
Abstract
The six types of pattern recognition molecules (PRMs) that initiate complement via the lectin pathway (LP) comprise collectins and ficolins. The importance of having various PRMs to initiate the LP is currently unclear. Mannan-binding lectin (MBL) is a collectin member of the LP PRMs. MBL deficiency is common with mild clinical consequence. Thus, the lack of MBL may be compensated for by the other PRMs. We hypothesized that variants FCN2 + 6424 and FCN3 + 1637delC that cause gene-dose-dependent reduction in ficolin-2 and ficolin-3 levels, respectively, may be rare in MBL-deficient individuals due to the importance of compensation within the LP. The aim of this study was to investigate the distribution and frequency of these variants among MBL2 genotypes in healthy subjects. The allele frequency of FCN2 + 6424 and FCN3 + 1637delC was 0.099 and 0.015, respectively, in the cohort (n = 498). The frequency of FCN2 + 6424 tended to be lower among MBL-deficient subjects (n = 53) than among MBL-sufficient subjects (n = 445) (0.047 versus 0.106, P = 0.057). In addition, individuals who were homozygous for FCN2 + 6424 were sufficient MBL producers. The frequency of FCN3 + 1637delC did not differ between the groups. The frequency of FCN2 + 6424 was similar in FCN3 + 1637delC carriers (n = 15) versus wild type (n = 498). Furthermore, subjects that were heterozygote carriers of both FCN2 + 6424 and FCN3 + 1637delC were sufficient MBL producers. In conclusion, FCN2 + 6424 carriers with MBL deficiency tend to be rare among healthy individuals. MBL-deficient individuals with additional LP PRM defects may be at risk to morbidity.
Similar content being viewed by others

Introduction
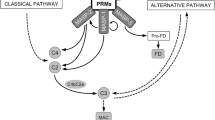
The lectin pathway (LP) is the most recently discovered of the three complement activation pathways. It is initiated by one of the six pattern recognition molecules (PRMs), either mannan-binding lectin (MBL), collectin kidney-1 (CL-K1), collectin liver-1 (CL-L1), ficolin-1 (M-ficolin), ficolin-2 (L-ficolin) or ficolin-3 (H-ficolin) (Henriksen et al. 2013; Kjaer et al. 2013). The PRMs bind specific molecular patterns on pathogens, apoptotic cells and cellular debris as they circulate in serum complexed with three serine proteases (MASP-1–3) and two non-enzymatic proteins with regulatory functions: MAP1 and sMAP (Dahl et al. 2001; Degn et al. 2009; Sato et al. 1994; Skjoedt et al. 2011; Stover et al. 1999; Takahashi et al. 1999; Thiel et al. 1997). Upon binding, MASP-1 activates MASP-2 which subsequently cleaves complement factors C2 and C4 resulting in the activation of the complement cascade with the generation of inflammatory mediators (C3a and C5a), assembly of the membrane attack complex (MAC) and opsonisation (Degn et al. 2012; Héja et al. 2012; Takahashi et al. 2008).
The collectins (MBL, CL-K1 and CL-L1) and ficolins are similar in structure (Kjaer et al. 2013). They are oligomers and share amino terminal collagen-like stalk that interacts with the MASPs. They differ at the carboxyl terminal where the ficolins contain fibrinogen-like recognition domain and the collectins contain C-type carbohydrate recognition domain. Despite having a common structure, ficolin-2 and ficolin-3 are only 48 % identical by sequence (Sugimoto et al. 1998). Both ficolin-2 and ficolin-3 have been shown to bind dying host cells and mediate their clearance through complement activation via the LP (Honoré et al. 2007; Jensen et al. 2007; Kuraya et al. 2005). Ficolin-2 binds various clinically relevant pathogens including those affecting the respiratory system, whereas ficolin-3 does not (Hummelshøj et al. 2012; Krarup et al. 2005; Pan et al. 2012). However, recently, it was shown that ficolin-3 binds enteropathogenic and enteropathoaggregative Escherichia coli (Sahagún-Ruiz et al. 2015). Furthermore, the first evidence of antimicrobial activity of ficolin-3 was recently reported against the enteric commensal and opportunistic gut bacteria Hafnia alvei (Michalski et al. 2015a). Thus, there appears to be a link between ficolin-3 and the gut. Notably, ficolin-3 is resistant to collagenases (whereas the other ficolins and collagens are not) and this may reflect the site of its antimicrobial activity, i.e. in the gastrointestinal tract (Hummelshoj et al. 2008).
