rapidly affects mitochondrial and synaptic signaling pathways in murine hippocampus and cortex. J Proteome Res 14(5):2055–2064. https://doi.org/10.1021/acs.jproteome.5b00114
Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A (2010) Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 65(10):1028–1041. https://doi.org/10.1093/gerona/glq113
Voghel G, Thorin-Trescases N, Farhat N, Mamarbachi AM, Villeneuve L, Fortier A, Perrault LP, Carrier M, Thorin E (2008) Chronic treatment with N-acetyl-cystein delays cellular senescence in endothelial cells isolated from a subgroup of atherosclerotic patients. Mech Ageing Dev 129(5):261–270. https://doi.org/10.1016/j.mad.2008.01.004
Voghel G, Thorin-Trescases N, Farhat N, Nguyen A, Villeneuve L, Mamarbachi AM, Fortier A, Perrault LP, Carrier M, Thorin E (2007) Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev 128(11–12):662–671. https://doi.org/10.1016/j.mad.2007.09.006
Campisi J, d’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8(9):729–740. https://doi.org/10.1038/nrm2233
Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37:614–636
Zhu H, Belcher M, van der Harst P (2011) Healthy aging and disease: role for telomere biology? Clinical science (London, England: 1979) 120(10):427–440. https://doi.org/10.1042/CS20100385
Ben-Porath I, Weinberg RA (2005) The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 37(5):961–976. https://doi.org/10.1016/j.biocel.2004.10.013
Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD (2004) Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci 117(Pt 11):2417–2426. https://doi.org/10.1242/jcs.01097
Minamino T, Komuro I (2007) Vascular cell senescence: contribution to atherosclerosis. Circ Res 100(1):15–26. https://doi.org/10.1161/01.RES.0000256837.40544.4a
El Assar M, Angulo J, Vallejo S, Peiro C, Sanchez-Ferrer CF, Rodriguez-Manas L (2012) Mechanisms involved in the aging-induced vascular dysfunction. Front Physiol 3:132. https://doi.org/10.3389/fphys.2012.00132
Erusalimsky JD (2009) Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol 106(1):326–332. https://doi.org/10.1152/japplphysiol.91353.2008
Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I (2002) Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105(13):1541–1544
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144
Sabatino L, Picano E, Andreassi MG (2012) Telomere shortening and ionizing radiation: a possible role in vascular dysfunction? Int J Radiat Biol 88(11):830–839. https://doi.org/10.3109/09553002.2012.709307
Vavrova J, Rezacova M (2011) The importance of senescence in ionizing radiation-induced tumour suppression. Folia Biol (Praha) 57(2):41–46
Lowe D, Raj K (2014) Premature aging induced by radiation exhibits pro-atherosclerotic effects mediated by epigenetic activation of CD44 expression. Aging Cell 13(5):900–910. https://doi.org/10.1111/acel.12253
Igarashi K, Miura M (2008) Inhibition of a radiation-induced senescence-like phenotype: a possible mechanism for potentially lethal damage repair in vascular endothelial cells. Radiat Res 170(4):534–539
Igarashi K, Sakimoto I, Kataoka K, Ohta K, Miura M (2007) Radiation-induced senescence-like phenotype in proliferating and plateau-phase vascular endothelial cells. Exp Cell Res 313(15):3326–3336. https://doi.org/10.1016/j.yexcr.2007.06.001
Kim KS, Kim JE, Choi KJ, Bae S, Kim DH (2014) Characterization of DNA damage-induced cellular senescence by ionizing radiation in endothelial cells. Int J Radiat Biol 90(1):71–80. https://doi.org/10.3109/09553002.2014.859763
Panganiban RA, Mungunsukh O, Day RM (2013) X-irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int J Radiat Biol 89(8):656–667. https://doi.org/10.3109/09553002.2012.711502
Oh CW, Bump EA, Kim JS, Janigro D, Mayberg MR (2001) Induction of a senescence-like phenotype in bovine aortic endothelial cells by ionizing radiation. Radiat Res 156(3):232–240
Rombouts C, Aerts A, Quintens R, Baselet B, El-Saghire H, Harms-Ringdahl M, Haghdoost S, Janssen A, Michaux A, Yentrapalli R, Benotmane MA, Van Oostveldt P, Baatout S (2014) Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int J Radiat Biol 90(7):560–574. https://doi.org/10.3109/09553002.2014.905724
Yentrapalli R, Azimzadeh O, Barjaktarovic Z, Sarioglu H, Wojcik A, Harms-Ringdahl M, Atkinson MJ, Haghdoost S, Tapio S (2013) Quantitative proteomic analysis reveals induction of premature senescence in human umbilical vein endothelial cells exposed to chronic low-dose rate gamma radiation. Proteomics 13(7):1096–1107. https://doi.org/10.1002/pmic.201200463
Yentrapalli R, Azimzadeh O, Sriharshan A, Malinowsky K, Merl J, Wojcik A, Harms-Ringdahl M, Atkinson MJ, Becker KF, Haghdoost S, Tapio S (2013) The PI3 K/Akt/mTOR pathway is implicated in the premature senescence of primary human endothelial cells exposed to chronic radiation. PLoS One 8(8):e70024. https://doi.org/10.1371/journal.pone.0070024
Slezak J, Kura B, Babal P, Barancik M, Ferko M, Frimmel K, Kalocayova B, Kukreja RC, Lazou A, Mezesova L, Okruhlicova L, Ravingerova T, Singal PK, Szeiffova Bacova B, Viczenczova C, Vrbjar N, Tribulova N (2017) Potential markers and metabolic processes involved in the mechanism of radiation-induced heart injury. Can J Physiol Pharmacol 95(10):1190–1203. https://doi.org/10.1139/cjpp-2017-0121
Slezak J, Kura B, Ravingerova T, Tribulova N, Okruhlicova L, Barancik M (2015) Mechanisms of cardiac radiation injury and potential preventive approaches. Can J Physiol Pharmacol 93(9):737–753. https://doi.org/10.1139/cjpp-2015-0006
Acknowledgements
This work was funded by European Union Seventh Framework Program DoReMi network of excellence (Grant #249689), European Union Seventh Framework Program project ProCardio (Grant #295823), the Belgian Federal Agency for Nuclear Control (FANC-AFCN; Grant #CO-90-13-3289-00) and the Belgian Fonds National de la Recherche Scientifique (F.R.S.-FNRS). Bjorn Baselet is supported by a doctoral SCK CEN grant. Pierre Sonveaux is a F.R.S.-FNRS Senior Research Associate.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Baselet, B., Sonveaux, P., Baatout, S. et al. Pathological effects of ionizing radiation: endothelial activation and dysfunction. Cell. Mol. Life Sci. 76, 699–728 (2019). https://doi.org/10.1007/s00018-018-2956-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2956-z