
Abstract
Real-time PCR (qPCR) is the principal technique for the quantification of pathogen biomass in host tissue, yet no generic methods exist for the determination of the limit of quantification (LOQ) and the limit of detection (LOD) in qPCR. We suggest using the Youden index in the context of the receiver operating characteristic (ROC) curve analysis for this purpose. The LOQ was defined as the amount of target DNA that maximizes the sum of sensitivity and specificity. The LOD was defined as the lowest amount of target DNA that was amplified with a false-negative rate below a given threshold. We applied this concept to qPCR assays for Fusarium verticillioides and Fusarium proliferatum DNA in maize kernels. Spiked matrix and field samples characterized by melting curve analysis of PCR products were used as the source of true positives and true negatives. On the basis of the analysis of sensitivity and specificity of the assays, we estimated the LOQ values as 0.11 pg of DNA for spiked matrix and 0.62 pg of DNA for field samples for F. verticillioides. The LOQ values for F. proliferatum were 0.03 pg for spiked matrix and 0.24 pg for field samples. The mean LOQ values correspond to approximately eight genomes for F. verticillioides and three genomes for F. proliferatum. We demonstrated that the ROC analysis concept, developed for qualitative diagnostics, can be used for the determination of performance parameters of quantitative PCR.
Similar content being viewed by others

Introduction
Real-time PCR (qPCR) is the standard analytical method for quantifying pathogen biomass in the tissue of host organisms. Standard performance parameters of an analytical method are the limit of detection (LOD) and the limit of quantification (LOQ). The LOD is defined as the lowest amount of the analyte detectable in a single reaction. The LOQ is the lowest amount of analyte that can be quantified. The methods commonly used in chemical analysis for determining LOD and LOQ values [1–3] are unsuitable for qPCR.
We suggest that the LOD and LOQ can be determined by use of receiver operating characteristic (ROC) curve analysis, which is a method used to evaluate the sensitivity and specificity of diagnostic tests. ROC is based on a comparison of the outcome of a series of assays (“positive” and “negative”) with the “true” status of the samples. The “true” status is either evaluated with a well-established test, which is called the “gold standard,” or known a priori because the samples were prepared by spiking a negative matrix with the target analyte. The central concept in ROC curve analysis is the cutoff point. The cutoff point is a threshold value of the analytical signals below which samples are regarded as negative and above which samples are regarded as positive. The ROC curve is a plot of the sensitivity (genuinely positive samples that are detected as positive, “true positives”) against 1 - specificity (negative samples that are detected as positive, “false positives”) for different cutoff points [4]. In qPCR, the cutoff point is the threshold cycle above which a sample is considered to be negative. If a cycle number is chosen as a cutoff point, the fraction of positive samples that reached the threshold of fluorescence intensity before this cycle is the “true-positive fraction.” The fraction of negative samples that reached the threshold of fluorescence intensity before this cycle is the “false-positive fraction.” If a higher cycle number is chosen as the cutoff point, more samples are likely to be rated as positive, increasing the sensitivity. At the same time, the false-positive rate is likely to grow and the specificity is likely to decrease. An optimal cutoff point corresponds to the desired trade-off between true-positive and false-negative rates. To balance the demands for sensitivity and specificity of a diagnostic assay, i.e., to determine the optimal cutoff point, the Youden index is often used [23].
Using artificially prepared, spiked samples for estimating an optimal cutoff value guarantees that the assignment of samples to true positives and true negatives is correct. The drawback is that the properties of a matrix spiked with target DNA may differ from the properties of samples obtained from the field. The optimal cutoff point determined with the help of spiked samples may therefore differ from the optimal cutoff point for field samples. In the current research, we investigated this dilemma by assigning field samples to true positive and true negative by melting curve analysis. We then compared cutoff values derived for field samples with those obtained for a spiked matrix. As a model system, we used the fungal plant pathogens Fusarium verticillioides and Fusarium proliferatum in maize kernels.