The concentration of MBL in serum ranges from <10–12,200 ng/ml in adults, with a median of 1340 ng/ml (Sallenbach et al. 2011). This large range span can be explained by combinations of functional single nuclear polymorphisms (SNPs) in exon 1 and in the promoter of the MBL2 gene (Garred et al. 2003). The SNPs in exon 1 have been named variant allele D (the codon 52 allele or p.Arg52Cys), B (the codon 54 allele or p.Gly54Asp) and C (the codon 57 allele or p.Gly57Glu). Collectively, they are referred to as the O allele, whereas the normal wild-type allele is referred to as the A allele. The O allele gives rise to dysfunctional forms of MBL that are unable to bind to their ligands (Lipscombe et al. 1995; Madsen et al. 1994; Sumiya et al. 1991). The SNPs in the MBL2 promoter region are located at position −50 (H/L) and −221 (X/Y) and at position +4 (P/W) in the 5′-untranslated portion of exon 1 (Madsen et al. 1995; Madsen et al. 1998; Steffensen et al. 2000). The variant at position −221 (X allele) has the strongest downregulating effect on MBL expression. A combination of the SNPs in exon 1 and MBL2 promoter determines various MBL2 genotypes. The XA/O and O/O genotypes are generally referred to as MBL-deficient genotypes as they give rise to undectable MBL levels (<10 ng/ml) (Eisen et al. 2008).
Ficolin-2 is expressed in the liver and serum concentration varies from 1340 to 14,700 ng/ml in adults, with a median of 3370 ng/ml (Sallenbach et al. 2011). The +6424 G > T variant in exon 8 of the FCN2 gene leads to amino acid substitution within the firbrinogen-like domain of ficolin-2, and the mutation has a gene-dose effect on ficolin-2 serum levels (Munthe-Fog et al. 2007). The median serum levels of wild-type (G/G), heterozygotes (G/T) and homozygotes (T/T) have been shown to be 5100, 2200 and 900 ng/ml, respectively (Kilpatrick et al. 2013; Munthe-Fog et al. 2007). Thus, homozygosity does not lead to absolute ficolin-2 deficiency but is generally referred to as ficolin-2 insufficiency (Kilpatrick and Chalmers 2012). Concomitant with the variant’s downregulating effect is increased binding affinity for GlcNAc (Hummelshoj et al. 2005). Allele frequency has been reported to be between 0.12 and 0.14 in Caucasians (Damman et al. 2012; Herpers et al. 2006; Kilpatrick et al. 2013; Munthe-Fog et al. 2007; Ojurongbe et al. 2012). Low levels of ficolin-2 have been associated with prematurity, low birth weight and infections in neonates, childhood infections combined with allergic diseases and preeclampsia (Atkinson et al. 2004; Cedzynski et al. 2009; Swierzko et al. 2009; Wang et al. 2007). In addition, the FCN2 + 6424 variant has been linked to the early onset of Pseudomonas aeruginosa in cystic fibrosis patients (Haerynck et al. 2012).
Ficolin-3 is the most abundant LP PRM in serum and the most potent LP activator in vitro (Hummelshoj et al. 2008). Its concentration varies tenfold (6100–60,300 ng/ml) in adults, with a median of 19,500 ng/ml (Sallenbach et al. 2011). The +1637delC variant in exon 5 of the FCN3 gene is a frameshift mutation leading to the truncation of the C-terminal end of the ficolin-3 protein (Munthe-Fog et al. 2008). The variant has gene-dose-dependent effect on ficolin-3 serum levels, and homozygotes have no detectable ficolin-3 in serum (Michalski et al. 2011; Munthe-Fog et al. 2009; Munthe-Fog et al. 2008). Gene frequency has been reported to be rare (0.01–0.02) suggesting the crucial functions of ficolin-3 (Michalski et al. 2011; Munthe-Fog et al. 2009). Congenital ficolin-3 deficiency (+1637delC homozygotes) has so far only been confirmed in six individuals including two prematures, one infant and three adults. These subjects were affected by various morbidities (though nothing in common), such as invasive necrotizing eneterocolitis (NEC), perinatal Streptococcus agalactiae infections, congenital heart disease, recurrent severe pulmonary infections, brain abscesses, and severe nephrotic syndrome (Michalski et al. 2011; Michalski et al. 2015b; Munthe-Fog et al. 2009; Schlapbach et al. 2011). One of the homozygous adult was found in a healthy control group (Metzger et al. 2015).
MASP-2 serum levels ranges from 125 to 1150 ng/ml, with a median of 416 ng/ml (Sallenbach et al. 2011). The p.D120G mutation results in impaired binding to both MBL and ficolins (Stengaard-Pedersen et al. 2003; Thiel et al. 2009) and influences MASP-2 serum levels in a gene-dose-dependent manner. Homozygotes for the p.D120G variant have no detectable MASP-2 in serum and no MBL-mediated activation of complement (Stengaard-Pedersen et al. 2003). Allele frequency is 0.039 in Danish Caucasians (Thiel et al. 2007). Interestingly, the allele is not found in other populations such as Hong Kong Chinese, Zambian Africans and Amerindians in Brazil (Thiel et al. 2007). A total of thirteen p.D120G homozygotes have been published from case-control studies since the first case that was reported in the year 2003 (Stengaard-Pedersen et al. 2003). Six were unaffected or healthy (Garcia-Laorden et al. 2006; Olszowski et al. 2014; Segat et al. 2008; Sokolowska et al. 2015; Stover et al. 2005). Seven were detected in cohorts of recurrent respiratory infections, cystic fibrosis, colorectal cancer, hepatocellular carcinoma and pulmonary tuberculosis (Cedzynski et al. 2009; Cedzynski et al. 2004; Olesen et al. 2006; Segat et al. 2008; Sokolowska et al. 2015). Thus, the impact of MASP-2 deficiency (p.D120G homozygosity) in disease association is uncertain and it appears that homozygotes are found by chance. One might conclude from this that the LP is dispensible or redundant for human health because the current knowledge is that MASP-2 is crucial for LP complement activation. There are probably unidentified molecules and functions involved in the LP that could explain why MASP-2 deficiency is relatively common in apparently healthy individuals. In certain situations, being a p.D120G carrier may be relevant such as in the medical intensive care unit where it has been reported that the p.D120G allele was associated with increased risk of early death (Henckaerts et al. 2009).
Numerous studies have linked MBL deficiency with increased susceptibility to a variety of infectious and autoimmune diseases (reviewed in (Bjarnadottir and Ludviksson 2010; Dommett et al. 2006; Eisen and Minchinton 2003; Kilpatrick 2002; Thiel et al. 2006; Worthley et al. 2005). Several recent case-control studies have been investigating alternations in the distribution patterns of serum concentration of the early LP components in disease pathogenesis such as in patient groups with systemic lupus erythematosis (SLE), hemodialysis, pulmonary tuberculosis and acute liver failure (Chalmers et al. 2015; Ishii et al. 2011; Laursen et al. 2015; Troldborg et al. 2015a; Troldborg et al. 2015b). However, there are various factors that can influence serum levels, such as acute phase response and medication. Thus, genetic analysis may in certain settings provide more lines of reliable information. Studies involving determination on the combination of LP variants within LP genotypes remain scarce. To our knowledge, this is the first study that describes the frequency and distribution of the downregulating variants in the FCN2, FCN3 and MASP2 genes among MBL2 genotypes in a healthy Caucasian population.
Material and methods
Subjects and samples
Between April and May 2010, EDTA blood was collected from a total of 498 volunteer blood donors from the Icelandic Blood Bank, and genomic DNA was isolated using high-salt procedure (Miller et al. 1988). The donors were Caucasians with age range of 18–60 years, and 125 were women and 375 were men. Informed consent was obtained from participants and only ethnic origin, gender and age was recorded. The study was approved by the Bioethics Committee of Iceland and the Data Protection Committee of Iceland.
Genotyping
A real-time polymerase chain reaction (PCR) with subsequent melting curve (T m ) analysis on amplicons was used to detect functional variants (rs1800450, rs1800451, rs5030737) in exon 1 and promoter (rs7096206) of the MBL2 gene with the LightCycler Instrument (Roche Diagnostics) using a previously described method (Steffensen et al. 2003). FCN2 genotyping was carried out using a sequence-specific primer PCR (SSP-PCR) to detect the functional variant (rs7851696) in exon 8 (Szala et al. 2013). Restriction fragment length polymorphism PCR (RFLP-PCR) was used to detect the base deletion 1637delC (rs28357092) in the FCN3 gene (Michalski et al. 2011). MASP2 genotyping was carried out using SSP-PCR to detect the mutation (rs72550870) in the MASP2 gene (Boldt et al. 2011).
Statistical analysis
Allele frequency was compared between two and three genotype subgroups within the blood donor cohort using the chi-square test. When comparing more than three genotype subgroups, the Kruskal-Wallis H test was used followed by the Dunn’s multiple comparison post hoc test. The level of significance was set at 0.05 and the program package SPSS 11.0 (SPSS, Inc, Chicago, Ill) was used for processing the data.
Results
Frequency of MBL2 genotypes and LP downregulating variants in the blood donor cohort
The frequencies of the structural MBL2 variant alleles and MBL2 genotypes are listed in Table 1. A total of 318 were wild-type A/A (63.9 %), 168 were A/O (33.7 %) and 12 were O/O (2.4 %). The A/O genotypes were genotyped for the downregulating X variant in the MBL2 promoter. Forty-one of the A/O genotypes were found to harbour the X variant, i.e. 25 XA/B, 1 XA/C and 15 XA/D. Thus, 53 of the blood donors (10.6 %) were MBL deficient (XA/O and O/O genotypes). The calculated allele frequencies of the downregulating variants FCN2 + 6424, FCN3 + 1637delC and MASP2 p.D120G in the blood donors are shown in Table 2. Six individuals were homozygous for the FCN2 + 6424 variant (T/T). In contrast, neither for the FCN3 + 1637delC variant nor the MASP2 p.D120G variant was detected in the healthy cohort.
Distributon of the FCN2 + 6424 variant among MBL2, FCN3 and MASP2 genotypes
A total of 93 subjects in the blood donor cohort carried the FCN2 + 6424 variant and the calculated gene frequency was 0.099 (Table 2). Figure 1a demonstrates how the variant was distributed among the five known MBL2 genotypes. Allele frequency did not significantly differ between the MBL2 genotypes; however, the frequency was substantially low in A/D genotype carriers (0.043) and the variant was absent in O/O genotypes (Fig. 1a). The comparison of FCN2 + 6424 frequency among O/O and A/D genotypes (n = 58) versus the A/A, A/B and A/C genotypes (n = 440) resulted in significant difference (0.034 versus 0.108, P = 0.013, chi-square test). Interestingly, the six FCN2 + 6424 homozygotes (T/T) (Table 2) were only found among wild-type MBL2 genotypes (A/A), whereas no homozygotes were found among individuals carrying the MBL2 structural alleles (A/O and O/O genotypes). The FCN2 + 6424 frequency did not differ between the heterozygous carriers of FCN3 + 1637delC versus wild-type (Fig. 1b) and the heterozygous carriers of MASP2p.D120G versus wild-type (Fig. 1c).

Frequency of the FCN2 + 6424 G > T variant among different genotypes in the blood donor cohort (n = 498). a Frequency among MBL2 genotypes; A/A (n = 318), A/B (n = 114), A/C (n = 8), A/D (n = 46), O/O (n = 12). b Frequency between wild type (WT) (n = 483) and carriers of the FCN3 + 1637delC variant (n = 15). c Frequency between wild type (n = 459) and carriers of the MASP2D120G variant (n = 39). d Frequency between MBL-sufficient genotypes (n = 445) and MBL-deficient genotypes (n = 53). Allele frequency was compared with the Kruskal-Wallis test followed by Dunn’s multiple comparison pos hoc test (a) and the chi-square test (b–d)
The FCN2 + 6424 variant in MBL deficiency
Previous studies indicate that ficolin-2 may be compensating for the lack of MBL (Ishii et al. 2011). Thus, we hypothesized that the FCN2 + 6424 variant may be rare in MBL deficiency due to its downregulating effect on ficolin-2 levels (Kilpatrick et al. 2013; Munthe-Fog et al. 2007). The study cohort was divided into those carrying MBL-sufficient genotypes (A/A and AY/O) (n = 445) and those carrying MBL-deficient genotypes (AX/O and O/O) (n = 53) according to previous suggestions (Eisen et al. 2008). Subsequently, the FCN2 + 6424G > T allele frequency was determined in each group. The observed frequency of the variant tended to be lower in the MBL-deficient group than in the MBL-sufficient group (0.047 versus 0.0106, P = 0.054, chi-square test) (Fig. 1d).
Distributon of the FCN3 + 1637delC variant among MBL2, FCN2 and MASP2 genotypes
There were 15 heterozygote carriers of the FCN3 + 1637delC in the blood donor cohort and calculated gene frequency was 0.015 (Table 2). The variant was equally distributed among the five known MBL2 genotypes (Fig. 2a). The high frequency that was observed in the A/C genotype subgroup may be explained by the small size of the group (n = 8). The variant was absent in O/O genotype carriers. No significant deviations were found in FCN3 + 1637delC frequency between FCN2 genotypes (Fig. 2b). Notably, four subjects were identified who were heterozygous for both downregulating variants in the ficolin genes, i.e. FCN3 + 1637delC (-/C) and FCN2 + 6424 (G/T) (central bar in Fig. 2b). Thus, healthy individuals have intermediate serum levels of both ficolin-3 and ficolin-2. Interestingly, these four individuals were all high producers of MBL (two AY/AY genotypes, one AY/B genotype and one AY/C genotype). In contrast, neither FCN3 + 1637delC nor FCN2 + 6424 were detected among O/O subjects which have undetectable MBL in serum. The FCN3 + 1637delC allele was equally distributed between wild-type and heterozygote MASP2p.D120G carriers (Fig. 2c).

Frequency of the FCN3 + 1637delC variant among different genotypes in the blood donor cohort (n = 498). a Frequency among MBL2 genotypes; A/A (n = 318), A/B (n = 114), A/C (n = 8), A/D (n = 46), O/O (n = 12). b Frequency among wild type (WT) (n = 483), heterozygous carriers of FCN2 + 6424 (G/T) (n = 87) and homozygous carriers of FCN2 + 6424 (T/T) (n = 6). c Frequency between wild type (n = 459) and carriers of the MASP2D120G variant (A/G) (n = 39). d Frequency between MBL-sufficient genotypes (n = 445) and MBL-deficient genotypes (n = 53). Allele frequency was compared with the Kruskal-Wallis test followed by Dunn’s multiple comparison pos hoc test (a) and the chi-square test (b–d)
The FCN3 + 1637delC variant in MBL deficiency
We hypothesized that the FCN3 + 1637delC variant would be rare in MBL deficiency due to its downregulating effect on ficolin-3 levels (Munthe-Fog et al. 2009). One individual was heterozygous in the MBL-deficient group whereas 14 were found in the MBL-sufficient group (Fig. 2d). The only FCN3 + 1637delC carrier in the MBL-deficient group was an AX/D genotype carrier.
Frequency of MASP2p.D120G among the MBL2, FCN2 and FCN3 genotypes
We included the MASP2p.D120G variant in our analysis. It causes downregulation of MASP-2 in a gene-dose-dependent manner that is the same effect of the FCN2 + 6424 and FCN3 + 1637delC variants. The MASP-2 protein is an enzyme activator of the LP and not one of the six recognition proteins of the LP. Thus, the compensation hypothesis would not be applied to this variant and one would not expect the frequency to differ. There were 39 MASP2p.D120G heterozygote carriers in the blood donor cohort and calculated gene frequency was 0.039 (Table 2). The observed gene frequency of MASP2p.D120G did not differ between MBL2 genotypes, FCN2 genotypes or FCN3 genotypes (Fig. 3a–c). The elevated frequency in the A/C (n = 8) and T/T (n = 6) subgroups (0.143 and 0.083, respectively) may be due to the small number of individuals in these groups. The variant was absent in O/O genotype carriers (Fig. 2a). The frequency was low in MBL-deficient subjects but not statistically different from MBL-sufficient subjects (Fig. 3d).

Frequency of the MASP2D120G variant among different genotypes in the blood donor cohort (n = 498). a Frequency among MBL2 genotypes; A/A (n = 318), A/B (n = 114), A/C (n = 8), A/D (n = 46), O/O (n = 12). b Frequency among wild type (n = 483), heterozygous carriers of FCN2 + 6424 (G/T) (n = 87) and homozygous carriers of FCN2 + 6424 (T/T) (n = 6). c Frequency between wild type (n = 483) and carriers of the FCN3 + 1637delC variant (-/C) (n = 15). d Frequency between MBL-sufficient genotypes (n = 445) and MBL-deficient genotypes (n = 53). Allele frequency was compared with the Kruskal-Wallis test followed by Dunn’s multiple comparison pos hoc test (a) and the chi-square test (b–d)
Discussion
The observed occurrence of MBL2 A/A and A/O genotypes in the blood donors is similar as has been observed among randomly selected individuals from a population of same ethnic origin (Dahl et al. 2004; Garred et al. 2006; Mead et al. 1997; Roy et al. 2002). However, only 2.4 % of the blood donors were of the O/O genotype whereas we have observed this ratio to be 5.6 % among randomly selected individuals from the Icelandic nation (n = 250, unpublished data). Similar findings have been observed in other studies. The ratio of O/O genotypes was 5–7 % when selected randomly (Dahl et al. 2004; Davies et al. 1995; Roy et al. 2002) but 2.5–2.8 % when selected for health (Kronborg et al. 2002; Lee et al. 2005). Furthermore, none of the O/O subjects in our healthy cohort were carriers of the downregulating variants (FCN2 + 6424, FCN3 + 1637delC or p.D120G). Thus, by selecting for health, one may expect to observe twofold fewer O/O genotypes than from a random selection and they may not be carriers of the downregulating LP variants. Therefore, being an O/O genotype carrier may be relevant when it comes to health and may be linked to morbidity, particularly when there is an additional LP component deficiency.
The observed gene frequency of the FCN2 + 6424 variant (0.099) in the Icelandic Caucasian blood donors (n = 498) is slightly lower than has been observed for Dutch (n = 188) (0.136) and Danish Caucasian (n = 214) (0.118) blood donors and Dutch Caucasian (n = 1268) (0.137) kidney donors (Damman et al. 2012; Herpers et al. 2006; Munthe-Fog et al. 2007). Our results (from this study and unpublished) demonstrate that the downregulating FCN2 + 6424 variant tends to be less frequent in MBL-deficient than in MBL-sufficient subjects. Supporting this observation are the findings of Ishii et al. who reported elevated levels of ficolin-2 among O/O genotypes (Ishii et al. 2011). Thus, ficolin-2 may be compensating for the lack of MBL. This conclusion is supported by the facts that ficolin-2 and MBL resemble in molecular structure, they are both produced in the liver, they both initiate the LP of complement via associated MASPs and they have been shown to bind a wide variety of pathogens some of which are in common (Degn and Thiel 2013; Kilpatrick and Chalmers 2012). It is conceivable that ficolin-2 is upregulated due to the lack of MBL, but we speculate, based on our results, that elevated ficolin-2 levels among O/O genotypes is due to the rarity of the downregulating FCN2 + 6424 variant among MBL-deficient individuals. Thus, there may be a certain beneficial balance between MBL and ficolin-2 serum concentration for the host that has been maintained throughout evolution, and this may be controlled by the genetic variants.
The low gene frequency of FCN3 + 1637delC and the small sample size of the blood donor cohort preclude significant conclusions regarding the FCN3 + 1637delC gene frequency in MBL deficiency or low producers of ficolin-2 (FCN2 + 6424 homozygotes). Therefore, further investigations in a cohort with a larger number of MBL-deficient genotypes and FCN2 + 6424 homozygotes are warranted. The only FCN3 + 1637delC carrier found in the MBL-deficient group of our study cohort was of the XA/D genotype. Notably, the D variant is believed to have less dramatic effect on MBL levels than the B or C variants. The D variant chain is partly incorporated into stable higher oligomerized MBL explaining why MBL levels are generally higher among A/D genotypes than A/B and A/C genotypes (Garred et al. 2003). Furthermore, some investigators state that D allele carriers are intermediate MBL producers and should not be grouped as deficient (Degn et al. 2011). We have screened additional MBL-deficient adults (n = 140) for the FCN3 + 1637delC variant and found only one heterozygote carrier, and this individual was also of the XA/D genotype (unpublished data), further supporting our findings demonstrated here. Interestingly, homozygosity for FCN3 + 1637delC accompanied with the AX/D genotype was recently confirmed in an 11-month-old infant with congenital heart disease, pneumonia (without recurrence) and no severe infections after cardiac surgery (Michalski et al. 2015b). Thus, being a carrier of the FCN3 + 1637delC deletion and of the AX/D genotype does not seem to be life-threatening and not associated with severe recurrent infections.
Remarkably, the FCN3 + 1637delC variant was not found among subjects carrying genotypes O/O, AX/B, and AX/C. Thus, we speculate that having intermediate ficolin-3 levels or ficolin-3 deficiency accompanied with severe MBL deficiency (excluding the D variant, i.e. genotypes O/O, AX/B and AX/C) may have a substantial impact as co-morbid factor. In fact, the only individual that has been reported to be both FCN3 + 1637delC homozygous and of the O/O genotype was a premature baby (born at 35 weeks of gestation) with perinatal Streptococcus agalactiae infections, microcephaly, growth and mental retardation, and was diagnosed with attention deficit hyperactivity disorder (ADHD) at 3 years of age (Michalski et al. 2011). It has been suggested that ficolin-3 may be relevant in controlling commensal intestinal flora (Michalski et al. 2015a), and our previous studies show that O/O individuals are highly susceptible to gastrointestinal (GI) symptoms (Bjarnadottir et al. 2014). Thus, ficolin-3 and MBL may be relevant in the establishment of the gut microbiota at infancy. Recently, much attention has been on the association of the gut microbiota and microbiome with various disorders (Dave et al. 2012; Wang and Kasper 2014). Given the potential role of the LP in these complex interactions of the gut microbiota with its host and our findings reported here warrant further investigations on the impact of combined ficolin-3 and MBL deficiencies on microbiota and human health.
The observed frequency of the p.D120G allele in the Icelandic blood donor group is compatible with other studies including healthy Caucasians (Thiel et al. 2007). What we found noteworthy was that among the MBL deficiency genotypes, the variant was only associated with AX/B subjects and not with AX/D and O/O subjects. Ishii et al. reported significantly elevated levels of MASP-2 among O/O genotypes (Ishii et al. 2011). It is conceivable that MASP-2 was elevated in O/O genotypes because the downregulating variant p.D120G is absent. Furthermore, none of the 14 published p.D120G homozygotes were MBL deficient (personal communication) (Cedzynski et al. 2009; Cedzynski et al. 2004; Garcia-Laorden et al. 2008; Olesen et al. 2006; Sokolowska et al. 2015; Stengaard-Pedersen et al. 2003). Thus, we speculate that it may not be beneficial for the host to be a carrier of p.D120G and MBL deficiency (excluding the AX/B genotype). In contrast, the p.D120G was found equally distributed among sufficient (G/G), intermediate (G/T) and low producers of ficolin-2 (T/T).
Our results demonstrate how various LP deficiency variants are distributed among various LP genotypes in healthy individuals. In continuation, this data may become useful as a reference in disease association studies. Employing LP variant distribution pattern analysis in case-control studies may not replace determining the patterns of serum levels, but it may be an alternative and perhaps an innovative approach to investigate the impact of the LP on human health.
References
Atkinson AP, Cedzynski M, Szemraj J, St Swierzko A, Bak-Romaniszyn L, Banasik M, Zeman K, Matsushita M, Turner ML, Kilpatrick DC (2004) L-ficolin in children with recurrent respiratory infections. Clin Exp Immunol 138:517–520
Bjarnadottir H, Ludviksson BR (2010) Inherited deficiency of the initiator molecules of the lectin-complement pathway. Laeknabladid 96:611–617
Bjarnadottir H, Thorsteinsdottir V, Jorgensen G, Arnardottir M, Ludviksson B (2014) Mannan-binding lectin (MBL) deficient individuals with the O/O genotype are highly susceptible to gastrointestinal diseases. J Clin Cell Immunol 5:182
Boldt AB, Grisbach C, Steffensen R, Thiel S, Kun JF, Jensenius JC, Messias-Reason IJ (2011) Multiplex sequence-specific polymerase chain reaction reveals new MASP2 haplotypes associated with MASP-2 and MAp19 serum levels. Hum Immunol 72:753–760
Cedzynski M, Atkinson AP, St Swierzko A, Macdonald SL, Szala A, Zeman K, Buczylko K, Bak-Romaniszyn L, Wiszniewska M, Matsushita M, Szemraj J, Banasik M, Turner ML, Kilpatrick DC (2009) L-ficolin (ficolin-2) insufficiency is associated with combined allergic and infectious respiratory disease in children. Mol Immunol 47:415–419
Cedzynski M, Szemraj J, Swierzko AS, Bak-Romaniszyn L, Banasik M, Zeman K, Kilpatrick DC (2004) Mannan-binding lectin insufficiency in children with recurrent infections of the respiratory system. Clin Exp Immunol 136:304–311
Chalmers JD, Matsushita M, Kilpatrick DC, Hill AT (2015) No strong relationship between components of the lectin pathway of complement and susceptibility to pulmonary tuberculosis. Inflammation 38:1731–1737
Dahl M, Tybjaerg-Hansen A, Schnohr P, Nordestgaard BG (2004) A population-based study of morbidity and mortality in mannose-binding lectin deficiency. J Exp Med 199:1391–1399
Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, Vorup-Jensen T, Jensenius JC (2001) MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity 15:127–135
Damman J, Kok JL, Snieder H, Leuvenink HG, van Goor H, Hillebrands JL, van Dijk MC, Hepkema BG, Reznichenko A, van den Born J, de Borst MH, Bakker SJ, Navis GJ, Ploeg RJ, Seelen MA (2012) Lectin complement pathway gene profile of the donor and recipient does not influence graft outcome after kidney transplantation. Mol Immunol 50:1–8
Dave M, Higgins PD, Middha S, Rioux KP (2012) The human gut microbiome: current knowledge, challenges, and future directions. Transl Res 160:246–257
Davies EJ, Snowden N, Hillarby MC, Carthy D, Grennan DM, Thomson W, Ollier WE (1995) Mannose-binding protein gene polymorphism in systemic lupus erythematosus. Arthritis Rheum 38:110–114
Degn SE, Hansen AG, Steffensen R, Jacobsen C, Jensenius JC, Thiel S (2009) MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J Immunol 183:7371–7378
Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, Thiel S (2012) Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol 189:3957–3969
Degn SE, Jensenius JC, Bjerre M (2011) The lectin pathway and its implications in coagulation, infections and auto-immunity. Curr Opin Organ Transplant 16:21–27
Degn SE, Thiel S (2013) Humoral pattern recognition and the complement system. Scand J Immunol 78:181–93
Dommett RM, Klein N, Turner MW (2006) Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens 68:193–209
Eisen DP, Dean MM, Boermeester MA, Fidler KJ, Gordon AC, Kronborg G, Kun JF, Lau YL, Payeras A, Valdimarsson H, Brett SJ, Ip WK, Mila J, Peters MJ, Saevarsdottir S, van Till JW, Hinds CJ, McBryde ES (2008) Low serum mannose-binding lectin level increases the risk of death due to pneumococcal infection. Clin Infect Dis 47:510–516
Eisen DP, Minchinton RM (2003) Impact of mannose-binding lectin on susceptibility to infectious diseases. Clin Infect Dis 37:1496–1505
Garcia-Laorden MI, Garcia-Saavedra A, de Castro FR, Violan JS, Rajas O, Blanquer J, Borderias L, Rodriguez-Gallego C (2006) Low clinical penetrance of mannose-binding lectin-associated serine protease 2 deficiency. J Allergy Clin Immunol 118:1383–1386
Garcia-Laorden MI, Sole-Violan J, Rodriguez de Castro F, Aspa J, