
Abstract
The core axoneme structure of both the motile cilium and sperm tail has the same ultrastructural 9 + 2 microtubular arrangement. Thus, it can be expected that genetic defects in motile cilia also have an effect on sperm tail formation. However, recent studies in human patients, animal models and model organisms have indicated that there are differences in components of specific structures within the cilia and sperm tail axonemes. Primary ciliary dyskinesia (PCD) is a genetic disease with symptoms caused by malfunction of motile cilia such as chronic nasal discharge, ear, nose and chest infections and pulmonary disease (bronchiectasis). Half of the patients also have situs inversus and in many cases male infertility has been reported. PCD genes have a role in motile cilia biogenesis, structure and function. To date mutations in over 40 genes have been identified cause PCD, but the exact effect of these mutations on spermatogenesis is poorly understood. Furthermore, mutations in several additional axonemal genes have recently been identified to cause a sperm-specific phenotype, termed multiple morphological abnormalities of the sperm flagella (MMAF). In this review, we discuss the association of PCD genes and other axonemal genes with male infertility, drawing particular attention to possible differences between their functions in motile cilia and sperm tails.
Similar content being viewed by others

Introduction
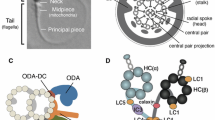
Motile cilia and flagella have a conserved axonemal structure consisting of a ring of nine microtubular doublets and a central pair of microtubules, giving the classical 9 + 2 microtubular arrangement (Fig. 1a). Central pair microtubules C1 and C2 are connected by a bridge-like structure and several projections are docked to C1 and C2 forming the central pair complex (CPC). Each outer doublet is composed of type A and B microtubules and connected by radial spokes (RS) to CPC (Fig. 1a). The force for motility is produced by inner and outer dynein arms (IDA and ODA, respectively), which are carried by the A-type tubule and project toward the B-tubule of the adjacent doublet. IDA and ODA are part of a specific protein complex of 96 nm repeat units, which contains four identical ODAs, one two-headed IDA (f/I1), six single-headed IDAs (a–e and g), three RS and a single nexin–dynein regulatory complex (N-DRC) (Fig. 1c) The N-DRC regulates and coordinates the activity of the dynein arms [43]. The current knowledge of motile cilia ultrastructure and protein content has been previously reviewed by Osinka et al. [96]. The sperm flagellum has an ultrastructurally comparable axonemal structure, but in addition the sperm tail contains accessory structures: the mitochondrial sheath (MS), fibrous sheath (FS) and outer dense fibres (ODFs) (Fig. 1b). These structures are specific to the sperm tail and are required for fertile sperm production providing, e.g. additional rigidity and energy for the movement of the sperm in the female reproductive tract [58]. Sperm tail is connected to the head by the head tail coupling apparatus (HTCA) and annulus forms a diffusion barrier between the mid-piece and principal piece (Fig. 1b). Any alteration in protein functions within these structures can be expected to have a sperm-specific phenotype. The formation of specific sperm tail structures and their associated proteins have been recently reviewed in detail [58]. Furthermore, although the axonemal structure appears to be highly conserved between sperm tail and motile cilia, several studies have suggested that differences exist based on tissue-specific gene and protein expression [1, 2, 6, 19, 63, 130, 135, 137].

Axonemal structure of respiratory cilia and sperm flagella. a The shared electron microscopic axonemal structure of the 9 + 2 motile respiratory cilia and sperm flagellum with major structural features indicated. Outer doublet microtubules are connected to the central pair (CP) by radial spokes (RS) and to each other by nexin links (nexin–dynein regulatory complex, N-DRC). Outer and inner dynein arms (ODA and IDA, respectively) provide the energy for the movement. b Accessory structures of the sperm tail. The 9 + 2 axoneme is surrounded by outer dense fibres (ODF) in the mid and principal piece of the sperm tail. Along the mid piece the ODFs are surrounded by mitochondria and along the principal piece the fibrous sheath replaces ODFs 3 and 8 and the transverse ribs (TR) encircle ODFs. The mid-piece and principal piece are separated by the annulus and the tail is connected to the head by the head tail coupling apparatus (HTCA). The formation of sperm tail structures and associated proteins have been previously reviewed [58]. c Schematic presentation of the protein complex 96 nm repeat units along the A doublet microtubule. The complex is formed of four identical ODAs, one two-headed IDA (f/I1), six single-headed IDAs (a–e and g), three RS and a single N-DRC
Motile cilia malfunction causes primary ciliary dyskinesia (PCD), which is a genetic condition affecting approximately 1:10,000 individuals worldwide [73, 106]. Symptoms frequently start in the neonatal period and include a chronic nasal discharge and wet cough, progressing in childhood to recurrent ear, nose and chest infections and eventual scarring of the lungs in the form of bronchiectasis. The cause of the symptoms is the malfunction of airway motile cilia, which are responsible for mucus clearance in the airways. Motile cilia are also present in the brain, oviduct, Eustachian tube and middle ear, where they function in fluid movement and in the embryonic node during development. Defects in the nodal cilia cause situs inversus approximately in half of PCD patients [7, 131]. PCD is a lifelong condition, the impact of symptoms changing as the patient reaches adulthood. Ear symptoms often attenuate, but bronchiectasis usually progresses and many patients experience fertility problems when trying to start a family [101, 129]. Clinical manifestations and management of PCD have been described in previous publications [36, 73, 106].
Despite extensive and increasing understanding of the spectrum of cilia structural defects in PCD, only few reports exist on the sperm structural defects affecting males with PCD. An early study of the association of male infertility with PCD was done at the Royal Brompton Hospital London in 1994 [86]. Twelve men with PCD had their respiratory cilia and sperm flagella analysed for motility and ultrastructure. Two patients had immotile sperm and one had normal motility with oligospermia (low sperm numbers; Table 1 explains terminology of sperm phenotypic defects). Three of these patients had normal sperm motility and ultrastructure and one had fathered a child. The twelfth patient had slowed sperm with ultrastructural dynein deficiency, but had also reportedly fathered a child. Five had azoospermia (no sperm), which the authors attributed at the time to defects in the ciliated portion of the vas deferens preventing normal transport. However, there are no cilia in the vas deferens, but motile cilia are present in the efferent ductules [20, 142]. Recent studies have shown that these cilia are not required for transport of sperm towards the cauda epididymis, but they agitate the seminal fluid to prevent blockage and to enable reabsorption of the fluid by a rotational swirling motion rather than forward ciliary beat [142]. Thus, the lack of sperm is unlikely to be caused by defects in sperm transport, but may arise from blockage of the efferent ductules. The role of motile cilia in the efferent ductules has not been studied in PCD patients, but mutant mouse models have shown that male infertility can be caused by reduced numbers of motile cilia in the efferent ductules [127, 142]. Furthermore, animal models have also shown that severe spermatid malformations can lead to very low sperm counts in the epididymis, due to sloughing of immature spermatids.
More recently, a 2017 study reported phenotype–genotype correlations for fertility in 46 male PCD patients, however neither the fertility status of the partner nor the sperm ultrastructure or motility were reported [130]. This study included the cilia structural defects and fertility data available for cases with mutations in 17 different PCD genes, which indicated that the PCD patients more likely to be infertile had cilia showing a loss of inner dynein arms with microtubular disorganization, or a lack of the outer and inner dynein arms. Recent advances in PCD genetics will increasingly enable investigations of the effect of specific mutations on male fertility and prediction of their effect based on the genetic test. However, prior to implementing fertility counselling in PCD clinics, the association of specific mutations with sperm phenotype needs to be elucidated. More comprehensive counselling for PCD would take into account different genetic effects on cilia versus sperm motilities. Therefore, in this review we present the current knowledge of PCD-associated male infertility and sperm tail phenotypes caused by mutations in axonemal genes.
Current PCD diagnostics
Primary ciliary dyskinesia is a genetically heterogeneous disorder of variable clinical impact and thus far there is no single or even combination of tests accurate enough for making a diagnosis under all circumstances [55, 72, 106, 112]. After suspecting a possible PCD phenotype from clinical history (neonatal respiratory distress, respiratory phenotypes, laterality defects), a series of tests can be used to confirm the diagnosis. Nasal nitric oxide gas is reduced in most patients with PCD [10]. Analysis of the respiratory epithelium by nasal brushing can be used for identification of ciliary structural defects by transmission electron microscopy (TEM) [113, 118] and defects of the ciliary beat pattern and frequency can be identified by high-speed video microscopy analysis (HSVMA) [107]. HSVMA has excellent sensitivity and specificity for PCD and particular beat patterns have been linked to specific ultrastructural defects [15]. Assisting analysis includes immunofluorescence to look at cilia protein defects [119]. This is becoming more routinely used, which in addition to more specialized electron tomography [116] can be used in difficult to diagnose cases.
Primary ciliary dyskinesia is caused by genetic variants in genes coding for proteins, which have a role in motile cilia structure, formation and function. Thus, genetics has become a more prominent component of the diagnostic pathway for PCD over recent years, with confirmation of PCD diagnosis now defined as being made through identification of an ultrastructural defect by transmission electron microscopy or the identification of bi-allelic mutations in a known PCD gene [72, 112]. Several ultrastructural defects have been identified in the respiratory cilia of patients with PCD, the majority involving absence, partial absence or shortening of the outer or both inner and outer dynein arms. Other defects include complete or partial absence of the central microtubular pair often accompanied by transposition of an outer microtubular doublet [13, 48, 91, 95], a variety of microtubular disarrangements associated with defects of the nexin–dynein regulator complex and inner dynein arm loss [3, 78, 138] or reduced numbers of cilia [11, 132]. A sizeable proportion of patients have no detectable ultrastructural defect, some explained by subtle defects visualized only at high resolution [23, 91, 117], or effects on the axoneme not directly affecting internal structures [9, 12]. To date, more than 40 genes with causative mutations for PCD have been identified (Table 2) and gene panels are available for genetic testing [84, 92, 136]. It is estimated that in about 70% of patients with a clinical PCD phenotype, a bi-allelic mutation in a known PCD gene can be found [105, 112]. Increased awareness of PCD and better testing mean that the average age of diagnosis is now in childhood [35] and therefore fertility assessment is rarely part of frontline PCD diagnostics, although it should be taken into account in adult PCD patients.
How do motile cilia and sperm differ?
The gross axonemal structure of the motile cilia and sperm tail may appear to be identical, but cell type-specific differences in axonemal proteins such as dynein arm components and in assembly of the axoneme exist [32]. This conclusion is supported by the fact that basic motility and aspects of morphology differ between cilia and sperm and also since not all mutations in PCD genes cause male infertility (Table 2). Differences exist in the length of the sperm flagellum and in the accessory structures surrounding the axoneme compared to cilia. This is linked to the distinct motility pattern and functions of motile cilia versus sperm tails. Cilia motility includes a forward power stroke to move the mucus in airway epithelia and a recovery return stroke to then produce another power stroke [123], but in sperm the bending waves of the tail produce a constant forward pushing motion [47]. The respiratory tract cilia are attached to the apical surfaces of the airway epithelium moving the overlying fluid, whereas the flagellum of spermatozoa has evolved to power movement of the gamete freely through fluid.