Fusarium species are among the most important pathogens of maize worldwide. Infection with Fusarium spp. reduces grain yield and quality [5], and infected grain, when used for the production of food and feedstuff, is often contaminated with mycotoxins that endanger the health of consumers and livestock [6]. Illness of farm animals and less frequently of humans caused by Fusarium mycotoxins has regularly been reported [7–9].
Fusarium species cause two types of ear rot in maize: red ear rot (Gibberella ear rot) caused by Fusarium spp. belonging to the Discolor section, and pink ear rot (Fusarium ear rot or ear mold) caused by species of the Liseola section. Fusarium species isolated from cobs exhibiting pink ear rot symptoms are usually Fusarium verticillioides, F. proliferatum, and F. subglutinans [5]. Apart from being found in maize [10] and asparagus [11], F. proliferatum has been found in wheat [12], sorghum [13], and rice [14], but only infection of the first two crops is considered economically relevant. F. verticillioides and F. proliferatum are producers of fumonisin mycotoxins. Fumonisin B1 (FB1) and fumonisin B2 (FB2) are the most abundant fumonisins in maize, and levels of FB1 are generally higher than those of FB2 [15]. FB1 causes leukoencephalomalacia in horses and pulmonary edemas in swine [16], and it is very likely that FB2 and fumonisin B3 have the same effects. Although toxicologically relevant amounts of fumonisins in maize are occasionally found in food products in countries with highly developed agriculture, serious health impacts of fumonisin contamination are thought to occur in areas with suboptimal growing and storage conditions and a high maize consumption [17]. Indeed, levels of FB1 and FB2 in maize used as staple food in South Africa correlated with the incidence of esophageal cancer [18]. Beside fumonisins, F. verticillioides produces the mycotoxins fusaric acid and fusarins, whereas F. proliferatum was reported to produce the mycotoxins beauvericin, enniatins, fusaproliferin, and moniliformin [19].
The relationship between the development of symptoms, the amount of fungal biomass in the plant tissue, and the production of mycotoxins is incompletely understood. Ramirez et al. [20] found that fumonisin contamination and the level of infection for Fusarium species of the Liseola section did not correlate. In contrast, Pascale et al. [21] found that fumonisin contamination was highly correlated with ear rot symptoms after inoculation of maize with F. verticillioides or F. proliferatum. Clarifying the relationship between the accumulation of fungal biomass in the plant, development of symptoms, and mycotoxin production requires a species-specific method to reliably quantify F. verticillioides and F. proliferatum biomass in plant tissue.
qPCR is useful for quantifying fungal colonization of crops while distinguishing among species. Species-specific PCR primers have been developed for most Fusarium species that cause ear rot [22–26].
In this work, we evaluated qPCR assays for quantification of F. verticillioides and F. proliferatum in maize kernels. Furthermore, we examined the use of the Youden index in the framework of ROC curve analysis for estimating the LOD and LOQ of qPCR assays.
Materials and methods
Fungal cultures
The fungal strains used in this study are listed in Table 1. Cultures for DNA extraction were grown in 100 ml of potato dextrose broth (24 g l-1; Scharlau, Barcelona, Spain) at room temperature and without shaking. The mycelium was harvested after 14 days by filtration and then freeze-dried.
DNA isolation from pure fungal cultures grown in liquid media
A variant of the cetyltrimethylammonium bromide method as described by Brandfass and Karlovsky [27] was used, and the quality and quantity of DNA were estimated by electrophoresis in 0.8% (w/v) agarose gels (Cambrex, Rockland, ME, USA) prepared in 40 mM tris(hydroxymethyl)aminomethane (Tris), 1 mM EDTA, pH adjusted to 8.5 with acetic acid. The electrophoresis was carried out at 4 V cm−1 for 90 min. The gel was stained with ethidium bromide (2 mg l−1) and documented with a digital imaging system (Vilber Lourmat, Marne la Vallee, France). The densitometry was performed using Multi-Analyst (Bio-Rad, Hercules, CA, USA). The concentration of fungal DNA was calculated by comparing a dilution series with defined amounts of DNA of lambda phage (methylated, from Escherichia coli host strain W3110).
DNA extraction from maize field samples
Maize kernels were dried at 60 °C for 24 h and ground in a cross hammer mill (SK 1 cross beater mill; bottom sieve 1 mm; Retsch, Haan, Germany). The DNA extraction from 1 g of maize meal was carried out following an upscaled protocol for DNA extraction from plant material as described by Brandfass and Karlovsky [28]. The quality and concentration of DNA were determined by agarose gel electrophoresis as described above. Total DNA from 1 g of starting material was dissolved in 200 μl of 10 mM Tris, 1 mM EDTA, pH adjusted to 8.0. The DNA solution was diluted tenfold, and 1 μl was used as the template for each reaction.
Primers
The primers used for F. verticillioides were VER1 (CTTCCTGCGATGTTTCTCC) and VER2 (AATTGGCCATTGGTATTATATATCTA), which were designed by Mule et al. [25] on the basis of the coding sequence of the calmodulin gene; these primers amplify a DNA fragment of 587 bp. The primers used for F. proliferatum were Fp3-F (CGGCCACCAGAGGATGTG) and Fp4-R (CAACACGAATCGCT TCCTGAC), which were designed by Jurado et al. [26] on the basis of the intergenic sequence of the ribosomal RNA gene cluster; these primers amplify a DNA fragment of 230 bp.
qPCR assays
The optimized conditions for qPCR assays were as follows. The reaction mixture for F. verticillioides (25 μl) contained reaction buffer amended with NH4 [67 mM Tris–HCl, 16 mM (NH4)2SO4, 0.01% (v/v) Tween 20, pH 8.8 at 25 °C; Bioline, Luckenwalde, Germany], 2.5 mM MgCl2, 0.1 mM concentration of each of the four deoxynucleoside triphosphates (dNTPs; Bioline, Luckenwalde, Germany), 0.3 μM concentration of each primer, 0.75 U of Taq DNA polymerase (BIOTaq, Bioline, Luckenwalde, Germany), 10 nM fluorescein (used for the calculation of well factors, see below), 0.1× SYBR Green I (Invitrogen, Karlsruhe, Germany), and 1 μl of template DNA.
The reaction mixture for F. proliferatum-specific PCR was identical except for the following components: 2 mM MgCl2, 0.6 μM concentration of each primer, and 0.4 U of Taq DNA polymerase.
qPCR was performed in an iCycler thermocycler (Bio-Rad, Hercules, CA, USA). The amplification for F. verticillioides consisted of an initial denaturation at 95 °C for 1.5 min, during which the well factors were collected (compensation for differences among optical properties of individual wells), followed by 40 cycles of 50 s denaturation at 94 °C, 50 s annealing at 62 °C, and 1 min elongation at 72 °C. The final elongation step was performed for 7 min at 72 °C. Fluorescence was measured in each cycle during the annealing phase. Melting curve analysis was performed after each PCR: samples were heated to 95 °C for 1 min, cooled to 55 °C for 1 min, and heated to 65 °C, and subsequently the temperature was ramped from 65 °C to 95 °C in steps of 0.5 °C every 10 s. Fluorescence was measured at each step.
The PCR for the quantification of F. proliferatum DNA was performed according to the following protocol: initial denaturation for 1.5 min at 95 °C, followed by 35 cycles with 35 s at 95 °C, 30 s at 64 °C, and 30 s at 72 °C, with fluorescence measurement during the annealing step of each cycle, and a final elongation of 5 min at 72 °C. The melting curve analysis was performed as described above.
Calibration curves and PCR efficiency
Dilution series were prepared containing purified fungal DNA in amounts of 0.05, 0.15, 0.5, 1, 5, 10, and 50 pg mixed with maize DNA. For F. proliferatum, two additional standards (1.5 and 15 pg of fungal DNA) were used. Every set of standards was analyzed ten times. Standard curves were generated by plotting threshold cycle values (Ct values) against the logarithm of starting DNA quantities. The slopes of the standard curves were used to calculate the reaction efficiency E of PCR assays, using the following equation:
These samples were also used as spiked positive samples for ROC curve analysis (see later).
Specificity of PCR primers
The specificity of both PCR assays was determined with DNA extracted from pure cultures of 81 fungal isolates (14 Fusarium species and 20 isolates of 12 other fungal species, Table 1). Samples were classified as positive when the melting point was identical with the melting point of the standard with a tolerance of 0.5 °C.
Sensitivity, specificity, ROC curves, and optimal cutoff points
ROC curve analysis was used to estimate the performance of qPCR assays [29]. ROC curves were constructed as plots of sensitivity versus 1 - specificity for a set of positive and negative samples. Sensitivity is the fraction of true-positive samples that score positive. Sensitivity was calculated for each PCR cycle by dividing the number of true-positive samples with equal or lower Ct value by the total number of true-positive samples. Specificity is the fraction of true-negative samples that score negative. Specificity was calculated for each PCR cycle by dividing the number of true-negative samples with higher or equal Ct value by the total number of true-negative samples. ROC curves show the relationship between sensitivity and specificity. They facilitate visual evaluation of the performance of an assay. The area under a ROC curve can be regarded as an aggregate quality indicator for a diagnostic assay.
The Youden index J is defined as [29]
where Se is sensitivity and Sp is specificity.
The optimal cutoff point is the PCR cycle with the highest value of the Youden index:
Samples with a threshold cycle higher than the chosen cutoff point are classified as negative, whereas samples with threshold cycle lower than the cutoff point are classified as positive [30]. ROCs, areas under ROC curves, and Youden indices were calculated with the ROC module of the package Sigma Plot 11.0 (Systat Software, San Jose, CA, USA). The same software package was used to generate graphics.
Determination of LOQ and LOD
The LOQ was determined as the amount of DNA corresponding to the threshold cycle at which the sum of specificity and sensitivity of the assay was maximized. For this purpose, the Youden index J was calculated for each PCR cycle. The cycle for which J reached the maximum was selected as the optimal cutoff point. The LOQ was then determined as the amount of DNA corresponding to the optimal cutoff point in the calibration curve.
The LOD was determined as the amount of DNA corresponding to the threshold cycle at which at most 5% of true-positive samples scored negative (selectivity of 0.95).
Determination of mycotoxin production
Polished rice (25 g) and 35 ml of tap water were autoclaved in 100-ml Erlenmeyer flasks and inoculated with a 100-μl spore suspension of the fungal strains. The cultures were incubated at 25 °C for 2 weeks. A 4-g portion of the colonized substrate (water content 15–20%) was extracted with 40 ml of acetonitrile. A 1-ml volume of the extract was dried in a vacuum, and the residue was dissolved in 1 ml of methanol/water (1:1), defatted with 1 ml of cyclohexane, and diluted 20 times with methanol/water (1:1). High-performance liquid chromatography was performed on a reverse-phase C18 column (Kinetex, 50.0 mm × 2.1 mm, particle size 2.6 μm; Phenomenex) with a gradient of methanol in water with 7 mM acetic acid at flow rate of 0.2 ml min−1. The analytes were ionized by electrospray and detected by tandem mass spectrometry with an ion trap detector (500 MS, Varian, Darmstadt, Germany).
Results
The first amplifications were performed under conditions for end-point PCR as described by Mule et al. [25] and Jurado et al. [26]. To improve the sensitivity, we reduced the reaction volume to 25 μl and optimized the following: the concentrations of dNTPs, MgCl2, and primers; the activity of Taq DNA polymerase; and the cycling parameters for qPCR conditions. For F. verticillioides, the most important changes in the conditions for PCR concerned the concentrations of dNTPs and MgCl2, which were increased from 50 to 100 μM and from 1.5 to 2.5 mM, respectively, as compared with the original publication. In contrast, the amount of Taq DNA polymerase could be reduced from 1.25 to 0.75 U. An annealing temperature of 62 °C yielded specific products, in contrast to the annealing temperature of 56 °C, which was suggested by the designers of the primers [25]. In the F. proliferatum assay, the amount of each primer could be reduced from 0.8 to 0.6 mM, the amount of dNTPs could be reduced from 1 mM to 100 μM, and the amount of Taq DNA polymerase could be reduced from 1.0 to 0.4 U per reaction. The annealing temperature was lowered from the recommended temperature of 69 °C [26] to 64 °C.
The optimized conditions were used for the ROC curve analysis with artificially prepared samples and field samples. Artificial negative samples consisted of nontarget DNA and blank plant matrix and artificial positive samples consisted of plant matrix spiked with known quantities of target DNA (0.05–50 pg). A total of 226 artificial samples for F. verticillioides assay and 224 samples for F. proliferatum assay were used. Field samples originated from monitoring and field trials carried out from 2005 to 2008 in Germany and Italy; 994 field samples for F. verticillioides assay and 436 field samples for F. proliferatum assay were used (Table 2). Melting curve analysis was used as the “gold standard” for classification of field samples as positive or negative. Unknown samples generating products with melting temperatures ±0.25 °C above/below the mean melting temperature of the standards and positive controls for a given PCR run were ranked as positive. Over a period of 3 years, the melting temperature among PCR runs fluctuated between 90.0 and 91.5 °C for F. verticillioides and between 91.5 and 92.5 °C for F. proliferatum. Within a single PCR run, melting temperatures for standards and positive controls were constant within a range of 0.5 °C.
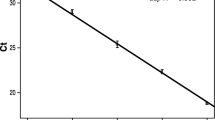
Calibration curves generated with spiked matrix revealed a linear relationship between Ct values and the logarithm of DNA amount down to at least 0.05 pg for F. proliferatum and 0.15 pg for F. verticillioides (Fig. 1). The average PCR efficiency of the assays was 0.92 for F. verticillioides and 0.98 for F. proliferatum. The Ct values for F. proliferatum DNA were consistently about four cycles lower than the values for the same amount of F. verticillioides DNA.

Linear standard curves obtained from dilution series of Fusarium verticillioides DNA (filled symbols) and F. proliferatum DNA (open symbols) in a range from 0.05 to 50 pg, with five to ten replications per quantity. The quantity of 0.05 pg of F. verticillioides DNA was excluded from the standard curve because of low reproducibility. The threshold cycle (Ct) is plotted against the decadic logarithm of starting DNA quantity in grams. Error bars represent standard deviation
With all 13 F. verticillioides isolates (formerly F. moniliforme) and 15 F. proliferatum isolates (Table 1), we obtained PCR products with the expected melting temperatures. As a confirmation of the taxonomic affiliation of these strains, we determined which mycotoxins they produced. Ten strains labeled as F. verticillioides and 12 strains labeled as F. proliferatum were grown in rice for 2 weeks. With one exception, only F. proliferatum strains produced F. proliferatum-specific depsipeptide beauvericin (Table 3). Furthermore, neither species produced enniatins, and all strains except one produced fumonisins.
Pure maize DNA and all isolates of 18 nontarget fungal species tested negatively (87 isolates for the F. proliferatum assay and 89 isolates for the F. verticillioides assay). Samples of nontarget fungal DNA generated no amplification products or unspecific products with melting temperatures lower than those of the target products by at least 4 °C (Fig. 2).

Melting curve analysis of PCR products obtained with primers specific for F. verticillioides (a) and F. proliferatum (b). Filled symbols indicate the negative first derivation of SYBR Green fluorescence for PCR products heated from 65 to 94 °C. Open symbols indicate melting curves of PCR products of negative controls (water and nontarget DNA). RFU relative fluorescence units
ROC curve analysis was performed for spiked maize matrix and field samples (Fig. 3). For both fungi, the areas under ROC curves were slightly higher for spiked matrix than for field samples. The ROC curves were used to determine the LOQ values of the assays. We defined the LOQ of the PCR assays as DNA amounts that maximized the sum of sensitivity and specificity. The corresponding Ct values (optimal cutoff points) were determined by maximizing the Youden index. Calibration curves (Fig. 1) were used to determine LOQs for both assays using these Ct values.

Receiver operating characteristic (ROC) curves for real-time PCR for F. proliferatum and F. verticillioides . The upper panels show the ROC curves resulting from maize flour spiked with F. verticillioides DNA (n = 226) and F. proliferatum DNA (n = 224). The lower panels show the ROC curves for field samples for F. verticillioides DNA (n = 994) and F. proliferatum DNA (n = 436)
For a given threshold of the false-positive rate, the LOD was defined as the lowest amount of target DNA that was amplified with a false-negative rate below or equal to this threshold. We selected a maximal acceptable false-negative rate of 5% and then used this threshold to determine the LOD values (Table 2).
Discussion
Using published PCR primers for F. verticillioides [25] and F. proliferatum [26], we developed qPCR assays for the quantification of the DNA of these species in maize kernels. Mule et al. [25] evaluated the specificity of their primers for F. verticillioides by testing 21 strains of F. verticillioides, 12 strains of F. proliferatum, and six strains of F. subglutinans, in addition to single isolates of F. graminearum, F. poae, Aspergillus flavus, and Acremonium strictum. Jurado et al. [26] tested the specificity of primers for F. proliferatum against 12 strains of F. graminearum, seven strains of F. culmorum, five strains of F. poae, six strains of F. sporotrichioides, and one or two strains of eight other Fusarium species and five other fungal species. The use of only one strain of F. verticillioides and F. subglutinans in the test of primers for F. proliferatum [26] appeared insufficient. We therefore extended the specificity tests for both primer pairs with additional 12 isolates of F. verticillioides, 15 isolates of F. proliferatum, 12 isolates of F. subglutinans, 42 isolates of nontarget Fusarium species, and 20 isolates of other fungal species. These tests, performed under qPCR conditions, generated positive signals only for the target species. Primer pairs Fp3-F/Fp4-R [26] and VER1/VER2 [25] can therefore be regarded as species-specific in real-time mode for F. proliferatum and F. verticillioides, respectively.
The qPCR assays described here are suitable for the estimation of F. verticillioides and F. proliferatum DNA in maize flour with LOQ values of 0.11 pg and 0.032 pg, respectively, which correspond to 3.8 and 1.05 μg of DNA per kilogram of flour, respectively. The mean LOQ values for field and spiked samples correspond to 8.5 genomes for F. verticillioides and 3.2 genomes for F. proliferatum, assuming that the genome size of both species is approximately 40 Mbp. The amount of genomic DNA determined by qPCR can be used as a measure of fungal content in studies of the relationships between Fusarium infection, mycotoxin production, and disease symptoms. Relative to classic end-point PCR, the sensitivity of the detection was increased significantly for both F. verticillioides and F. proliferatum. Furthermore, the costs of the modified assays were reduced because optimized PCR uses less Taq DNA polymerase and a lower concentration of dNTPs than classic end-point PCR.
The Ct values for F. proliferatum DNA were consistently lower than those for the same amount of F. verticillioides DNA. This observation is reasonable because the primers for F. proliferatum were derived from a multicopy sequence [26], whereas the primers for F. verticillioides were based on a single-copy calmodulin gene [25]. The difference in the copy number of targets also explains why the F. proliferatum assay was more sensitive than the F. verticillioides assay.
ROC curve analysis of a dilution series of target DNA and nontarget DNA generated areas under ROC curves of 0.98 for the F. verticillioides assay and 0.99 for the F. proliferatum assay, which are close to the optimal value of 1. Occasionally, nontarget DNA caused unspecific amplification. On the basis of cutoff points calculated according to the Youden index (Table 2), the sensitivity was 97% for the F. proliferatum assay and 94% for the F. verticillioides assay, whereas the specificity was 97% in the F. verticillioides assay and 96% in the F. proliferatum assay. Therefore, automatic processing of the results based merely on Ct values (without melting curve analysis) is possible. Melting curve analysis is recommended when the content of target DNA approaches LOQ values.
Adejumo et al. [31] compared PCR analysis with an agar plating method for detection of F. verticillioides in maize samples from a Nigerian market. They found that only 71% of the maize samples that were positive for F. verticillioides by agar plating were confirmed positive by species-specific PCR. Part of this contradiction can probably be explained by the morphological similarity between F. verticillioides and F. proliferatum, highlighting the difficulty in distinguishing between these species on the basis of morphology. Other work by these authors [32] demonstrated an even greater difficulty in differentiating between F. verticillioides and F. proliferatum on the basis of morphology: F. verticillioides was found to be the dominant species in Nigerian maize, followed by eight other Fusarium species, but F. proliferatum was not found. It is likely that F. proliferatum isolates were confused with F. verticillioides in this work and that 29% of isolates morphologically identified as F. verticillioides but not confirmed by PCR were F. proliferatum. The use of PCR for differentiating F. proliferatum from F. verticillioides is therefore highly recommended [33].
To confirm the taxonomical affiliation of strains used in this work, we determined the production of beauvericin, enniatins, fumonisins, and moniliformin by 12 isolates each of F. verticillioides and F. proliferatum. Whereas fumonisins are produced by both F. verticillioides and F. proliferatum, moniliformin is produced only by F. proliferatum [19] and beauvericin is produced by F. proliferatum but is not produced or is produced in only low amounts by F. verticillioides [34–36]. That F. proliferatum produces enniatins was affirmed in an authoritative review [19] but this was rejected in other publications [37, 38]. We did not find enniatins in any of the F. verticillioides or F. proliferatum cultures in the current study.
Our laboratory has extensively used the qPCR assays described here for quantifying F. verticillioides and F. proliferatum. We have used qPCR to analyze maize kernels artificially infected with F. verticillioides or F. proliferatum, naturally infected samples from the field, and maize cobs inoculated with mixtures of F. verticillioides, F. proliferatum, and other fungal species in the greenhouse.
ROC curve analysis was developed for the assessment of qualitative diagnostic assays. Turechek et al. [44] used ROC curve analysis to compare the performance of PCR primers [45]. Inspired by their work, we used the ROC concept to establish performance parameters for quantitative PCR assays. LOQ and LOD, which are fundamental parameters in analytical chemistry, thus became available for quantitative PCR.
References
Haeckel R, Hänecke P, Hänecke M, Haeckel H (1998) J Lab Med 22:273–280
Mikkelsen SR, Cortón E (2004) Bioanalytical chemistry. Wiley, Hoboken
Kaus R (1998) Accred Qual Assur 3:150–154
Greiner M, Pfeiffer D, Smith R (2000) Prev Vet Med 45:23–41
Logrieco A, Mule G, Moretti A, Bottalico A (2002) Eur J Plant Pathol 108:597–609
Yazar S, Omurtag G (2008) Int J Mol Sci 9:2062–2090
D’Mello J, Placinta C, Macdonald A (1999) Anim Feed Sci Technol 80:183–205
Yiannikouris A, Jouany J (2002) Anim Res 51:81–99
Zimmer I, Dietrich R, Märtlbauer E, Usleber E, Klaffke H, Tiebach R, Weber R, Majerus P, Otteneder H (2004) Mycotox Res 24:40–52
Chulze S, Ramirez M, Farnochi M, Pascale M, Visconti A, March G (1996) J Agric Food Chem 44:2797–2801
Logrieco A, Doko B, Moretti A, Frisullo S, Visconti A (1998) J Agric Food Chem 46:5201–5204
Leslie J, Plattner R, Desjardins A, Klittich C (1992) Phytopathology 82:341–345
Gonzalez HHL, Martinez E, Resnik S (1997) Mycopathologia 139:35–41
Abbas H, Cartwright R, Shier W, Abouzied M, Bird C, Rice L, Ross P, Sciumbato G, Meredith F (1998) Plant Dis 82:22–25
Chulze SN, Etcheverry MG, Lecumberry SE, Magnoli CE, Dalcero AM, Ramirez ML, Pascale M, Rodriguez MI (1999) J Food Prot 62:814–817
Thiel P, Marasas W, Sydenham E, Shephard G, Gelderblom W (1992) Mycopathologia 117:3–9
Dutton MF (2009) Mycotox Res 25:29–39
Rheeder J, Marasas W, Thiel P, Sydenham E, Shephard G, Van Schalkwyk DJ (1992) Phytopathology 82:353–357
Desjardins AE (2006) In: Desjardins AE (ed) Fusarium mycotoxins: chemistry, genetics and biology. APS Press, St Paul
Ramirez M, Pascale M, Chulze S, Reynoso M, March G, Visconti A (1996) Mycopathologia 135:29–34
Pascale M, Visconti A, Chelkowski J (2002) Eur J Plant Pathol 108:645–651
Murillo I, Cavallarin L, Segundo B (1998) Eur J Plant Pathol 104:301–311
Moeller E, Chelkowski J, Geiger H (1999) J Phytopathol 147:497–508
Patino B, Mirete S, Gonzalez-Jaen M, Mule G, Rodriguez M, Vazquez C (2004) J Food Prot 67:1278–1283
Mule G, Susca A, Stea G, Moretti A (2004) Eur J Plant Pathol 110:495–502
Jurado M, Vazquez C, Marin S, Sanchis V, Gonzalez-Jaen M (2006) Syst Appl Microbiol 29:681–689
Brandfass C, Karlovsky P (2006) BMC Microbiol. doi:10.1186/1471-2180-6-4
Brandfass C, Karlovsky P (2008) Int J Mol Sci 9:2306–2321
Fluss R, Faraggi D, Reiser B (2005) Biom J 47:458–472
Schaefer H (1989) Stat Med 8:1381–1391
Adejumo T, Hettwer U, Nutz S, Karlovsky P (2009) J Plant Prot Res 49:399–404
Adejumo T, Hettwer U, Karlovsky P (2007) Int J Food Microbiol 116:350–357
Visentin I, Tamietti G, Valentino D, Portis E, Karlovsky P, Moretti A, Cardinale F (2009) Mycol Res 113:1137–1145
Shephard GS, Sewram V, Nieuwoudt TW, Marasas WF, Ritieni A (1999) J Agric Food Chem 47:5111–5115
Srobarova A, Moretti A, Ferracane R, Ritieni A, Logrieco A (2002) Eur J Plant Pathol 108:299–306
Bottalico A, Logrieco A, Ritieni A, Moretti A, Randazzo G, Corda P (1995) Food Addit Contam 12:599–607
Nicholson P, Simpson DR, Wilson AH, Chandler E, Thomsett M (2004) Eur J Plant Pathol 110:503–514
Jestoi M (2008) Crit Rev Food Sci Nutr 48:21–49
Miedaner T, Bolduan C, Melchinger AE (2010) Eur J Phytopathol 127:113–123
Bacon CW, Porter JK, Norred WP, Leslie JF (1996) Appl Environ Microbiol 62:4039–4043
Leslie JF, Summerell BA, Doe FJ (2005) Mycologia 97:718–724
Munkvold G, Carlton WM (1997) Phytopathology 81:211–216
Desjardins AE, Plattner RD, Nelson PE (1994) Appl Environ Microbiol 60:1695–1697
Turechek WW, Hartung JS, McCallister JM (2007) Phytopathology 98:359–368
Acknowledgements
We thank KWS SAAT AG and the University of Hohenheim for providing us with maize samples. The study was supported by the Ministry for Science and Culture of Lower Saxony within the network KLIFF—climate impact and adaptation research in Lower Saxony, by FAEN Joint Project “Quality-related plant production under modified basic conditions: mycotoxins in the context of production, quality and processing” funded by the Ministry of Science and Culture of Lower Saxony, Germany, and by the Ministry of Science and Education (BMBF) and KWS SAAT AG within the German–French–Spanish consortium PG-CEREHEALTH of the Era-NET network.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The first two authors contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nutz, S., Döll, K. & Karlovsky, P. Determination of the LOQ in real-time PCR by receiver operating characteristic curve analysis: application to qPCR assays for Fusarium verticillioides and F. proliferatum . Anal Bioanal Chem 401, 717–726 (2011). https://doi.org/10.1007/s00216-011-5089-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-5089-x