
Abstract
Dendritic cell (DC)-based immunotherapy is an attractive approach to induce long lasting antitumor effector cells aiming to control cancer progression. DC targeting is a critical step in the design of DC vaccines in order to optimize delivery and processing of the antigen, and several receptors have been characterized for this purpose. In this study, we employed the FcγRs to target DCs both in vitro and in vivo. We designed a recombinant molecule (HER2-Fc) composed of the immunogenic sequence of the human tumor-associated antigen HER2 (aa 364–391) and the Fc domain of a human IgG1. In a mouse model, HER2-Fc cDNA vaccination activated significant T cell-mediated immune responses towards HER2 peptide epitopes as detected by IFN-γ ELIspot and induced longer tumor latency as compared to Ctrl-Fc-vaccinated control mice. Human in vitro studies indicated that the recombinant HER2-Fc immunogen efficiently targeted human DCs through the FcγRs resulting in protein cross-processing and in the activation of autologous HER2-specific CD8+ T cells from breast cancer patients.
Similar content being viewed by others


Introduction
The design of cancer vaccines is finalized to control tumor growth by activating long lasting antitumor immune responses and removing the immunosuppressive networks that are established during tumor progression. So far, the harnessing of dendritic cells (DCs) in order to modulate immune system activity is an experimental approach gaining increasing therapeutic significance [1].
DCs are the most powerful antigen presenting cells (APCs) and key regulators for the coordination and balance of the innate and acquired immunity leading to T cell activation and finally to tumor cell killing. A heterogeneous array of receptors is employed by DCs to sense danger signals, triggering DC maturation, and licensing [2]. The targeting of these receptors has been proposed as an efficacious strategy to deliver antigen to DCs both in vivo and ex vivo [3]. The family of Fc receptors for IgGs (FcγRs) is an example of such molecules [4]. In vivo uptake of IgG-soluble antigen complexes through FcγRs leads to DC activation, secretion of pro-inflammatory cytokines, and steers antigen processing towards MHCI and MHCII pathways, inducing CD4+ and CD8+ antigen-specific T cell responses [5].
DC targeting through activatory FcγRs has been proposed as optimal strategy to induce efficacious antitumor immune responses [3, 6]. DCs pulsed with immunocomplexed antigen have shown to be superior to soluble antigen-pulsed DCs as antitumor vaccine [7]. Moreover, in vivo studies demonstrate that the combined administration of antitumor antibody and tumor vaccine enhanced antitumor CD8+ T cell response by FcγRs-mediated activation of DCs [8]. Vaccination with plasmids coding for immunogenic sequences fused to the Fc fragment of human IgG1 (hIgG1 Fc) is also a strategy to target DCs in vivo [9, 10]. So far, however, the employment of FcγR targeting in antitumor clinical setting is limited to the treatment of hematological malignancies [11].
In this study, we designed an HER2-Fc chimera to target DCs both in vivo and ex vivo as cancer vaccine. HER2 is a growth factor receptor overexpressed in epithelial cancers and is optimal target for passive and active immunotherapy [12]. In cancer patients, natural specific immune responses can be observed and peptide vaccination has been correlated to a better disease outcome [13].
Results obtained in our experimental model indicate that HER2-Fc cDNA vaccination activated HER2 specific T cell-mediated responses and induced a significantly longer tumor latency in mice than the control Ctrl-Fc cDNA. The recombinant immunogen was able to target human DCs through the FcγR, and antigen processing occurred both in human leukocyte antigens (HLA)II and HLAI compartments. HER2-Fc-pulsed DCs induced a significant interferon gamma (IFN-γ) secretion by HER2 specific CD8+ T cells obtained from HER2+/HLAI-A2+ breast cancer patients.
Material and methods
Cell cultures
CHO-K1 cells were cultured in RPMI 1640 (Hyclone, UT, USA) with penicillin 100 U/ml, streptomycin 100 μg/mL, 2 mM L-glutamine, (Sigma Company, St. Louis, MO) and 10% fetal calf serum (FCS) (Hyclone), 5% CO2 at 37°C. For CHO-K1 transfectants, 400 μg/ml Geneticin (Invitrogen, Germany) were added to the medium. D2F2/E2 cells, BALB/c mammary tumor cotransfected with pRSV/neo and human HER2 were cultured in DMEM with Glutamax1, 20% FCS, 800 μg/ml Geneticin. They were kindly provided by Dr. Wei, Karmanos Cancer Center, Detroit.
Cloning of HER2-Fc and Ctrl-cDNA constructs
The leader sequence (LS) and HER2 cDNAs were generated by oligonucleotide annealing. After purification, the annealed oligonucleotides were cloned into pBS-KS+ (Stratagene, USA) (LS: ApaI/XhoI sites; HER2: EcoRI/PstI sites). This sequence was used as a template to amplify HER2 cDNA with XhoI-BamHI ends (primer A1xA2). The HER2 cDNA was cloned at the 3′ end of the LS (cloning sites: 5′-XhoI, 3′-BamHI) (construct pBS-KS+LS-HER2). Fc was amplified (B1xB2 primers) using as template the pUCγ1 and fused to LS-HER2 (BamHI-NotI) to generate the HER2-Fc cDNA. For the Ctrl-Fc cDNA, Fc sequence was amplified with C1xB2 primers and cloned at the 3′ of LS in pBS-KS+ in XhoI-NotI sites. Both cDNAs were cloned HindIII-NotI into pcDNA3.1 Neo (Invitrogen). Large-scale preparation was carried out with EndoFree Plasmid Giga kits (Qiagen). The oligo sequences and experimental conditions are reported in Supplementary Table 1.
Protein production
The CHO-K1 cells were transfected using Lipofectamine™ 2000 (Invitrogen) in a 10-cm tissue culture disk following the manufacturer’s instructions.
Transfectant supernatant was loaded onto protein A sepharose 4B conjugate (Invitrogen). The bound protein was eluted with 0.1 M glycine hydrochloride, pH 2.6, neutralized (1 M Tris) and dialyzed against 0.1× PBS using SnakeSkin® Pleated Dialysis Tubing (3.5 MWCO) (Pierce, Rockford, USA).
Western blotting
Purified HER2-Fc and Ctrl-Fc proteins were separated on 4–12% SDS-PAGE and blotted onto nitrocellulose membrane (Schleicher and Schuell, Dassel, Germany). After 5% BSA blocking (16 h, 4°C), membranes were incubated with anti-human IgG peroxidase-conjugated antibody (1:10,000) (Sigma) for 1 h at room temperature and protein bands detected by chemiluminescence (Amersham Pharmacia, Buckinghamshire, UK).
Mice, immunization, and tumor growth
BALB/c (H-2d) female mice were from Charles River Laboratory and were treated according to the European Union guidelines. The mice were anesthetized and vaccinated as previously described [14], i.e., i.m. injections of plasmid (50 μg) followed by electroporation. The mice were vaccinated two times at 14-day intervals. The group of five to 11 mice were used for tumor challenge experiments, while the group of three mice were used for the analysis of immune response. Experiments were repeated three times. For tumor rejection studies, 1 week after the second vaccination, the mice were challenged in the mammary pad with a lethal dose of transfected D2F2/E2 cells with human HER2 (2 × 105). The mammary pad was inspected weekly by palpation. Progressively growing masses >2 mm in diameter were regarded as tumors. The mice were sacrificed when one of the tumors exceeded 10 mm in diameter. Differences in the time required for the tumor to reach 2 or 10 mm in mean diameter in each experimental group was calculated.
Mouse IFN-γ enzyme-linked immunospot assay
Splenocytes (SPCs), 1 × 106, from vaccinated mice 2 weeks after the second vaccination were plated in triplicate on nitrocellulose 96-well HTS IP plates (Millipore, Bedford, MA) precoated with 5 μg/mL of rat anti-mouse IFN-γ antibody (clone R4-6A2, BD Biosciences, Milan, Italy). SPCs were stimulated for 16 h at 37°C with 15 μg/mL of each peptide (Proimmune, Oxford, UK). Plates were developed according to the manufacturer’s instruction (BD Biosciences). Briefly, after washing with PBS 0.05% Tween 20, plates were incubated for 24 h at 4°C with 50 μl/well of biotin-conjugated rat anti-mouse IFN-γ (BD Biosciences) diluted in assay buffer. Plates were incubated with streptavidin horseradish peroxidase conjugate (HRP, BD Biosciences). After extensive washing, AEC substrate solution was added until spot development. Plates were washed, and IFN-γ spots were enumerated as previously described [15].
DC generation
DCs were generated as previously described [16]. Peripheral blood mononuclear cells (PBMCs) were isolated from the buffy coat of healthy donors or from blood samples of HLAI-A2+/HER2+ breast cancer patients and separated by Ficoll-Hypaque (1,077 g/mL; Pharmacia LKB, Upsala, Sweden) gradient. Immunoselected CD14+ monocytes were plated (5 × 105 cells per milliliter) in CellGro® DC medium (CellGenix, Freiburg, Germany), with rhGM-CSF (50 ng/ml), and rhIL4 (2000 U/ml). On day 6, immature DCs (iDCs) were matured for 16 h (mDCs) with pro-inflammatory cytokines (R&D System). The study was approved by the institution regulatory board and by the ethical committee. Informed consent was obtained by cancer patients.
Fusion protein binding to DCs
iDCs (2 × 104/sample) were incubated with HER2-Fc protein or MOPC21 (Sigma) (10 μg/ml, 2 h on ice). After washing, DCs were incubated with fluorescein isothiocynate (FITC)-conjugated F(ab)2 specific for anti-human Fcγ (Jackson ImmunoResearch Laboratories, Inc., West Grove, USA) (30 min, 4°C). To block Fcγ-R, iDCs were incubated with anti-human FcγRIIa MoAb (StemStep) (5 μg/ml, 30 min, 4°C) before the binding procedure. Cells of 5 × 104 were evaluated using FACSCanto II flow cytometer running CellQuest software (Becton Dickinson).
Immunofluorescence microscopy
iDCs were pulsed with HER2-Fc (10 μg/ml; 30 min on ice), washed and incubated at 37°C (2 or 12 h). After washing, the cells were cytospun onto SuperFrost® Plus Microscope Slides (Menzel-Glaser, Germany) (8 × 104/sample), fixed with cold acetone/methanol (Carlo Erba Reagents) (1:1). After the blocking of unspecific sites, the samples were incubated for 45 min with compartment marker-specific antibodies: MoAb anti-lysosomal-associated membrane protein 1 (LAMP1) (E-5, Santa Cruz Biotechnology), anti-HLAI MoAb W6.32, and rabbit polyclonal anti-calreticulin (Stressgene, Ann Arbor, MI). Texas Red-conjugated anti-mouse (1:100) or anti-rabbit (1:300) antibodies were used as secondary Abs (30 min). After the blocking of aspecific sites by Superblock (20 min), the samples were incubated with FITC-anti-human Fcγ-specific donkey F(ab)2. The secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. Cover slips were mounted with VECTASHIELD (Vector Laboratories Inc., Burlingame, CA). To assess the extent of the colocalization of fluorescence signals, the cells were scanned in a series of 0.5-μm sequential sections with an ApoTome System (Zeiss, Oberkochen, Germany) connected with an Axiovert 200 inverted microscope (Zeiss); image analysis and 3D reconstruction were then performed by the Axiovision software (Zeiss). A quantitative analysis of the extent of colocalization was performed using the Zeiss KS300 3.0 Image Processing system (Zeiss). The mean ± standard error (SE) percent of colocalization was calculated analyzing a minimum of 30 cells for each treatment randomly taken from three independent experiments.
E75-specific CD8+ T cell enrichment
PBMCs were isolated from six HER2+/HLAI-A2+ breast cancer patients. CD8+ T cells were purified using CD8 MicroBeads (Miltenyi Biotech). CD8− cells were pulsed with the E75 HER2 HLAI-A2 restricted epitope (KIFGSLAFL) (ClinAlfa, Germany) (50 μg/ml) and β2-microglobulin (Sigma) (5 μg/ml). After overnight incubation, irradiated E75-pulsed CD8− cells were added to CD8+ T cells (1:1). Interleukin (IL)-2 (50 U/ml) and IL-7 (10 ng/ml) were added to the culture. A second stimulation was performed after 9 days. E75-specific CD3+/CD8+ T cells were analyzed by staining with PE-HLA-A*0201 Pro5-pentamer containing the E75 epitope (ProImmune, Oxford, UK).
IFN-γ ELIspot
iDCs were pulsed with HER2-Fc and Ctrl-Fc proteins (10 μg/ml) and E75 peptide (20 μg/ml) with β2-microglobulin (5 μg/ml) (Sigma) for 4–8 h and matured. Pulsed mDCs (2 × 104 cells per milliliter) were plated with autologous E75-stimulated CD8+ T cells (1 × 105 cells per milliliter) onto plates (Millipore, Bedford, MA), precoated with the anti-IFN-γ MoAb (NIB42, Pharmingen, San Diego, CA) for 24 h. Cells were removed and IFN-γ production was detected with biotinylated anti-IFN-γ MoAb (4S.B3, Pharmingen) (5 μg/ml), revealed with streptavidin–alkaline phosphatase (Pharmingen) (1:2,000), and BCIP/NBT alkaline phosphatase substrate (50 μl/well, Sigma). Unpulsed mDCs plated with CD8+ cells were used as negative control (IFN-γ spots <6/105 cells). Spots were counted using the ImmunoSpot Image Analyzer (Aelvis, Germany). Results were plotted as median of spot values among triplicates.
Statistical analysis
Descriptive statistics (average and standard deviation) were used to describe various groups of data. A two-tailed Student’s t test was adopted to compare the different groups of data. A statistical significance was set at a p value less than 0.05.
Results
Generation of the recombinant HER2-Fc and Ctrl-Fc chimera
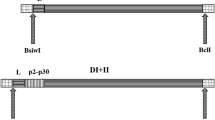
The human IgG1 Fc fragment was used as cell-binding domain to deliver HER2 tumor antigen to DCs. The HER2-Fc cDNA was made by cloning the leader sequence of a murine κ chain (LS) in front of the HER2(364–391) coding sequence containing the immunogenic epitope E75 (KIFGSLAFL) (Fig. 1a). The HER2 sequence was mutated (I370 → V370; L377 → V377) to generate a modified E75 epitope with higher binding affinity for HLAI-A2 allele (KVFGSLAFV). This fragment was then linked in frame to the amplified human Fc IgG1 cDNA. The Ctrl-Fc construct was made by linking the LS and Fc sequences (Fig. 1a). Stable CHO-K1 transfectants efficiently secreted the proteins. HER2-Fc, and Ctrl-Fc displayed the expected molecular weight (59.4 and 53.4 kDa, respectively) as detected by Western blotting (Fig. 1b). In reducing conditions, HER2-Fc and Ctrl-Fc migrated as bands corresponding to 29.7 and 26.7 kDa, respectively, indicating that both recombinant proteins were secreted as dimers (Fig. 1b).

Generation and characterization of HER2-Fc and Ctrl-Fc proteins. a Diagrams illustrating the cDNA structures of the two chimeric cDNAs. The coded amino acid sequence of the HER2 region used is shown (modified amino acids are underlined) (LS, leader sequence; HER2 indicates the amino acid stretch 364–391). b Western blot analysis of HER2-Fc and Ctrl-Fc. Proteins (1 μg/sample) were fractionated in nonreducing and reducing conditions (±β-mercaptoethanol) in SDS-PAGE 4–12% gradient gel and blotted onto nitrocellulose. Western blotting was performed using peroxidase-conjugated MoAb anti-Fcγ revealed by chemiluminescence
HER2-Fc vaccination induces longer survival in BALB/c mice transplanted with HER2+ mammary tumor
Immunogenicity of HER2-Fc cDNA was evaluated in vivo in tumor rejection mouse model. BALB/c mice were vaccinated twice at 2-week intervals and 1 week after the second vaccination challenged s.c. with mouse mammary D2F2/E2 tumor cells, transfected with the human HER2 antigen. A growing tumor was displayed by all mice vaccinated with the empty pcDNA3.1 plasmid, whereas about 50% protection was afforded by both Ctrl-Fc and HER2-Fc plasmids (Fig. 2a). A delay in tumor growth was observed in three out of five mice vaccinated with HER2-Fc as compared to those vaccinated with Ctrl-Fc or with pcDNA3.1 (Fig. 2b). Moreover, the tumor latency in mice vaccinated with HER2-Fc was higher as compared to those vaccinated with Ctrl-Fc (44.3 ± 3.9 days vs 26.8 ± 2.7 days). Indeed, the mean time required for a carcinoma to exceed a 10-mm threshold was 91 ± 10.4 days in mice vaccinated with HER2-Fc, and it was significantly longer (p = 0.02) as compared to the one measured in mice vaccinated with Ctrl-Fc (58 ± 3.9 days).

HER2-Fc cDNA vaccination and antitumor response elicited in BALB/c mice. Groups of mice were vaccinated twice at 2 weeks interval with empty pcDNA3.1 (n = 5; gray line), Ctrl-Fc (n = 10; dotted black line), and HER2-Fc (n = 11; black line) and challenged 1 week after the second vaccination with a lethal dose of D2F2/E2 cells transfected with the human HER2. a The percentage of tumor-free mice in function of time is represented. b Ability of vaccination to delay tumor growth. Each line refers to an individual tumor (pcDNA3.1, n = 5; Ctrl-Fc, n = 5; HER2-Fc, n = 5)
HER2-Fc vaccination promotes CD8+ T cell-mediated IFN-γ secretion in BALB/c-vaccinated mice
To test if the delay in tumor growth observed in HER2-Fc-vaccinated mice was due to the ability of the vaccine to induce HER2-specific T cell responses, we evaluated in vitro IFN-γ secretion of splenocytes from vaccinated mice by ELIspot assay. The HER2(364–391) sequence coded by HER2-Fc chimera (FAGCKKVFGSLAFVPESFDGDPASNTAP) and the corresponding native HER2 sequence (FAGCKKIFGSLAFLPESFDGDPASNTAP) were scanned for H2d class I binding peptides using the prediction algorithm used by the software “SYFPEITHI” [17], in order to select human HER2 immunogenic epitopes presented by mouse APCs in the context of H2d. Five nonapeptides with high score for H2d binding (>13) were selected: three nonapeptides derived from the modified HER2 peptide sequence of the vaccine (GCKKVFGSL, KVFGSLAFV and KKVFGSLAF) and two nonapeptides contained in the wt HER2 sequence (GCKKIFGSL and KIFGSLAFL) (Table 1). SPCs of the cDNA-vaccinated mice were incubated overnight with each of the epitopes derived from the HER2-modified sequence encoded by the vaccine, and T cell-mediated response was measured by IFN-γ ELIspot assay. Following HER2 peptide stimulation, SPCs from HER2-Fc-vaccinated mice displayed a significantly higher number of IFN-γ producing T cells as compared to those elicited by Ctrl-Fc vaccination (Fig. 3a, c, e; p = 0.0004 for A and E, p = 0,008 for C). Also, a significant HER2-specific T cell-mediated IFN-γ response was elicited by stimulation with the corresponding native HER2 epitopes (Fig. 3b, d; p = 0.008 and p = 0.0004, respectively). These results suggested that cDNA vaccination induced specific HER2 T cell responses reacting with the modified HER2 sequences and could cross-react with the native HER2 peptides.

CD8+ T cell-mediated IFN-γ response in BALB/c vaccinated mice. ELIspot analysis of IFN-γ-producing cells after restimulation of spleen cells with H-2d peptides derived from the HER2 modified sequence encoded by the vaccine (a, c, e) or from the native HER2 sequence (b, d). IFN-γ secretion by SPCs from mice (n = 3) vaccinated twice with the Ctrl-Fc (gray) or with the HER2-Fc (black) was expressed as spot-forming unit and mean of triplicate plotted as histograms. Data were analyzed using the two-tailed Student’s t test (**p = 0.008; ***p = 0.0004)
HER2-Fc chimera protein internalized through FcγR is cross-processed by DCs
In order to evaluate whether HER2-Fc protein could be a valid immunogen to load DCs, uptake and processing of HER2-Fc were investigated employing DCs differentiated from human PBMCs. DCs generated in serum-free medium for ex vivo use, expressed preferentially the activatory FcγRIIa (Supplementary Data, Fig. 1). HER2-Fc binding to DC surface was completely inhibited by the treatment with a blocking antibody towards this FcγR (Supplementary Data, Fig. 2). After binding at 4°C, HER2-Fc intracellular processing was characterized 2 and 12 h after uptake by fluorescence microscopy (Fig. 4). The HER2-Fc protein was visualized with FITC-conjugated F(ab)2 anti-human IgGγ Fc-specific Ab (green), while compartment markers were labeled in red.

Intracellular localization of HER2-Fc in iDCs after FcγR-mediated internalization. Immunofluorescence analysis of HER2-Fc intracellular localization in DCs after 2 or 12 h from the uptake. The HER2-Fc protein is visualized in green, while the HLAI compartment (identified by calreticulin and HLAI staining) or the endolysosomal HLAII compartment (LAMP1 staining) are immunolabeled in red. 3D reconstruction of a selection of three central sections crossing the nucleus out of the total number of the serial optical sections is shown in each image. A quantitative analysis of the percentage of colocalization was performed by serial optical sectioning and 3D reconstruction. Results are expressed as mean values ± SE (standard errors); the percentage of colocalization was calculated analyzing a minimum of 30 cells for each treatment randomly taken from three independent experiments. After 2 h of internalization, the HER2-Fc staining appears in dots clustered in the perinuclear area of the cells. HER2-positive spots colocalize with the HLAI compartment (57% colocalization with HLAI and 29% colocalization with calreticulin) and only partially with the HLAII compartment (17% colocalization with LAMP1). Arrows point to yellow double-positive spots. After 12 h of internalization, HER2-positive dots are more scattered throughout the entire cytoplasm and the colocalization with HLAI compartment appears only slightly increased. Bar, 10 μm
The HLAI molecule was employed to identify HLA class I compartment (ER+ and HLAI+ vesicles), while calreticulin was used to specifically define ER+ compartment. LAMP1 distribution marked endolysosomal/HLAII compartment.
Two hours after internalization, HER2-Fc was localized in discrete granules in the 85–90% of the analyzed cells and appeared mostly to be associated to HLAI compartment. In fact, absolute percentage values of colocalization indicated that the processed protein predominantly colocalized with HLAI in the perinuclear area (57%) (orange/yellow signal) (Fig. 4, first and second columns) and partially overlapped with calreticulin (29%). HER2-Fc staining was also associated, but to a less extent, to LAMP1+ granules (17%).
After 12 h, HER2-Fc cytoplasmic staining was more diffuse, maintaining the colocalization pattern with a slight increase in HLAI compartment (absolute values: HLAI 63%, calreticulin 20%, LAMP1 7%) (Fig. 4, third and fourth columns). These results indicated that HER2-Fc was processed in both HLAI and HLAII compartments and that already after 2 h it could be found predominantly associated to HLAI+ areas.
HER2-Fc-pulsed DCs induce CD8+ T cell-mediated IFN-γ secretion in breast cancer patients
We further studied HER2-Fc ability to stimulate HER2-specific CD8+ T cells in HER2+/HLAI-A2+ breast cancer patients. In vitro-enriched E75-specific CD8+ T cells, were plated with autologous DCs pulsed with HER2-Fc or Ctrl-Fc proteins (DCHER2-Fc or DCCtrl-Fc, respectively), and T cell-mediated response was measured by IFN-γ ELIspot assay (Fig. 5). Each of the CD8+ T cell cultures secreted IFN-γ in response to the DCHER2-Fc (black bars) in a significantly higher amount than DCCtrl-Fc (light gray bars) (p < 0.05). In three patients, stimulation of CD8+ T cells by E75-pulsed DCs (10 μg/mL; DCE75-dark gray histograms) resulted in an IFN-γ response lower than DCHER2-Fc and above DCCtrl-Fc.

CD8+ T cell-mediated IFN-γ response to HER2-Fc in HLAI-A2+/HER2+ breast cancer. ELIspot analysis of IFN-γ production by CD8+ T cells plated with antigen-pulsed autologous DCs (50:1). IFN-γ secretion was evaluated as number of spots and mean of triplicate plotted as histograms (black, HER2-Fc-pulsed DCs; light gray, Ctrl-Fc-pulsed DCs; dark gray, E75-pulsed DCs). Data were analyzed using the two-tailed Student’s t test (*p < 0.05; **p ≤ 0.005)
Discussion
DC-based immunotherapy is an attractive therapeutic approach to be combined with standard therapies for the induction of long lasting antitumor immunity, aiming at the control of cancer progression. The clinical effectiveness of such an approach is documented by an increasing number of clinical trials and has been confirmed by the recent FDA approval of the first DC-based autologous vaccine [18]. A critical key step in the DC vaccine design is the modality of antigen delivery to DCs in vivo as well ex vivo [6]. Several experimental evidences indicate that antigen DC targeting through FcγR is an optimal strategy for anticancer immunotherapy [3]. However, so far, such clinical approach is limited to idiotype-pulsed DC vaccines, employed with controversial results in the therapy of hematological malignancies [11]. In mouse models, vaccination with cDNA coding viral/tumor-associated antigen fused with human Fc has been shown to exert prophylactic and therapeutic efficacy, although with a broad heterogeneity in the vaccination schedules and in the choice of therapeutic endpoints [9, 19, 20].
Objective of this work was to generate a HER2-Fc chimera to load DCs in vivo and ex vivo as antitumor vaccine. We used the HER2 antigen because it is a clinically relevant target for both passive and active immunotherapies. The administration of humanized HER2-specific antibody Trastuzumab is nowadays a consolidation treatment for the management of breast cancer as well as other epithelial cancers [21, 22]. Moreover, HER2 peptide vaccinations have been shown to induce HER2-specific immune responses and to be associated with a better survival of the vaccinated patients [13, 23]. We selected the HER2(364–391) region, spanning the E75 HLA-A2-restricted epitope, modifying the anchor residues to increase binding affinity [24] and fusing this sequence to the Fc fragment of human IgG1. The immunogenicity of the HER2-Fc chimera was tested both as a cDNA vaccine in a mouse model and as a protein in an in vitro human setting utilizing DCs that represent the optimal target for this immunogen.
In mice, two HER2-Fc cDNA vaccinations were sufficient to activate an anti-HER2 specific immune response and to increase tumor latency with a significantly longer survival than the Ctrl-Fc cDNA. SPCs from HER2-Fc-vaccinated mice produced significant levels of IFN-γ in response to human HER2 epitopes, selected as strong binders to H2d MHC class I molecules. Almost no IFN-γ secretion was detected in Ctrl-Fc-vaccinated mice. These results indicated that HER2-Fc cDNA was correctly translated, the antigenic sequence processed and efficiently presented in vivo.
It is interesting to note that the vaccination with the modified HER2 sequences induced significative response against the epitopes contained in the vaccine as well as the corresponding native HER2 sequences [24]. A third cDNA vaccination did not significantly modify such a response (data not shown). Recent experimental evidences have highlighted the key role of antigen-specific immunosurveillance in limiting tumor outgrowth by cytolytic and cytostatic T cell-mediated mechanisms [25]. We assume that the delayed tumor growth and the longer survival observed in mice vaccinated with HER2-Fc cDNA could be due to the anti-HER2 T cell responses specifically elicited by the cDNA vaccine. Both HER2-Fc and Ctrl-Fc vaccination induced 50% tumor protection (at day 100 from challenge), while empty vector-vaccinated mice displayed 100% tumor growth, suggesting that the Fc sequence could exert an unspecific antitumor effects at early stage of tumor engraftment. It is well-known that innate immunity plays a fundamental role during cancer immunosurveillance at early stages, when innate immune mechanisms are involved in the recognition and killing of transformed cells thus preventing the development of malignancy [26]. The Fc portion of the chimera vaccine, could most probably interact with FcγR expressed in vivo by DCs and by other innate immune cells thus exerting an aspecific antitumor effects. In this context, the Fc domain in the cDNA vaccine could be regarded as an adjuvant component, contributing to the activation of the innate immune network. We are currently investigating this hypothesis in FcγR KO mice.
The immunogenicity of the HER2-Fc molecule was also evaluated as a protein in human experimental settings. The HER2-Fc chimera, secreted as a dimer, could homogenously target DCs grown in clinical grade conditions through the FcγRIIa, the activatory receptor preferentially expressed by DCs differentiated in serum-free media (Supplementary Data). The bound HER2-Fc protein was readily internalized and colocalized within 2 h in HLAII and, most predominantly, in HLAI and calreticulin compartments as shown by immunofluorescence studies, suggesting that the vast majority of the protein could be cross-processed.
Intracellular processing of soluble tumor antigens is often blocked in HLAII compartment, likely inducing an exclusive CD4+ T cell-mediated response [27, 28]. The delivery of soluble antigens through FcR could redirect antigen processing also into HLAI pathway. To our knowledge, this is the first demonstration that a monovalent chimeric ligand for FcγR is efficiently processed as polyvalent immunocomplexes are [3].
The ability of DCs pulsed with HER2-Fc to induce a specific anti-HER2 immune response was tested in autologous setting in vitro, using E75-specific CD8+ T cells from six HER2+/HLA-A2+ patients. For each patient, the enriched E75 CD8+ T cells secreted a significant higher amount of IFN-γ in response to HER2-Fc loaded DCs (p < 0.05) as compared to DCs pulsed with the Ctrl-Fc protein as evaluated in ELIspot assay. The consistent IFN-γ response to DCCtrl-Fc observed in some patients may be due to allotypic residues present in the Fc domain. When the HER2 epitope was targeted to DCs through FcγR, the amount of antigen required to induce a specific IFN-γ response was 80 fold less as compared to the E75 peptides (0.025 μmol HER2-protein vs 2 μmol E75 peptide). These results confirmed that HER2-Fc was efficiently processed and HER2 presentation occurred resulting in the activation of specific IFN-γ CD8+ T cell-mediated responses.
It is now clear that the standard treatments such as surgery, radio, and chemotherapy can have a strong interplay with the immune system [29, 30], and that immune activation contributes significantly to the efficacy of classical cancer treatments. Following this line of evidence, the use of distinct immunotherapeutic interventions to induce, reinforce, and maintain a long lasting immune response is strongly encouraged. Targeting DCs through FcγRs can foresee the combined use of the same antigen in two distinct immunogen formulations both as protein and as cDNA.
In conclusion, we have produced a recombinant HER2-Fc chimera for targeting DCs in vivo and in vitro. cDNA vaccination activated anti-HER2 epitope-specific CD8+ T cells and afforded a longer survival in mice immunized with HER2-Fc plasmid. In vitro, DCs pulsed with HER2-Fc protein induced a significant IFN-γ secretion by HER2-specific CD8+ T cells, as detected in breast cancer patients. This immunogen has several advantages as compared to protein/peptide-based formulation. The antigen domain can be engineered to contain multivalent sequences of the same or different antigens, providing a multi-epitope vaccination. This would overcome the requirement of knowledge of patients’ haplotype and supplying epitopes for a concomitant HLAI and HLAII presentation as long peptides do [31]. Engineering of the Fc domain could tune the FcγR binding affinity with huge impact on the enhancement of antigen cross-processing and on DCs activation [3, 32]. Moreover this antigenic formulation offers the option to be efficaciously employed as cDNA and as protein formulation in prime boost strategy in patient tailored, integrated therapies.
References
Palucka K, Banchereau J, Mellman I (2010) Designing vaccines based on biology of human dendritic cell subsets. Immunity 33:464–478
Pulendran B (2005) Variegation of the immune response with dendritic cells and pathogen recognition receptors. J Immunol 174:2457–2465
Tacken PJ, de Vries IJ, Torensma R, Figdor CG (2007) Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol 7:790–802
Nimmerjahn F, Ravetch JV (2008) Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8:34–47
Schuurhuis DH, Ioan-Facsinay A, Nagelkerken B, van Schip JJ, Sedlik C, Melief CJ, Verbeek JS, Ossendorp F (2002) Antigen-antibody immune complexes empower dendritic cells to efficiently prime specific CD8+ CTL responses in vivo. J Immunol 168:2240–2246
Gilboa E (2007) DC-based cancer vaccines. J Clin Invest 117:1195–1203
Schuurhuis DH, van Montfoort N, Ioan Facsinay A, Jiawan R, Camps M, Nouta J, Melief CJ, Verbeek JS, Ossendorp F (2006) Immune complex loaded dendritic cells are superior to soluble immune complexes as antitumor vaccine. J Immunol 176:4573–4580
Kim PS, Armstrong TD, Song H, Wolpoe ME, Weiss V, Manning EA, Huang LQ, Murata S, Sgouros G, Emens LA (2008) Antibody association with HER-2/neu-targeted vaccine enhances CD8 T cell responses in mice through Fc-mediated activation of DCs. J Clin Invest 118:1700–1711
You Z, Huang X, Hester J, Toh HC, Chen SY (2001) Targeting dendritic cells to enhance DNA vaccine potency. Cancer Res 61:3704–3711
Lowe DB, Shearer MH, Jumper CA, Bright RK, Kennedy RC (2007) Fc gamma receptors play a dominant role in protective tumor immunity against a virus-encoded tumor-specific antigen in a murine model of experimental pulmonary metastases. J Virol 81:1313–1318
Hollander N (2009) Current vaccination strategies for the treatment of B-cell lymphoma and multiple myeloma. Crit Rev Immunol 29:399–418
Ladjemi MZ, Jacot W, Chardès T, Pèlegrin A, Navarro-Teulon I (2010) Anti-HER2 vaccines: new prospects for breast cancer therapy. Cancer Immunol Immunother 59:1295–1312
Peoples GE, Gurney JM, Hueman MT, Woll MM, Ryan GB, Storrer CE, Fisher C, Shriver CD, Ioannides CG, Ponniah S (2005) Clinical trial results of a HER2/neu (E75) vaccine to prevent recurrence in high-risk breast cancer patients. J Clin Oncol 23:7536–7545
Quaglino E, Mastini C, Amici A, Marchini C, Iezzi M, Lanzardo S, De Giovanni C, Montani M, Lollini PL, Masucci G et al (2010) A better immune reaction to Erbb-2 tumors is elicited in mice by DNA vaccines encoding rat/human chimeric proteins. Cancer Res 70(7):2604–12
Mastini C, Becker PD, Iezzi M, Curcio C, Musiani P, Forni G, Cavallo F, Guzman CA (2008) Intramammary application of non-methylated-CpG oligodeoxynucleotides (CpG) inhibits both local and systemic mammary carcinogenesis in female BALB/c Her-2/neu transgenic mice. Curr Cancer Drug Targets 8:230–42
Napoletano C, Pinto D, Bellati F, Taurino F, Rahimi H, Tomao F, Panici PB, Rughetti A, Frati L, Nuti M (2007) A comparative analysis of serum and serum-free media for generation of clinical grade DCs. J Immunother 30:567–576
Rammensee HG, Friede T, Stevanoviíc S (1995) MHC ligands and peptide motifs: first listing. Immunogenetics 41:178–228
Hovden AO, Appel S (2010) The first dendritic cell-based therapeutic cancer vaccine is approved by the FDA. Scand J Immunol 72:554
Sun W, Qian H, Zhang X, Zhou C, Liang X, Wang D, Fu M, Ma W, Zhang S, Lin C (2006) Induction of protective and therapeutic antitumour immunity using a novel tumour-associated antigen-specific DNA vaccine. Immunol Cell Biol 84:440–447
Wang M, Xie Z, Shi M, Lu H, Yu M, Hu M, Lu F, Ma Y, Shen B, Guo N (2008) A new strategy to induce effective antitumour response in vitro and in vivo. Scand J Immunol 68:287–296
Mariani G, Fasolo A, De Benedictis E, Gianni L (2009) Trastuzumab as adjuvant systemic therapy for HER2-positive breast cancer. Nat Clin Pract Oncol 6:93–104
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, ToGa Trial Investigators et al (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376:687–697
Mittendorf EA, Holmes JP, Ponniah S, Peoples GE (2008) The E75 HER2/neu peptide vaccine. Cancer Immunol Immunother 57:1511–1521
Vertuani S, Sette A, Sidney J, Southwood S, Fikes J, Keogh E, Lindencrona JA, Ishioka G, Levitskaya J, Kiessling R (2004) Improved immunogenicity of an immunodominant epitope of the HER-2/neu protooncogene by alterations of MHC contact residues. J Immunol 172:3501–3508
Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y (2010) Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 120:2030–2039
Vesely MD, Kershaw MH, Schreiber RD, Smyth M (2011) Natural innate and adaptive immunity to cancer. Ann Rev Immunol 29:235–71
Hiltbold EM, Vlad AM, Ciborowski P, Watkins SC, Finn OJ (2000) The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J Immunol 165:3730–3741
Napoletano C, Rughetti A, Agervig Tarp MP, Coleman J, Bennett EP, Picco G, Sale P, Denda-Nagai K, Irimura T, Mandel U et al (2007) Tumor-associated Tn-MUC1 glycoform is internalized through the macrophage galactose-type C-type lectin and delivered to the HLA class I and II compartments in dendritic cells. Cancer Res 67:8358–8367
Ma Y, Kepp O, Ghiringhelli F, Apetoh L, Aymeric L, Locher C, Tesniere A, Martins I, Ly A, Haynes NM et al (2010) Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol 22:113–124
Napoletano C, Bellati F, Landi R, Pauselli S, Marchetti C, Visconti V, Sale P, Liberati M, Rughetti A, Frati L et al (2010) Ovarian cancer cytoreduction induces changes in T cell population subsets reducing immunosuppression. J Cell Mol Med 14:2748–2759
Melief CJ, van der Burg SH (2008) Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 8:351–360
Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR (2008) Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 7:2517–2527
Acknowledgment
We are most grateful to Dr. D. Snary (CRT-UK, London, UK), Dr. J.M. Burchell (King’s College of London, UK), Dr. Wei (Karmanos Cancer Center, Detroit) for kindly providing the pγUC plasmid, the CHO-K1 cells, and D2F2/E2 cells, respectively. This work has been founded by M.I.U.R. (A.R., M.N., F.B., M.R.T.); AIRC (M.N., M.R.T.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
I.G. Zizzari and F. Veglia equally contributed to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1183 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Zizzari, I.G., Veglia, F., Taurino, F. et al. HER2-based recombinant immunogen to target DCs through FcγRs for cancer immunotherapy. J Mol Med 89, 1231–1240 (2011). https://doi.org/10.1007/s00109-011-0794-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0794-7