Because of the role of motile cilia in mucus removal in the airways, multiciliogenesis is required for production of multiple cilia in the epithelial cells [17]. This differentiation programme is not necessary for sperm tail formation [58] or laterality determination [39] and thus mutations in genes encoding proteins with a role in multiciliogenesis (e.g. CCNO and MCIDAS) are not expected to affect male germ cell development. However, recent studies indicate that multiciliogenesis is required for correct function of the efferent ductules [20, 142]. In mice it has been shown that mutations in multiciliogenesis genes, Gemc1, Mcidas and Ccno, cause male infertility and complete lack of sperm in the epididymis [127].
Conversely, defects in sperm tail-specific structures can cause infertility with no airway phenotype. The long axoneme within the sperm flagellum is supported by the ODFs and FS and therefore malformations in these structures may also cause instability of the axonemal structure, as has been seen in patients with mutations in the FS protein, FSIP2 (fibrous sheath interacting protein 2) [28]. Depletion of another FS protein, AKAP4 (A-kinase anchoring protein 4), gives rise to lowered sperm numbers and short sperm tails with disruptions to the principal piece formation of the sperm tail (asthenoteratospermia), but the axoneme appears intact [5, 79]. Male infertility has also been reported to be caused by mutations in outer dense fibre gene ODF1, where the head tail connection was weakened in addition to disorganization of the ODFs and MS [41, 139]. Sperm tail-specific proteins may also be involved in attachment of the axoneme to the ODFs and FS [19] and unidentified or poorly characterized differences affecting the structural components may also contribute to the specific requirements of the sperm tail. This has been demonstrated by the identification of the tail axoneme intra-lumenal spiral (TAILS), a structure which binds directly to 11 protofilaments on the internal microtubule wall, along the end piece of the sperm tail [70, 143].
Differences in tissue distribution of dynein arm genes and the effect of known mutations
There is evidence that different motility patterns of cilia and sperm may require cell specific dynein arm complexes. In respiratory cilia mutations in ODA genes affect the ciliary beating and ODA ultrastructure, but no mutations in IDA genes have been identified in PCD patients to date. ODAs and IDAs are required for motility in both cilia and sperm, but differences in protein content of these complexes may contribute to the specialized motility-specific patterns required for forward swimming sperm, cilia mucus clearance in airways and the rotational movement of nodal cilia. In 9 + 0 nodal cilia, their lack of central pair microtubules and radial spokes seems to be the key for their unidirectional rotational movement [115], but the structural differences explaining the differences in motility between 9 + 2 cilia and sperm are not immediately obvious.
Recent expression studies in Drosophila indicate that specific ODA and IDA components are required for different waveforms in sperm and neuronal cilia [148]. Previously published transcriptome profiles during the first wave of spermatogenesis in mice [56] show the PCD gene expression pattern during male germ cell development. This dataset consist of testis tissue samples at five different postnatal (PND) time points, which contain specific male germ cell populations (Fig. 2a). Furthermore, analysis of RNAseq data from Fagerberg et al. [29] suggests that the cilia ODA dynein axonemal heavy chain (DNAH) proteins DNAH5 and DNAH11 may be replaced in sperm by DNAH8 and DNAH17 (Fig. 3a). This is supported by sperm-specific symptoms in patients with mutations in DNAH17 [137]. DNAH8 is not expressed in the lung and low level of expression is detected for DNAH17 (Fig. 3b). Very low levels of expression are detected for DNAH5 and DNAH11 in the human testis, but DNAH9 appeared present (Fig. 3B). PCD mutations in DNAH9 have recently been identified and for one patient very low sperm motility (5% normal progressive motility) was reported [31, 68]. This suggests that DNAH9 may have a role in sperm motility. During mouse spermatogenesis Dnah5, Dnah9 and Dnah11 expression was extremely low (Fig. 3c), indicating that there may also be species specific differences. Similar results have been reported for testis-specific expression on RNA and proteomic levels of DNAH8 and DNAH17 and motile cilia-specific expression of DNAH5, DNAH9 and DNAH11 [137].

PCD gene expression during the first wave of mouse spermatogenesis. a Cell type appearance of mouse sperm during the first wave of spermatogenesis. In addition to specific populations of male germ cells, all samples contain somatic cells. PND post-natal day. b PCD gene expression changes during the progression of mouse spermatogenesis, divided up according to the different functional categories of PCD genes. RNAseq was conducted on testis tissue samples collected at specific postnatal time points as indicated in 3A [56]. Red circles indicate the detected levels of genes Dnah5, Dnah9, Dnah11 and Rsph4a, showing their very low levels of expression as discussed in the text, which supports a potentially lesser role in sperm compared to their known motile cilia-specific functions. In the mouse testis, Dnaaf3 expression was also limited. All identified MMAF genes show a pattern of increasing expression during the progression of spermatogenesis. The highest reads per kilobase million (RPKM) expression value is indicated in brackets

Expression of axonemal dynein arm genes in motile cilia and during spermatogenesis. A model of suggested differences in components of ODA (a) and IDA (b) in motile cilia versus sperm, based on evidence of expression studies from Drosophila [128], human [26] and mouse [50]. The IDA is a suggested model, because in humans there are approximately 7 isoforms [1 double headed dynein (I1/f) and 6 single dynein IDA complexes (a–e, g)] which are thought to be structurally different within an axonemal 96-nm repeating pattern [104]. The human IDA structure is much less resolved in terms of its possible subunit components and more complicated than the human ODA. ODA (c) and IDA (d) DNAH gene expression in the human testis and lung [26]. ODA component DNAH8 is not expressed in the lung and DNAH17 shows low expression (grey box). In contrast, DNAH5 and DNAH11 have low expression in the testis (black box) compared to the lung. DNAH2, DNAH10 and DNAH binding gene WDR63 show low transcript levels in the lung, but DNAH1 expression in relatively high compared to axonemal gene expression in general. RPKM reads per kilobase million. ODA (e) and IDA (f) gene expression during the first wave of mouse spermatogenesis (see Fig. 2 for graph of cell content during the first wave of spermatogenesis in mouse). Dnah5, Dnah9 and Dnah11 show very low expression during mouse spermatogenesis [black box, fragments per kilobase million (FPKM) expression levels indicated in brackets]. All IDA genes show high expression at PND21 during the axoneme formation, except Dnah3 may be required later during the spermatid elongation
Immunofluorescence results from PCD patients with mutations in ODA genes show a lack of DNAH9 along the cilium [33, 69, 81], but DNAH9 was reported to be present in sperm from patients with DNAH5 and DNAL1 (dynein axonemal light chain 1) mutations [33]. This result suggests that DNAH5 or DNAL1 are not required for DNAH9 localization along the sperm tail, although DNAL1 is highly expressed in the testes (Fig. 4). DNAL1 mutations are a very rare cause of PCD [77], but male infertility has been identified in a patient with a mutation in the dynein axonemal intermediate chain gene (DNAI2), which was also missing from the sperm tail in a male PCD patient with mutations in dynein axonemal assembly factor 2 (DNAAF2) [93].

PCD gene expression in the human testis and MMAF gene expression in the lung. PCD genes are divided up into functional categories in each graph. DNAH5, DNAH11 and RSPH4A show markedly low expression (RPKM < 3) in the testis (boxes) compared to other PCD genes. The high RPKM value for DNAAF3, ZMYND10 and TTC21A is indicated above the bar (73, 210 and 71, respectively). Some expression is also detected for MMAF genes in the lung. The data was produced by RNAseq of human tissues samples [29]
In general, a limited amount of information is available about the effects of dynein arm mutations on male fertility. Amongst the most common PCD-causing mutations are those affecting the ODA genes DNAH5 and DNAH11 [45, 144]. The effect of DNAH5 mutations on male fertility has been reported in a small number of individuals, but with contradictory results. Oligo- and azoospermia was reported for two men with DNAH5 mutations [33] and fertility reported for three men and infertility for one, in a separate study [129]. In oligo- and azoospermic patients, localization of DNAH5 along the sperm tail was reported in patient and control sperm, although it was reduced/absent in the cilia [33]. Since a lack of staining of sperm DNAH5 was previously published [137], we suspect that the identified DNAH5 staining along the sperm tail [33] is unspecific. In Dnahc11iv mice homozygous for a Glu2271Lys missense mutation in Dnah11, there are defects in sperm motility, but without fertility being affected [71]. The Dnahc11iv sperm axonemal ultrastructure was also undisturbed in TEM analysis, however it is known that in cilia DNAH11 mutations confer a very subtle ultrastructure defect to the ODAs which so far can only be visualized using the much higher resolution of 3D tomography [23, 117]. In agreement with these variable outcomes, several male patients with DNAH11 mutations have been reported to father children without assisted techniques, whereas others have been diagnosed as infertile [109, 129]. Thus, it appears that complete function of DNAH5 and DNAH11 is not always crucial for male fertility.
In addition to ODA components, there might be differences in IDA proteins between motile cilia and sperm. This has been suggested by studies in Drosophila, where the IDA proteins DNAH2, DNAH10, WDR63 (WD repeat domain 63) and CASC1 (cancer susceptibility 1) appear to be testis specific [148] and some indication of low expression of DNAH2, DNAH10 and WDR63 was also seen in the transcriptomic data of the human lung (Fig. 3b, [29]). However, identification of the role of different proteins of the ODA and IDA in human motile cilia versus sperm requires further studies. Interestingly, although no mutations in IDA genes have yet been identified for PCD, a sperm-specific phenotype called multiple morphological abnormalities of flagella (MMAF), where sperms are severely malformed (teratozoospermia), has been reported in affected individuals carrying mutations in DNAH1 [2, 6, 19, 135] and DNAH2 [63].
The effect of PCD mutations in dynein arm assembly genes and structural components on sperm development
Apart from dyneins, mutations in a number of other structural proteins of the axoneme also cause PCD, including mutations in CCDC65, SPAG1 (sperm-associated antigen 1) and NME8 (NME/NM23 family member 8) which have all been localized along the sperm tail [82, 88, 108, 145]. Axonemal disorganization was also described in sperm from patients with PCD-causing mutations in the ‘ruler genes’ CCDC39 and CCDC40 [90], which is a comparable phenotype to that of the patient’s respiratory multiple motile cilia. Subtle ODA defects have been reported in patients with mutations in MNS1 (meiosis specific nuclear structural 1), accompanied by male infertility, situs inversus and mild PCD symptoms [126]. MNS1 is an axonemal protein found to dimerize and interact with the ODA docking complex component CCDC114, which is also mutated in PCD [94, 126, 133]. Based on previous publications, fertility is not affected in PCD patients with CCDC114 mutations [94] and the expression level is low in the mouse, but relatively high in the human testis. CCDC114 is therefore probably not crucial for sperm function, or as previously proposed, it can be compensated by other gene products such as CCDC63 during spermatogenesis [94]. Mutations in several central pair complex and radial spoke head proteins have been identified in PCD patients. For patients with mutations in the C2 central pair projection gene HYDIN, infertility has been reported in affected men [59, 91]. Subfertility caused by sperm motility defects have also been reported for male PCD patients with mutations in the radial spoke genes DNAJB13 (DnaJ heat shock protein family (Hsp40) member B13), RSPH3 (radial spoke head 3 homolog) and RSPH9 [13, 27, 37, 48, 62]. Interestingly, the PCD protein RSPH4A seems to be dispensable for spermatogenesis, since male PCD patients with RSPH4A mutations have been reported to be fertile and its expression levels in the testis are very low ([130], Fig. 4). These contrasting effects on male fertility suggest that differences between sperm flagella and cilia exist for the radial spoke head proteins, since mutations in any of the RSPH genes seems to have a similar consequence in the cilia; central pair loss and motility defects. A compensatory gene for RSPH4A in the testis may be RSPH6A, which has not been associated with PCD, but depletion of the mouse Rsph6a is a reported cause of male infertility [1].
Mutations in axonemal dynein complex preassembly genes have often been reported to cause male infertility in PCD (Table 2). In addition to mutations in DNAAF2 [93], PCD-causing mutations in DNAAF1, DNAAF3 and DNAAF4 have been reported to cause male infertility, but the patient number is minimal (Table 2). Mutations in their interacting partner, the dynein complex assembly factor PIH1 domain containing 3 (PIH1D3) have also been shown to result in lack of IDA and ODA [92, 98]. Male infertility has also been detected in PCD patients with mutations in axonemal dynein assembly factors LRRC6 (leucine-rich repeat containing 6), ZMYND10 (zinc finger MYND-type containing 10) and cilia and flagella-associated protein 300 (CFAP300). PCD with a lack of dynein arms in both the cilia and sperm is reported for LRRC6, ZMYND10, CFAP300 and axonemal dynein complex gene CCDC103 (coiled-coil domain containing 103) [16, 44, 54, 65, 97, 99, 100]. However, for CCDC103 mutations variable male fertility phenotype has been reported; one patient showed a lack of DA and DRC comparable to the motile cilia phenotype, but the other was fertile without DA loss [100].
Genetic mutations causing sperm tail-specific phenotypes
Mutations in a number of human genes result in sperm motility defects of a structural nature that is within the same spectrum as those caused by mutations in PCD genes. In these cases, associated with malformation of the sperm tail and low sperm count, mutations in a number of genes not connected to the respiratory PCD phenotype are reported. These apparently only affect the sperm tail axoneme and not cilia functions, having been identified as the cause of MMAF. The main findings in MMAF are absent, short, bent, coiled, and/or irregular sperm flagella with very low numbers of normal spermatozoa (0–2%) associated with low sperm concentrations [6]. A MMAF sperm tail phenotype of short, stumpy tails has also previously been described as dysplasia of the fibrous sheath (DFS) [5, 14, 102]. DFS has been reported to be caused by mutations in three genes encoding components of the sperm tail specific fibrous sheath: AKAP3, AKAP4 and FSIP2 [5, 75]. AKAP3 and 4 are the main components of the fibrous sheath and FSIP2 is required for localization of AKAP4 to the FS [75].
Sperm tail accessory structures are also often disorganized in MMAF patients with mutations in axonemal genes, such as DNAH1, which is the most common identified cause of MMAF [2, 6, 135]. DNAH1 is a component of the IDAs, which are completely disorganized in sperm tail axonemes of MMAF patients with DNAH1 mutations [6]. Mutations in another IDA gene, DNAH2, have also been identified as a cause for MMAF [63]. The sperm tail phenotype is similar in patients with DNAH1 and DNAH2 mutations with lack of the CP or complete disorganization of axonemal and ODF structures in addition to IDA loss [2, 6, 63, 135] indicating that IDAs are crucial for sperm tail axoneme integrity. Infertility has been the only PCD symptom identified in MMAF affected men with DNAH1 and DNAH2 mutations suggesting that these IDA components are not required for motile cilia function. However, DNAH1 mutations have also been reported in one family with siblings affected by PCD [46] and DNAH1 appears to be expressed in the lung (Fig. 3B). The possible role of DNAH1 and other IDA genes in PCD awaits to be revealed. IDA gene mutations may cause milder PCD symptoms due to the role of IDAs in controlling the wave form [52].
There seem to be differences in the severity of the sperm tail phenotype caused by IDA mutations between species in addition to differences between motile cilia and sperm. Dnah1 knock-out (KO) mice only show motility, but no structural sperm defects (asthenozoospermia) [87], whereas in humans DNAH1 truncating mutations induce a MMAF phenotype [2, 135]. Decreased cilia beat frequency was also detected in the mutant mice, which suggests a comparable role of DNAH1 in mouse sperm tail and motile cilia. In humans the lack of DNAH1 in motile cilia could be compensated by other DNAH genes such as DNAH12, which is the closest paralog to DNAH1 [74].
A report on the effect of sperm-specific ODA heavy chain DNAH17 mutations was recently published [137]. DNAH17 mutations were identified in five MMAF patients with sperm lacking ODAs. Some sperm tail cross sections showed missing CP and even disorganized outer doublet microtubules, but no defects were seen in the patient’s respiratory cilia. Thus, DNAH17 mutations only appear to affect sperm tail ODA formation, a finding also supported by expression and protein localization studies [137].
The cilia and flagella-associated protein (CFAP) CFAP251, also known as WDR66, a component of the calmodulin- and radial-spoke-associated complex [24, 25] is located adjacent to DNAH1 in T. thermophila at the base of the RS3 [42, 53] radial spoke complex. The amount of DNAH1 homolog IDA g was reduced in T. thermophila CFAP251 KO [128] and a direct physical link between CFAP251, IDA3, and RS3 has been shown by co-immunoprecipitation experiments [4]. Mutations in WDR66 cause a typical MMAF phenotype with immotile, malformed sperm tails in which all the sperm tail structures are disorganized [4, 51, 61].
The main axonemal ultrastructural finding in MMAF patient sperm seems to be the lack of central pairs from the 9 + 2 microtubular arrangement (Table 2). Mutations in ARMC2 (armadillo repeat containing 2), CFAP43 and CFAP44 cause male infertility associated with sperm axonemal central pair defects [18, 19]. The main defect based on the human sperm EM appears to be the lack of central pair, although some completely disorganized axonemal and accessory structures were also observed. The Armc2 KO mouse model shows a similar phenotype and immunofluoresence experiments indicated the presence of other axonemal components except the central pair [18]. In patients with CFAP43 and CFAP44 mutations, some variable axonemal phenotypes were also observed [19]. In some sperm tail cross sections, the missing central pair was accompanied by disorganized outer doublets and in patients with CFAP43 mutations the central pair was misoriented when present. In KO mouse models, mutations in Cfap43 showed a more dramatic sperm tail phenotype than for the Cfap44 KO. Only short disorganized tails were detected in Cfap43 mutant mice while Cfap44 KO showed normal flagellar length, but with irregular mid-pieces. Another study reported short and immotile tail phenotypes arising from mutations in both genes [124]. CFAP43 and CFAP44 are suggested to have a role in connecting specific doublet microtubules to periaxonemal structures in the sperm tail and thus may not be required for motile cilia. The trypanosome ciliate homologs were localized on the outer surface of the doublet microtubules [19].
Mutations in CFAP69 were reported in two MMAF patients with typical short immotile sperm tail characteristics that were replicated in Cfap69 KO mice, thereby confirming a role for CFAP69 in spermiogenesis [22]. In mature human sperm, CFAP69 is localized in the mid-piece, thus it may not be a structural component of the axoneme. However, all sperm tail structures were completely disorganized in the Cfap69 mutant mouse model. Human patients with CFAP69 mutations have a variable sperm tail phenotype depending on their mutation. Sperm-specific phenotypes have also been suggested for mutations in CEP135 (centrosomal protein 135) and AK7 (adenylate kinase 7). In one MMAF patient with short or absent sperm tails, a missense mutation in CEP135 was identified as a probable cause for infertility [111]. A sperm-specific phenotype with incomplete mitochondrial sheath, dysplasia of the fibrous sheath and lack of central pair microtubules with axonemal disorganization was also reported for AK7 [70]. PCD-associated mutations and decreased AK7 expression have also been reported in PCD patients [76, 80]. Tissue specific effects of AK7 mutations are likely, since a missense mutation was reported to affect the expression in the testis, but not in multiciliated tissues [70].
Recently, mutations in SPEF2 (sperm flagellar 2) have been reported to cause MMAF and interestingly some of the patients show mild PCD-like symptoms [64, 67]. Variable SPEF2 mutant phenotypes have also been demonstrated in animal models, where depletion of different gene variants causes sperm-specific or PCD-like symptoms [57, 121, 122]. The variability of symptoms can potentially be explained by the nature of the gene variants and severity of their effect on protein function and/or the nature of the proteins themselves. It has been suggested that SPEF2 has a role in protein transport and it interacts with IFT20 (intraflagellar transport 20) [120]. Another IFT-associated protein TTC21A (tetratricopeptide repeat domain 21A) was also reported to cause MMAF with malformed sperm tails and additional abnormalities in the sperm HTCA [66]. Thus, sperm or motile cilia-specific IFT-associated proteins may also be required for transport of proteins in these specialized cilia types.
For most MMAF patients, no PCD-associated respiratory symptoms have been identified. Associated motile cilia defects and disease symptoms if any could be mild, variable and hard to detect in some cases. However, phenotypes rather tend to suggest that roles of at least some of the identified gene products are potentially restricted to the male germ cells. This could have something to do with involvement in stabilizing the long sperm tail axoneme or with connection of the axoneme to the accessory structures. In addition, it seems clear that different dynein arm proteins can be more crucial for sperm tail functions than for motile cilia. Thus, the different motion patterns of cilia and sperm may require specific protein components. These differences require further investigation in larger numbers of patients in order to elucidate the exact differences between human cilia and sperm tail formation. Most mutations for MMAF have been identified in North African and Chinese populations and a few single French patients, hence human data is sparse.
Clinical implications of sperm defects in PCD and fertility treatment
Male fertility can be affected by many genetic and non-genetic factors, but infertility arising from axonemal defects is a clearly testable phenotype [30]. For PCD patients presenting at respiratory clinics, age-appropriate genetic and fertility counselling should be available or in place. Arrangements for fertility testing should be made available if required and this is especially important around the time of transition from paediatric care to the adult clinic in PCD males. Knowledge is increasing with regards to which PCD genes may or may not be associated with sperm defects and male infertility (Table 2). Currently, due to preserved sperm function in some cases of PCD, the fertility phenotype should be assessed by laboratory confirmation alongside or ideally prior to counselling of patients. Knowledge of male infertility phenotype/genotype correlations is the prerequisite for the most appropriate counselling of patients, based on their result from genetic testing for PCD.
The diagnostics and family counselling of PCD should be informed by the genetic background of the defect. Indeed, PCD patients seeking in vitro fertilization (IVF) privately without knowledge of their condition and the effect on sperm motility may face the problem of sperm viability (related to motility) being a prerequisite for being sent forward for IVF; they will benefit from the fullest knowledge around their condition. Thus far, an estimated 70% of PCD patients can be expected to have mutations in known causative genes, but the effect of these mutations on male fertility is largely unknown. Future studies will establish novel PCD mutations, which can be assisted by identification of teratozoospermic or asthenozoospermic patients, who may have mild PCD symptoms. In rare cases azoospermia has also been reported for PCD patients, thus these patients may also have mutations in cilia/flagella related genes. Although some implications of the effect of PCD mutations on male infertility have emerged, the comprehensive analysis of association of sperm phenotype with specific PCD mutations requires future studies. On the other hand, identification of the effects on male fertility of specific PCD mutations in specific genes is of great importance and enables the inclusion of PCD genes on wider gene panels for male infertility.
For those PCD patients who are not able to conceive naturally, several studies report successful intracytoplasmic sperm injection (ICSI) based fertilization of eggs and consequent IVF births with sperm from PCD patients. Successful ICSI outcomes of PCD sperm have been reported for 28% of cases with live births [50], although the fertilization rate was much higher, at 80%. The low live birth rate compared to fertilization rate may imply developmental problems during embryogenesis. This may be due to poor embryo quality, which could result from functional failure of the sperm-derived centrosome. Centrosome dysfunction often leads to irregular cleavage during embryo development or chromosomal aberrations, resulting in delays or arrest of embryo development [103]. The specific mutations within the PCD cases were not reported, thus the effect of the genetic cause on ICSI outcome requires further studies. Reports for MMAF patients show that spermatozoa with mutations in the centrosomal gene CEP135 produced no pregnancy after embryo transplantation [111]. However, the ICSI outcome for MMAF patient sperm from DNAH1 mutation carriers shows good prognoses, possibly due to these mutations primarily affecting the structure of the sperm axonemal inner dynein arms [134]. Successful ICSI outcomes were also shown for CFAP43 and CFAP44 patients [110]. Outcomes of ICSI are reported to be improved through testicular sperm extraction and with the addition of pentoxifylline treatment [34, 141]. Further studies on the effects of specific gene mutations on male fertility and ICSI outcome are required to improve the diagnosis and treatment of male infertility in PCD patients.
Conclusion
Although it appears that many PCD genes have an effect on male fertility, it is clear that the comparative disease phenotype between cilia and sperm does not correlate in all cases. For example, DNAH11 and DNAH5 may be cilia specific and DNAH17 and DNAH8 have a role only in the sperm axoneme, with consequent cilia- and sperm-restricted defects seen in affected patients. Thus far, male infertility specific phenotype (MMAF) has been reported for mutations in 13 potentially axonemal genes, DNAH1, DNAH2, DNAH17, CEP135, WDR66, SPEF2, ARMC2, TTC21A, QRICH2 and CFAP genes 43, 44, 69 and 251. Expression of some of these genes is limited to the testis, however some may also have roles in motile cilia as suggested for SPEF2 [64, 67]. Testis- and cilia-specific compensatory genes could prevent the effect of specific mutations in spermio- or ciliogenesis, respectively, as has been speculated for CCDC114 [94]. Male infertility in PCD has been documented for 22 PCD genes (Table 2), but the patient numbers are very low and the exact effect of mutations on sperm tail structures is not known in most cases. Therefore it is of high importance to investigate the exact effect of PCD mutations on male fertility. Information on the spermatogenesis specific roles of ciliary genes will enable counselling of patients about their fertility status and possible clinical treatments required for conceiving.
References
Abbasi F, Miyata H, Shimada K, Morohoshi A, Nozawa K, Matsumura T, Xu Z, Pratiwi P, Ikawa M (2018) RSPH6A is required for sperm flagellum formation and male fertility in mice. J Cell Sci. https://doi.org/10.1242/jcs.221648
Amiri-Yekta A, Coutton C, Kherraf ZE, Karaouzene T, Le Tanno P, Sanati MH, Sabbaghian M, Almadani N, Sadighi Gilani MA, Hosseini SH, Bahrami S, Daneshipour A, Bini M, Arnoult C, Colombo R, Gourabi H, Ray PF (2016) Whole-exome sequencing of familial cases of multiple morphological abnormalities of the sperm flagella (MMAF) reveals new DNAH1 mutations. Hum Reprod 31:2872–2880
Antony D, Becker-Heck A, Zariwala MA, Schmidts M, Onoufriadis A, Forouhan M, Wilson R, Taylor-Cox T, Dewar A, Jackson C, Goggin P, Loges NT, Olbrich H, Jaspers M, Jorissen M, Leigh MW, Wolf WE, Daniels ML, Noone PG, Ferkol TW, Sagel SD, Rosenfeld M, Rutman A, Dixit A, O’Callaghan C, Lucas JS, Hogg C, Scambler PJ, Emes RD, Uk10k, Chung EM, Shoemark A, Knowles MR, Omran H, Mitchison HM (2013) Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum Mutat 34:462–472
Auguste Y, Delague V, Desvignes JP, Longepied G, Gnisci A, Besnier P, Levy N, Beroud C, Megarbane A, Metzler-Guillemain C, Mitchell MJ (2018) Loss of calmodulin- and radial-spoke-associated complex protein CFAP251 leads to immotile spermatozoa lacking mitochondria and infertility in men. Am J Hum Genet 103:413–420
Baccetti B, Collodel G, Estenoz M, Manca D, Moretti E, Piomboni P (2005) Gene deletions in an infertile man with sperm fibrous sheath dysplasia. Hum Reprod 20:2790–2794
Ben Khelifa M, Coutton C, Zouari R, Karaouzene T, Rendu J, Bidart M, Yassine S, Pierre V, Delaroche J, Hennebicq S, Grunwald D, Escalier D, Pernet-Gallay K, Jouk PS, Thierry-Mieg N, Toure A, Arnoult C, Ray PF (2014) Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet 94:95–104
Best S, Shoemark A, Rubbo B, Patel MP, Fassad MR, Dixon M, Rogers AV, Hirst RA, Rutman A, Ollosson S, Jackson CL, Goggin P, Thomas S, Pengelly R, Cullup T, Pissaridou E, Hayward J, Onoufriadis A, O’Callaghan C, Loebinger MR, Wilson R, Chung EM, Kenia P, Doughty VL, Carvalho JS, Lucas JS, Mitchison HM, Hogg C (2019) Risk factors for situs defects and congenital heart disease in primary ciliary dyskinesia. Thorax 74:203–205
Blanchon S, Legendre M, Copin B, Duquesnoy P, Montantin G, Kott E, Dastot F, Jeanson L, Cachanado M, Rousseau A, Papon JF, Beydon N, Brouard J, Crestani B, Deschildre A, Desir J, Dollfus H, Leheup B, Tamalet A, Thumerelle C, Vojtek AM, Escalier D, Coste A, de Blic J, Clement A, Escudier E, Amselem S (2012) Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J Med Genet 49:410–416
Bonnefoy S, Watson CM, Kernohan KD, Lemos M, Hutchinson S, Poulter JA, Crinnion LA, Berry I, Simmonds J, Vasudevan P, O’Callaghan C, Hirst RA, Rutman A, Huang L, Hartley T, Grynspan D, Moya E, Li C, Carr IM, Bonthron DT, Leroux M, Care4Rare Canada Consortium, Boycott KM, Bastin P, Sheridan EG (2018) Biallelic mutations in LRRC56, encoding a protein associated with intraflagellar transport, cause mucociliary clearance and laterality defects. Am J Hum Genet 103:727–739
Boon M, Meyts I, Proesmans M, Vermeulen FL, Jorissen M, De Boeck K (2014) Diagnostic accuracy of nitric oxide measurements to detect primary ciliary dyskinesia. Eur J Clin Invest 44:477–485
Boon M, Wallmeier J, Ma L, Loges NT, Jaspers M, Olbrich H, Dougherty GW, Raidt J, Werner C, Amirav I, Hevroni A, Abitbul R, Avital A, Soferman R, Wessels M, O’Callaghan C, Chung EM, Rutman A, Hirst RA, Moya E, Mitchison HM, Van Daele S, De Boeck K, Jorissen M, Kintner C, Cuppens H, Omran H (2014) MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun 5:4418
Bustamante-Marin XM, Yin WN, Sears PR, Werner ME, Brotslaw EJ, Mitchell BJ, Jania CM, Zeman KL, Rogers TD, Herring LE, Refabert L, Thomas L, Amselem S, Escudier E, Legendre M, Grubb BR, Knowles MR, Zariwala MA, Ostrowski LE (2019) Lack of GAS2L2 causes PCD by impairing cilia orientation and mucociliary clearance. Am J Hum Genet 104:229–245
Castleman VH, Romio L, Chodhari R, Hirst RA, de Castro SC, Parker KA, Ybot-Gonzalez P, Emes RD, Wilson SW, Wallis C, Johnson CA, Herrera RJ, Rutman A, Dixon M, Shoemark A, Bush A, Hogg C, Gardiner RM, Reish O, Greene ND, O’Callaghan C, Purton S, Chung EM, Mitchison HM (2009) Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am J Hum Genet 84:197–209
Chemes HE, Brugo S, Zanchetti F, Carrere C, Lavieri JC (1987) Dysplasia of the fibrous sheath: an ultrastructural defect of human spermatozoa associated with sperm immotility and primary sterility. Fertil Steril 48:664–669
Chilvers MA, Rutman A, O’Callaghan C (2003) Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol 112:518–524
Cho KJ, Noh SH, Han SM, Choi WI, Kim HY, Yu S, Lee JS, Rim JH, Lee MG, Hildebrandt F, Gee HY (2018) ZMYND10 stabilizes intermediate chain proteins in the cytoplasmic pre-assembly of dynein arms. PLoS Genet 14:e1007316
Choksi SP, Lauter G, Swoboda P, Roy S (2014) Switching on cilia: transcriptional networks regulating ciliogenesis. Development 141:1427–1441
Coutton C, Martinez G, Kherraf ZE, Amiri-Yekta A, Boguenet M, Saut A, He X, Zhang F, Cristou-Kent M, Escoffier J, Bidart M, Satre V, Conne B, Fourati Ben Mustapha S, Halouani L, Marrakchi O, Makni M, Latrous H, Kharouf M, Pernet-Gallay K, Bonhivers M, Hennebicq S, Rives N, Dulioust E, Toure A, Gourabi H, Cao Y, Zouari R, Hosseini SH, Nef S, Thierry-Mieg N, Arnoult C, Ray PF (2019) Bi-allelic mutations in ARMC2 lead to severe astheno-teratozoospermia due to sperm flagellum malformations in humans and mice. Am J Hum Genet 104:331–340
Coutton C, Vargas AS, Amiri-Yekta A, Kherraf ZE, Ben Mustapha SF, Le Tanno P, Wambergue-Legrand C, Karaouzene T, Martinez G, Crouzy S, Daneshipour A, Hosseini SH, Mitchell V, Halouani L, Marrakchi O, Makni M, Latrous H, Kharouf M, Deleuze JF, Boland A, Hennebicq S, Satre V, Jouk PS, Thierry-Mieg N, Conne B, Dacheux D, Landrein N, Schmitt A, Stouvenel L, Lores P, El Khouri E, Bottari SP, Faure J, Wolf JP, Pernet-Gallay K, Escoffier J, Gourabi H, Robinson DR, Nef S, Dulioust E, Zouari R, Bonhivers M, Toure A, Arnoult C, Ray PF (2018) Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in Trypanosoma and human. Nat Commun 9:686
Danielian PS, Hess RA, Lees JA (2016) E2f4 and E2f5 are essential for the development of the male reproductive system. Cell Cycle 15:250–260
Diggle CP, Moore DJ, Mali G, zur Lage P, Ait-Lounis A, Schmidts M, Shoemark A, Garcia Munoz A, Halachev MR, Gautier P, Yeyati PL, Bonthron DT, Carr IM, Hayward B, Markham AF, Hope JE, von Kriegsheim A, Mitchison HM, Jackson IJ, Durand B, Reith W, Sheridan E, Jarman AP, Mill P (2014) HEATR2 plays a conserved role in assembly of the ciliary motile apparatus. PLoS Genet 10:e1004577
Dong FN, Amiri-Yekta A, Martinez G, Saut A, Tek J, Stouvenel L, Lores P, Karaouzene T, Thierry-Mieg N, Satre V, Brouillet S, Daneshipour A, Hosseini SH, Bonhivers M, Gourabi H, Dulioust E, Arnoult C, Toure A, Ray PF, Zhao H, Coutton C (2018) Absence of CFAP69 causes male infertility due to multiple morphological abnormalities of the flagella in human and mouse. Am J Hum Genet 102:636–648
Dougherty GW, Loges NT, Klinkenbusch JA, Olbrich H, Pennekamp P, Menchen T, Raidt J, Wallmeier J, Werner C, Westermann C, Ruckert C, Mirra V, Hjeij R, Memari Y, Durbin R, Kolb-Kokocinski A, Praveen K, Kashef MA, Kashef S, Eghtedari F, Haffner K, Valmari P, Baktai G, Aviram M, Bentur L, Amirav I, Davis EE, Katsanis N, Brueckner M, Shaposhnykov A, Pigino G, Dworniczak B, Omran H (2016) DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am J Respir Cell Mol Biol 55:213–224
Dymek EE, Heuser T, Nicastro D, Smith EF (2011) The CSC is required for complete radial spoke assembly and wild-type ciliary motility. Mol Biol Cell 22:2520–2531
Dymek EE, Smith EF (2007) A conserved CaM- and radial spoke associated complex mediates regulation of flagellar dynein activity. J Cell Biol 179:515–526
Edelbusch C, Cindric S, Dougherty GW, Loges NT, Olbrich H, Rivlin J, Wallmeier J, Pennekamp P, Amirav I, Omran H (2017) Mutation of serine/threonine protein kinase 36 (STK36) causes primary ciliary dyskinesia with a central pair defect. Hum Mutat 38:964–969
El Khouri E, Thomas L, Jeanson L, Bequignon E, Vallette B, Duquesnoy P, Montantin G, Copin B, Dastot-Le Moal F, Blanchon S, Papon JF, Lores P, Yuan L, Collot N, Tissier S, Faucon C, Gacon G, Patrat C, Wolf JP, Dulioust E, Crestani B, Escudier E, Coste A, Legendre M, Toure A, Amselem S (2016) Mutations in DNAJB13, encoding an HSP40 family member, cause primary ciliary dyskinesia and male infertility. Am J Hum Genet 99:489–500
ElInati E, Fossard C, Okutman O, Ghedir H, Ibala-Romdhane S, Ray PF, Saad A, Hennebicq S, Viville S (2016) A new mutation identified in SPATA16 in two globozoospermic patients. J Assist Reprod Genet 33:815–820
Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, Szigyarto CA, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, Nilsson P, Schwenk JM, Lindskog C, Danielsson F, Mardinoglu A, Sivertsson A, von Feilitzen K, Forsberg M, Zwahlen M, Olsson I, Navani S, Huss M, Nielsen J, Ponten F, Uhlen M (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13:397–406
Fainberg J, Kashanian JA (2019) Recent advances in understanding and managing male infertility. F1000Res. https://doi.org/10.12688/f1000research.17076.1(eCollection 2019)
Fassad MR, Shoemark A, Legendre M, Hirst RA, Koll F, le Borgne P, Louis B, Daudvohra F, Patel MP, Thomas L, Dixon M, Burgoyne T, Hayes J, Nicholson AG, Cullup T, Jenkins L, Carr SB, Aurora P, Lemullois M, Aubusson-Fleury A, Papon JF, O’Callaghan C, Amselem S, Hogg C, Escudier E, Tassin AM, Mitchison HM (2018) Mutations in outer dynein arm heavy chain DNAH9 cause motile cilia defects and situs inversus. Am J Hum Genet 103:984–994
Fliegauf M, Benzing T, Omran H (2007) When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 8:880–893
Fliegauf M, Olbrich H, Horvath J, Wildhaber JH, Zariwala MA, Kennedy M, Knowles MR, Omran H (2005) Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med 171:1343–1349
Gerber PA, Kruse R, Hirchenhain J, Krussel JS, Neumann NJ (2008) Pregnancy after laser-assisted selection of viable spermatozoa before intracytoplasmatic sperm injection in a couple with male primary cilia dyskinesia. Fertil Steril 89:1826.e9–1826.e12
Goutaki M, Eich MO, Halbeisen FS, Barben J, Casaulta C, Clarenbach C, Hafen G, Latzin P, Regamey N, Lazor R, Tschanz S, Zanolari M, Maurer E, Kuehni CE, Swiss PCD Registry (CH-PCD) Working Group (2019) The Swiss Primary Ciliary Dyskinesia registry: objectives, methods and first results. Swiss Med Wkly. https://doi.org/10.4414/smw.2019.20004(eCollection 2019 Jan 1)
Goutaki M, Meier AB, Halbeisen FS, Lucas JS, Dell SD, Maurer E, Casaulta C, Jurca M, Spycher BD, Kuehni CE (2016) Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J 48:1081–1095
Guan J, Yuan L (2008) A heat-shock protein 40, DNAJB13, is an axoneme-associated component in mouse spermatozoa. Mol Reprod Dev 75:1379–1386
Guo T, Tan ZP, Chen HM, Zheng DY, Liu L, Huang XG, Chen P, Luo H, Yang YF (2017) An effective combination of whole-exome sequencing and runs of homozygosity for the diagnosis of primary ciliary dyskinesia in consanguineous families. Sci Rep 7:7905
Hamada H (2016) Roles of motile and immotile cilia in left-right symmetry breaking. In: Markwald RR, Baldwin HS, Keller BB, Srivastava D, Yamagishi H, Nakanishi T (eds) Etiology and morphogenesis of congenital heart disease: from gene function and cellular interaction to morphology. The Author(s), Tokyo, pp 57–65
He X, Li W, Wu H, Lv M, Liu W, Liu C, Zhu F, Li C, Fang Y, Yang C, Cheng H, Zhang J, Tan J, Chen T, Tang D, Song B, Wang X, Zha X, Wang H, Wei Z, Yang S, Saiyin H, Zhou P, Jin L, Wang J, Zhang Z, Zhang F, Cao Y (2018) Novel homozygous CFAP69 mutations in humans and mice cause severe asthenoteratospermia with multiple morphological abnormalities of the sperm flagella. J Med Genet 56:96–103
Hetherington L, Schneider E, Scott C, DeKretser D, Muller CH, Hondermarck H, Velkov T, Baker MA (2016) Deficiency in outer dense fiber 1 is a marker and potential driver of idiopathic male infertility. Mol Cell Proteom 15:3685–3693
Heuser T, Dymek EE, Lin J, Smith EF, Nicastro D (2012) The CSC connects three major axonemal complexes involved in dynein regulation. Mol Biol Cell 23:3143–3155
Heuser T, Raytchev M, Krell J, Porter ME, Nicastro D (2009) The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J Cell Biol 187:921–933
Hoben IM, Hjeij R, Olbrich H, Dougherty GW, Nothe-Menchen T, Aprea I, Frank D, Pennekamp P, Dworniczak B, Wallmeier J, Raidt J, Nielsen KG, Philipsen MC, Santamaria F, Venditto L, Amirav I, Mussaffi H, Prenzel F, Wu K, Bakey Z, Schmidts M, Loges NT, Omran H (2018) Mutations in C11orf70 cause primary ciliary dyskinesia with randomization of left/right body asymmetry due to defects of outer and inner dynein arms. Am J Hum Genet 102:973–984
Hornef N, Olbrich H, Horvath J, Zariwala MA, Fliegauf M, Loges NT, Wildhaber J, Noone PG, Kennedy M, Antonarakis SE, Blouin JL, Bartoloni L, Nusslein T, Ahrens P, Griese M, Kuhl H, Sudbrak R, Knowles MR, Reinhardt R, Omran H (2006) DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med 174:120–126
Imtiaz F, Allam R, Ramzan K, Al-Sayed M (2015) Variation in DNAH1 may contribute to primary ciliary dyskinesia. BMC Med Genet 16:14
Ishimoto K, Gadelha H, Gaffney EA, Smith DJ, Kirkman-Brown J (2017) Coarse-graining the fluid flow around a human sperm. Phys Rev Lett 118:124501
Jeanson L, Copin B, Papon JF, Dastot-Le Moal F, Duquesnoy P, Montantin G, Cadranel J, Corvol H, Coste A, Desir J, Souayah A, Kott E, Collot N, Tissier S, Louis B, Tamalet A, de Blic J, Clement A, Escudier E, Amselem S, Legendre M (2015) RSPH3 mutations cause primary ciliary dyskinesia with central-complex defects and a near absence of radial spokes. Am J Hum Genet 97:153–162
Jeanson L, Thomas L, Copin B, Coste A, Sermet-Gaudelus I, Dastot-Le Moal F, Duquesnoy P, Montantin G, Collot N, Tissier S, Papon JF, Clement A, Louis B, Escudier E, Amselem S, Legendre M (2016) Mutations in GAS8, a gene encoding a nexin-dynein regulatory complex subunit, cause primary ciliary dyskinesia with axonemal disorganization. Hum Mutat 37:776–785
Kawasaki A, Okamoto H, Wada A, Ainoya Y, Kita N, Maeyama T, Edamoto N, Nishiyama H, Tsukamoto S, Joraku A, Waku N, Yoshikawa H (2015) A case of primary ciliary dyskinesia treated with ICSI using testicular spermatozoa: case report and a review of the literature. Reprod Med Biol 14:195–200
Kherraf ZE, Amiri-Yekta A, Dacheux D, Karaouzene T, Coutton C, Christou-Kent M, Martinez G, Landrein N, Le Tanno P, Fourati Ben Mustapha S, Halouani L, Marrakchi O, Makni M, Latrous H, Kharouf M, Pernet-Gallay K, Gourabi H, Robinson DR, Crouzy S, Blum M, Thierry-Mieg N, Toure A, Zouari R, Arnoult C, Bonhivers M, Ray PF (2018) A homozygous ancestral SVA-insertion-mediated deletion in WDR66 induces multiple morphological abnormalities of the sperm flagellum and male infertility. Am J Hum Genet 103:400–412
King SM (2016) Axonemal dynein arms. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a028100
King SM (2013) A solid-state control system for dynein-based ciliary/flagellar motility. J Cell Biol 201:173–175
Kott E, Duquesnoy P, Copin B, Legendre M, Dastot-Le Moal F, Montantin G, Jeanson L, Tamalet A, Papon JF, Siffroi JP, Rives N, Mitchell V, de Blic J, Coste A, Clement A, Escalier D, Toure A, Escudier E, Amselem S (2012) Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am J Hum Genet 91:958–964
Kuehni CE, Lucas JS (2017) Diagnosis of primary ciliary dyskinesia: summary of the ERS Task Force report. Breathe (Sheff) 13:166–178
Laiho A, Kotaja N, Gyenesei A, Sironen A (2013) Transcriptome profiling of the murine testis during the first wave of spermatogenesis. PLoS One 8:e61558
Lehti MS, Henriksson H, Rummukainen P, Wang F, Uusitalo-Kylmala L, Kiviranta R, Heino TJ, Kotaja N, Sironen A (2018) Cilia-related protein SPEF2 regulates osteoblast differentiation. Sci Rep 8:859
Lehti MS, Sironen A (2017) Formation and function of sperm tail structures in association with sperm motility defects. Biol Reprod 97:522–536
Li L, Sha YW, Xu X, Mei LB, Qiu PP, Ji ZY, Lin SB, Su ZY, Wang C, Yin C, Li P (2018) DNAH6 is a novel candidate gene associated with sperm head anomaly. Andrologia 50:e12953
Li W, He X, Yang S, Liu C, Wu H, Liu W, Lv M, Tang D, Tan J, Tang S, Chen Y, Wang J, Zhang Z, Wang H, Jin L, Zhang F, Cao Y (2019) Biallelic mutations of CFAP251 cause sperm flagellar defects and human male infertility. J Hum Genet 64:49–54
Li W, He X, Yang S, Liu C, Wu H, Liu W, Lv M, Tang D, Tan J, Tang S, Chen Y, Wang J, Zhang Z, Wang H, Jin L, Zhang F, Cao Y (2018) Biallelic mutations of CFAP251 cause sperm flagellar defects and human male infertility. J Hum Genet 64:49
Li W, Liu G (2014) DNAJB13, a type II HSP40 family member, localizes to the spermatids and spermatozoa during mouse spermatogenesis. BMC Dev Biol 14:38
Li Y, Sha Y, Wang X, Ding L, Liu W, Ji Z, Mei L, Huang X, Lin S, Kong S, Lu J, Qin W, Zhang X, Zhuang J, Tang Y, Lu Z (2019) DNAH2 is a novel candidate gene associated with multiple morphological abnormalities of the sperm flagella (MMAF). Clin Genet 95:590–600
Liu C, Lv M, He X, Zhu Y, Amiri-Yekta A, Li W, Wu H, Kherraf ZE, Liu W, Zhang J, Tan Q, Tang S, Zhu YJ, Zhong Y, Li C, Tian S, Zhang Z, Jin L, Ray P, Zhang F, Cao Y (2019) Homozygous mutations in SPEF2 induce multiple morphological abnormalities of the sperm flagella and male infertility. J Med Genet. https://doi.org/10.1136/jmedgenet-2019-106011
Liu L, Luo H (2018) Whole-exome sequencing identified a novel compound heterozygous mutation of LRRC6 in a Chinese primary ciliary dyskinesia patient. Biomed Res Int 2018:1854269
Liu W, He X, Yang S, Zouari R, Wang J, Wu H, Kherraf ZE, Liu C, Coutton C, Zhao R, Tang D, Tang S, Lv M, Fang Y, Li W, Li H, Zhao J, Wang X, Zhao S, Zhang J, Arnoult C, Jin L, Zhang Z, Ray PF, Cao Y, Zhang F (2019) Bi-allelic mutations in TTC21A induce asthenoteratospermia in humans and mice. Am J Hum Genet 104:738–748
Liu W, Sha Y, Li Y, Mei L, Lin S, Huang X, Lu J, Ding L, Kong S, Lu Z (2019) Loss-of-function mutations in SPEF2 cause multiple morphological abnormalities of the sperm flagella (MMAF). J Med Genet 56(10):678–684
Loges NT, Antony D, Maver A, Deardorff MA, Gulec EY, Gezdirici A, Nothe-Menchen T, Hoben IM, Jelten L, Frank D, Werner C, Tebbe J, Wu K, Goldmuntz E, Cuturilo G, Krock B, Ritter A, Hjeij R, Bakey Z, Pennekamp P, Dworniczak B, Brunner H, Peterlin B, Tanidir C, Olbrich H, Omran H, Schmidts M (2018) Recessive DNAH9 loss-of-function mutations cause laterality defects and subtle respiratory ciliary-beating defects. Am J Hum Genet 103:995–1008
Loges NT, Olbrich H, Fenske L, Mussaffi H, Horvath J, Fliegauf M, Kuhl H, Baktai G, Peterffy E, Chodhari R, Chung EM, Rutman A, O’Callaghan C, Blau H, Tiszlavicz L, Voelkel K, Witt M, Zietkiewicz E, Neesen J, Reinhardt R, Mitchison HM, Omran H (2008) DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet 83:547–558
Lores P, Coutton C, El Khouri E, Stouvenel L, Givelet M, Thomas L, Rode B, Schmitt A, Louis B, Sakheli Z, Chaudhry M, Fernandez-Gonzales A, Mitsialis A, Dacheux D, Wolf JP, Papon JF, Gacon G, Escudier E, Arnoult C, Bonhivers M, Savinov SN, Amselem S, Ray PF, Dulioust E, Toure A (2018) Homozygous missense mutation L673P in adenylate kinase 7 (AK7) leads to primary male infertility and multiple morphological anomalies of the flagella but not to primary ciliary dyskinesia. Hum Mol Genet 27:1196–1211
Lucas JS, Adam EC, Goggin PM, Jackson CL, Powles-Glover N, Patel SH, Humphreys J, Fray MD, Falconnet E, Blouin JL, Cheeseman MT, Bartoloni L, Norris DP, Lackie PM (2012) Static respiratory cilia associated with mutations in Dnahc11/DNAH11: a mouse model of PCD. Hum Mutat 33:495–503
Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, Dell S, Eber E, Escudier E, Hirst RA, Hogg C, Jorissen M, Latzin P, Legendre M, Leigh MW, Midulla F, Nielsen KG, Omran H, Papon JF, Pohunek P, Redfern B, Rigau D, Rindlisbacher B, Santamaria F, Shoemark A, Snijders D, Tonia T, Titieni A, Walker WT, Werner C, Bush A, Kuehni CE (2017) European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. https://doi.org/10.1183/13993003.01090-2016(print 2017 Jan)
Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C (2014) Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child 99:850–856
Maiti AK, Mattei MG, Jorissen M, Volz A, Zeigler A, Bouvagnet P (2000) Identification, tissue specific expression, and chromosomal localisation of several human dynein heavy chain genes. Eur J Hum Genet 8:923–932
Martinez G, Kherraf ZE, Zouari R, Fourati Ben Mustapha S, Saut A, Pernet-Gallay K, Bertrand A, Bidart M, Hograindleur JP, Amiri-Yekta A, Kharouf M, Karaouzene T, Thierry-Mieg N, Dacheux-Deschamps D, Satre V, Bonhivers M, Toure A, Arnoult C, Ray PF, Coutton C (2018) Whole-exome sequencing identifies mutations in FSIP2 as a recurrent cause of multiple morphological abnormalities of the sperm flagella. Hum Reprod 33:1973–1984
Mata M, Lluch-Estelles J, Armengot M, Sarrion I, Carda C, Cortijo J (2012) New adenylate kinase 7 (AK7) mutation in primary ciliary dyskinesia. Am J Rhinol Allergy 26:260–264
Mazor M, Alkrinawi S, Chalifa-Caspi V, Manor E, Sheffield VC, Aviram M, Parvari R (2011) Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am J Hum Genet 88:599–607
Merveille AC, Davis EE, Becker-Heck A, Legendre M, Amirav I, Bataille G, Belmont J, Beydon N, Billen F, Clement A, Clercx C, Coste A, Crosbie R, de Blic J, Deleuze S, Duquesnoy P, Escalier D, Escudier E, Fliegauf M, Horvath J, Hill K, Jorissen M, Just J, Kispert A, Lathrop M, Loges NT, Marthin JK, Momozawa Y, Montantin G, Nielsen KG, Olbrich H, Papon JF, Rayet I, Roger G, Schmidts M, Tenreiro H, Towbin JA, Zelenika D, Zentgraf H, Georges M, Lequarre AS, Katsanis N, Omran H, Amselem S (2011) CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet 43:72–78
Miki K, Willis WD, Brown PR, Goulding EH, Fulcher KD, Eddy EM (2002) Targeted disruption of the Akap4 gene causes defects in sperm flagellum and motility. Dev Biol 248:331–342
Milara J, Armengot M, Mata M, Morcillo EJ, Cortijo J (2010) Role of adenylate kinase type 7 expression on cilia motility: possible link in primary ciliary dyskinesia. Am J Rhinol Allergy 24:181–185
Milisav I, Jones MH, Affara NA (1996) Characterization of a novel human dynein-related gene that is specifically expressed in testis. Mamm Genome 7:667–672
Miranda-Vizuete A, Tsang K, Yu Y, Jimenez A, Pelto-Huikko M, Flickinger CJ, Sutovsky P, Oko R (2003) Cloning and developmental analysis of murid spermatid-specific thioredoxin-2 (SPTRX-2), a novel sperm fibrous sheath protein and autoantigen. J Biol Chem 278:44874–44885
Mitchison HM, Schmidts M, Loges NT, Freshour J, Dritsoula A, Hirst RA, O’Callaghan C, Blau H, Al Dabbagh M, Olbrich H, Beales PL, Yagi T, Mussaffi H, Chung EM, Omran H, Mitchell DR (2012) Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat Genet 44(381–9):S1–S2
Mitchison HM, Valente EM (2017) Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol 241:294–309
Moore DJ, Onoufriadis A, Shoemark A, Simpson MA, zur Lage PI, de Castro SC, Bartoloni L, Gallone G, Petridi S, Woollard WJ, Antony D, Schmidts M, Didonna T, Makrythanasis P, Bevillard J, Mongan NP, Djakow J, Pals G, Lucas JS, Marthin JK, Nielsen KG, Santoni F, Guipponi M, Hogg C, Antonarakis SE, Emes RD, Chung EM, Greene ND, Blouin JL, Jarman AP, Mitchison HM (2013) Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am J Hum Genet 93:346–356
Munro NC, Currie DC, Lindsay KS, Ryder TA, Rutman A, Dewar A, Greenstone MA, Hendry WF, Cole PJ (1994) Fertility in men with primary ciliary dyskinesia presenting with respiratory infection. Thorax 49:684–687
Neesen J, Kirschner R, Ochs M, Schmiedl A, Habermann B, Mueller C, Holstein AF, Nuesslein T, Adham I, Engel W (2001) Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum Mol Genet 10:1117–1128
Neesse A, Gangeswaran R, Luettges J, Feakins R, Weeks ME, Lemoine NR, Crnogorac-Jurcevic T (2007) Sperm-associated antigen 1 is expressed early in pancreatic tumorigenesis and promotes motility of cancer cells. Oncogene 26:1533–1545
Nunez-Olle M, Jung C, Terre B, Balsiger NA, Plata C, Roset R, Pardo-Pastor C, Garrido M, Rojas S, Alameda F, Lloreta J, Martin-Caballero J, Flores JM, Stracker TH, Valverde MA, Munoz FJ, Gil-Gomez G (2017) Constitutive Cyclin O deficiency results in penetrant hydrocephalus, impaired growth and infertility. Oncotarget 8:99261–99273
Oda T, Yanagisawa H, Kamiya R, Kikkawa M (2014) A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 346:857–860
Olbrich H, Schmidts M, Werner C, Onoufriadis A, Loges NT, Raidt J, Banki NF, Shoemark A, Burgoyne T, Al Turki S, Hurles ME, UK10 K Consortium, Kohler G, Schroeder J, Nurnberg G, Nurnberg P, Chung EM, Reinhardt R, Marthin JK, Nielsen KG, Mitchison HM, Omran H (2012) Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am J Hum Genet 91:672–684
Olcese C, Patel MP, Shoemark A, Kiviluoto S, Legendre M, Williams HJ, Vaughan CK, Hayward J, Goldenberg A, Emes RD, Munye MM, Dyer L, Cahill T, Bevillard J, Gehrig C, Guipponi M, Chantot S, Duquesnoy P, Thomas L, Jeanson L, Copin B, Tamalet A, Thauvin-Robinet C, Papon JF, Garin A, Pin I, Vera G, Aurora P, Fassad MR, Jenkins L, Boustred C, Cullup T, Dixon M, Onoufriadis A, Bush A, Chung EM, Antonarakis SE, Loebinger MR, Wilson R, Armengot M, Escudier E, Hogg C, UK10 K Rare Group, Amselem S, Sun Z, Bartoloni L, Blouin JL, Mitchison HM (2017) X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat Commun 8:14279
Omran H, Kobayashi D, Olbrich H, Tsukahara T, Loges NT, Hagiwara H, Zhang Q, Leblond G, O’Toole E, Hara C, Mizuno H, Kawano H, Fliegauf M, Yagi T, Koshida S, Miyawaki A, Zentgraf H, Seithe H, Reinhardt R, Watanabe Y, Kamiya R, Mitchell DR, Takeda H (2008) Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 456:611–616
Onoufriadis A, Paff T, Antony D, Shoemark A, Micha D, Kuyt B, Schmidts M, Petridi S, Dankert-Roelse JE, Haarman EG, Daniels JM, Emes RD, Wilson R, Hogg C, Scambler PJ, Chung EM, UK10 K, Pals G, Mitchison HM (2013) Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am J Hum Genet 92:88–98
Onoufriadis A, Shoemark A, Schmidts M, Patel M, Jimenez G, Liu H, Thomas B, Dixon M, Hirst RA, Rutman A, Burgoyne T, Williams C, Scully J, Bolard F, Lafitte JJ, Beales PL, Hogg C, Yang P, Chung EM, Emes RD, O’Callaghan C, UK10K, Bouvagnet P, Mitchison HM (2014) Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects. Hum Mol Genet 23:3362–3374
Osinka A, Poprzeczko M, Zielinska MM, Fabczak H, Joachimiak E, Wloga D (2019) Ciliary proteins: filling the gaps. Recent advances in deciphering the protein composition of motile ciliary complexes. Cells. https://doi.org/10.3390/cells8070730
Ozkavukcu S, Celik-Ozenci C, Konuk E, Atabekoglu C (2018) Live birth after laser assisted viability assessment (LAVA) to detect pentoxifylline resistant ejaculated immotile spermatozoa during ICSI in a couple with male Kartagener’s syndrome. Reprod Biol Endocrinol 16:10
Paff T, Loges NT, Aprea I, Wu K, Bakey Z, Haarman EG, Daniels JMA, Sistermans EA, Bogunovic N, Dougherty GW, Hoben IM, Grosse-Onnebrink J, Matter A, Olbrich H, Werner C, Pals G, Schmidts M, Omran H, Micha D (2017) Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am J Hum Genet 100:160–168
Pereira R, Oliveira J, Ferraz L, Barros A, Santos R, Sousa M (2015) Mutation analysis in patients with total sperm immotility. J Assist Reprod Genet 32:893–902
Pereira R, Oliveira ME, Santos R, Oliveira E, Barbosa T, Santos T, Goncalves P, Ferraz L, Pinto S, Barros A, Oliveira J, Sousa M (2019) Characterization of CCDC103 expression profiles: further insights in primary ciliary dyskinesia and in human reproduction. J Assist Reprod Genet 36:1683–1700
Raidt J, Werner C, Menchen T, Dougherty GW, Olbrich H, Loges NT, Schmitz R, Pennekamp P, Omran H (2015) Ciliary function and motor protein composition of human fallopian tubes. Hum Reprod 30:2871–2880
Rawe VY, Galaverna GD, Acosta AA, Olmedo SB, Chemes HE (2001) Incidence of tail structure distortions associated with dysplasia of the fibrous sheath in human spermatozoa. Hum Reprod 16:879–886
Rawe VY, Terada Y, Nakamura S, Chillik CF, Olmedo SB, Chemes HE (2002) A pathology of the sperm centriole responsible for defective sperm aster formation, syngamy and cleavage. Hum Reprod 17:2344–2349
Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA (2013) Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol 14:713–726
Robson EA, Chetcuti P, Hirst RA, Mitchison H, Moya E, Peckham D, Robinson PJ, Rutman A, O’Callaghan C (2017) Update on primary ciliary dyskinesia. Paediatr Child Health 27(7):337–342
Rubbo B, Lucas JS (2017) Clinical care for primary ciliary dyskinesia: current challenges and future directions. Eur Respir Rev. https://doi.org/10.1183/16000617.0023-2017(print 2017 Sep 30)
Rubbo B, Shoemark A, Jackson CL, Hirst R, Thompson J, Hayes J, Frost E, Copeland F, Hogg C, O’Callaghan C, Reading I, Lucas JS (2019) Accuracy of high-speed video analysis to diagnose primary ciliary dyskinesia. Chest 155:1008–1017
Sadek CM, Damdimopoulos AE, Pelto-Huikko M, Gustafsson JA, Spyrou G, Miranda-Vizuete A (2001) Sptrx-2, a fusion protein composed of one thioredoxin and three tandemly repeated NDP-kinase domains is expressed in human testis germ cells. Genes Cells 6:1077–1090
Schwabe GC, Hoffmann K, Loges NT, Birker D, Rossier C, de Santi MM, Olbrich H, Fliegauf M, Failly M, Liebers U, Collura M, Gaedicke G, Mundlos S, Wahn U, Blouin JL, Niggemann B, Omran H, Antonarakis SE, Bartoloni L (2008) Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat 29:289–298
Sha YW, Wang X, Su ZY, Mei LB, Ji ZY, Bao H, Li P (2018) Patients with multiple morphological abnormalities of the sperm flagella harbouring CFAP44 or CFAP43 mutations have a good pregnancy outcome following intracytoplasmic sperm injection. Andrologia 51:e13151
Sha YW, Xu X, Mei LB, Li P, Su ZY, He XQ, Li L (2017) A homozygous CEP135 mutation is associated with multiple morphological abnormalities of the sperm flagella (MMAF). Gene 633:48–53
Shapiro AJ, Davis SD, Polineni D, Manion M, Rosenfeld M, Dell SD, Chilvers MA, Ferkol TW, Zariwala MA, Sagel SD, Josephson M, Morgan L, Yilmaz O, Olivier KN, Milla C, Pittman JE, Daniels MLA, Jones MH, Janahi IA, Ware SM, Daniel SJ, Cooper ML, Nogee LM, Anton B, Eastvold T, Ehrne L, Guadagno E, Knowles MR, Leigh MW, Lavergne V, American Thoracic Society Assembly on Pediatrics (2018) Diagnosis of primary ciliary dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med 197:e24–e39
Shapiro AJ, Leigh MW (2017) Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: genetic defects with normal and non-diagnostic ciliary ultrastructure. Ultrastruct Pathol 41:373–385
Shen Y, Zhang F, Li F, Jiang X, Yang Y, Li X, Li W, Wang X, Cheng J, Liu M, Zhang X, Yuan G, Pei X, Cai K, Hu F, Sun J, Yan L, Tang L, Jiang C, Tu W, Xu J, Wu H, Kong W, Li S, Wang K, Sheng K, Zhao X, Yue H, Yang X, Xu W (2019) Loss-of-function mutations in QRICH2 cause male infertility with multiple morphological abnormalities of the sperm flagella. Nat Commun 10:433
Shinohara K, Chen D, Nishida T, Misaki K, Yonemura S, Hamada H (2015) Absence of radial spokes in mouse node cilia is required for rotational movement but confers ultrastructural instability as a trade-off. Dev Cell 35:236–246
Shoemark A (2017) Applications of emerging transmission electron microscopy technology in PCD research and diagnosis. Ultrastruct Pathol 41:408–414
Shoemark A, Burgoyne T, Kwan R, Dixon M, Patel MP, Rogers AV, Onoufriadis A, Scully J, Daudvohra F, Cullup T, Loebinger MR, Wilson R, Chung EMK, Bush A, Mitchison HM, Hogg C (2018) Primary ciliary dyskinesia with normal ultrastructure: three-dimensional tomography detects absence of DNAH11. Eur Respir J. https://doi.org/10.1183/13993003.01809-2017(print 2018 Feb)
Shoemark A, Dixon M, Corrin B, Dewar A (2012) Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J Clin Pathol 65:267–271
Shoemark A, Frost E, Dixon M, Ollosson S, Kilpin K, Patel M, Scully J, Rogers AV, Mitchison HM, Bush A, Hogg C (2017) Accuracy of immunofluorescence in the diagnosis of primary ciliary dyskinesia. Am J Respir Crit Care Med 196:94–101
Sironen A, Hansen J, Thomsen B, Andersson M, Vilkki J, Toppari J, Kotaja N (2010) Expression of SPEF2 during mouse spermatogenesis and identification of IFT20 as an interacting protein. Biol Reprod 82:580–590
Sironen A, Kotaja N, Mulhern H, Wyatt TA, Sisson JH, Pavlik JA, Miiluniemi M, Fleming MD, Lee L (2011) Loss of SPEF2 function in mice results in spermatogenesis defects and primary ciliary dyskinesia. Biol Reprod 85:690–701
Sironen A, Thomsen B, Andersson M, Ahola V, Vilkki J (2006) An intronic insertion in KPL2 results in aberrant splicing and causes the immotile short-tail sperm defect in the pig. Proc Natl Acad Sci USA 103:5006–5011
Smith DJ, Gaffney EA, Blake JR (2008) Modelling mucociliary clearance. Respir Physiol Neurobiol 163:178–188
Tang S, Wang X, Li W, Yang X, Li Z, Liu W, Li C, Zhu Z, Wang L, Wang J, Zhang L, Sun X, Zhi E, Wang H, Li H, Jin L, Luo Y, Wang J, Yang S, Zhang F (2017) Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. Am J Hum Genet 100:854–864
Tarkar A, Loges NT, Slagle CE, Francis R, Dougherty GW, Tamayo JV, Shook B, Cantino M, Schwartz D, Jahnke C, Olbrich H, Werner C, Raidt J, Pennekamp P, Abouhamed M, Hjeij R, Kohler G, Griese M, Li Y, Lemke K, Klena N, Liu X, Gabriel G, Tobita K, Jaspers M, Morgan LC, Shapiro AJ, Letteboer SJ, Mans DA, Carson JL, Leigh MW, Wolf WE, Chen S, Lucas JS, Onoufriadis A, Plagnol V, Schmidts M, Boldt K, UK10K, Roepman R, Zariwala MA, Lo CW, Mitchison HM, Knowles MR, Burdine RD, Loturco JJ, Omran H (2013) DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat Genet 45:995–1003
Ta-Shma A, Hjeij R, Perles Z, Dougherty GW, Abu Zahira I, Letteboer SJF, Antony D, Darwish A, Mans DA, Spittler S, Edelbusch C, Cindric S, Nothe-Menchen T, Olbrich H, Stuhlmann F, Aprea I, Pennekamp P, Loges NT, Breuer O, Shaag A, Rein AJJT, Gulec EY, Gezdirici A, Abitbul R, Elias N, Amirav I, Schmidts M, Roepman R, Elpeleg O, Omran H (2018) Homozygous loss-of-function mutations in MNS1 cause laterality defects and likely male infertility. PLoS Genet 14:e1007602
Terre B, Lewis M, Gil-Gomez G, Han Z, Lu H, Aguilera M, Prats N, Roy S, Zhao H, Stracker TH (2019) Defects in efferent duct multiciliogenesis underlie male infertility in GEMC1-, MCIDAS- or CCNO-deficient mice. Development. https://doi.org/10.1242/dev.162628
Urbanska P, Song K, Joachimiak E, Krzemien-Ojak L, Koprowski P, Hennessey T, Jerka-Dziadosz M, Fabczak H, Gaertig J, Nicastro D, Wloga D (2015) The CSC proteins FAP61 and FAP251 build the basal substructures of radial spoke 3 in cilia. Mol Biol Cell 26:1463–1475
Vanaken GJ, Bassinet L, Boon M, Mani R, Honore I, Papon JF, Cuppens H, Jaspers M, Lorent N, Coste A, Escudier E, Amselem S, Maitre B, Legendre M, Christin-Maitre S (2017) Infertility in an adult cohort with primary ciliary dyskinesia: phenotype-gene association. Eur Respir J. https://doi.org/10.1183/13993003.00314-2017(print 2017 Nov)
Vanaken GJ, Bassinet L, Boon M, Mani R, Honore I, Papon JF, Cuppens H, Jaspers M, Lorent N, Coste A, Escudier E, Amselem S, Maitre B, Legendre M, Christin-Maitre S (2017) Infertility in an adult cohort with primary ciliary dyskinesia: phenotype-gene association. Eur Respir J. https://doi.org/10.1183/13993003.00314-2017(print 2017 Nov)
Van’s Gravesande KS, Omran H (2005) Primary ciliary dyskinesia: clinical presentation, diagnosis and genetics. Ann Med 37:439–449
Wallmeier J, Al-Mutairi DA, Chen CT, Loges NT, Pennekamp P, Menchen T, Ma L, Shamseldin HE, Olbrich H, Dougherty GW, Werner C, Alsabah BH, Kohler G, Jaspers M, Boon M, Griese M, Schmitt-Grohe S, Zimmermann T, Koerner-Rettberg C, Horak E, Kintner C, Alkuraya FS, Omran H (2014) Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet 46:646–651
Wallmeier J, Shiratori H, Dougherty GW, Edelbusch C, Hjeij R, Loges NT, Menchen T, Olbrich H, Pennekamp P, Raidt J, Werner C, Minegishi K, Shinohara K, Asai Y, Takaoka K, Lee C, Griese M, Memari Y, Durbin R, Kolb-Kokocinski A, Sauer S, Wallingford JB, Hamada H, Omran H (2016) TTC25 deficiency results in defects of the outer dynein arm docking machinery and primary ciliary dyskinesia with left-right body asymmetry randomization. Am J Hum Genet 99:460–469
Wambergue C, Zouari R, Fourati Ben Mustapha S, Martinez G, Devillard F, Hennebicq S, Satre V, Brouillet S, Halouani L, Marrakchi O, Makni M, Latrous H, Kharouf M, Amblard F, Arnoult C, Ray PF, Coutton C (2016) Patients with multiple morphological abnormalities of the sperm flagella due to DNAH1 mutations have a good prognosis following intracytoplasmic sperm injection. Hum Reprod 31:1164–1172
Wang X, Jin H, Han F, Cui Y, Chen J, Yang C, Zhu P, Wang W, Jiao G, Wang W, Hao C, Gao Z (2017) Homozygous DNAH1 frameshift mutation causes multiple morphological anomalies of the sperm flagella in Chinese. Clin Genet 91:313–321
Wheway G, Mitchison HM (2019) Opportunities and challenges for molecular understanding of ciliopathies—the 100,000 Genomes Project. Front Genet 10:127
Whitfield M, Thomas L, Bequignon E, Schmitt A, Stouvenel L, Montantin G, Tissier S, Duquesnoy P, Copin B, Chantot S, Dastot F, Faucon C, Barbotin AL, Loyens A, Siffroi JP, Papon JF, Escudier E, Amselem S, Mitchell V, Toure A, Legendre M (2019) Mutations in DNAH17, encoding a sperm-specific axonemal outer dynein arm heavy chain, cause isolated male infertility due to asthenozoospermia. Am J Hum Genet 105(1):198–212
Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS, Loges NT, Pennekamp P, Lindberg S, Stenram U, Carlen B, Horak E, Kohler G, Nurnberg P, Nurnberg G, Porter ME, Omran H (2013) The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat Genet 45:262–268
Yang K, Meinhardt A, Zhang B, Grzmil P, Adham IM, Hoyer-Fender S (2012) The small heat shock protein ODF1/HSPB10 is essential for tight linkage of sperm head to tail and male fertility in mice. Mol Cell Biol 32:216–225
Yang L, Banerjee S, Cao J, Bai X, Peng Z, Chen H, Huang H, Han P, Feng S, Yi N, Song X, Wu J (2018) Compound heterozygous variants in the coiled-coil domain containing 40 gene in a chinese family with primary ciliary dyskinesia cause extreme phenotypic diversity in cilia ultrastructure. Front Genet 9:23
Yildirim G, Ficicioglu C, Akcin O, Attar R, Tecellioglu N, Yencilek F (2009) Can pentoxifylline improve the sperm motion and ICSI success in the primary ciliary dyskinesia? Arch Gynecol Obstet 279:213–215
Yuan S, Liu Y, Peng H, Tang C, Hennig GW, Wang Z, Wang L, Yu T, Klukovich R, Zhang Y, Zheng H, Xu C, Wu J, Hess RA, Yan W (2019) Motile cilia of the male reproductive system require miR-34/miR-449 for development and function to generate luminal turbulence. Proc Natl Acad Sci USA 116:3584–3593
Zabeo D, Heumann JM, Schwartz CL, Suzuki-Shinjo A, Morgan G, Widlund PO, Hoog JL (2018) A lumenal interrupted helix in human sperm tail microtubules. Sci Rep 8:2727
Zariwala MA, Leigh MW, Ceppa F, Kennedy MP, Noone PG, Carson JL, Hazucha MJ, Lori A, Horvath J, Olbrich H, Loges NT, Bridoux AM, Pennarun G, Duriez B, Escudier E, Mitchison HM, Chodhari R, Chung EM, Morgan LC, de Iongh RU, Rutland J, Pradal U, Omran H, Amselem S, Knowles MR (2006) Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am J Respir Crit Care Med 174:858–866
Zheng Y, Zhang J, Wang L, Zhou Z, Xu M, Li J, Sha JH (2006) Cloning and characterization of a novel sperm tail protein, NYD-SP28. Int J Mol Med 18:1119–1125
Zhou J, Yang F, Leu NA, Wang PJ (2012) MNS1 is essential for spermiogenesis and motile ciliary functions in mice. PLoS Genet 8:e1002516
Zuccarello D, Ferlin A, Cazzadore C, Pepe A, Garolla A, Moretti A, Cordeschi G, Francavilla S, Foresta C (2008) Mutations in dynein genes in patients affected by isolated non-syndromic asthenozoospermia. Hum Reprod 23:1957–1962
Zur Lage P, Newton FG, Jarman AP (2019) Survey of the ciliary motility machinery of drosophila sperm and ciliated mechanosensory neurons reveals unexpected cell-type specific variations: a model for motile ciliopathies. Front Genet 10:24
Acknowledgements
We acknowledge funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Grant Agreement PCDFert: Identification of novel genes and mechanisms for PCD and male infertility No. 800556 (MSCA Individual Fellowship awarded to AS). HMM is supported by the NIHR Biomedical Research Center at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London and Great Ormond Street Children’s Charity Leadership Grant awards (V1299, V2217). The authors participate in the COST Action BEAT-PCD: Better Evidence to Advance Therapeutic options for PCD network (BM1407).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sironen, A., Shoemark, A., Patel, M. et al. Sperm defects in primary ciliary dyskinesia and related causes of male infertility. Cell. Mol. Life Sci. 77, 2029–2048 (2020). https://doi.org/10.1007/s00018-019-03389-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03389-7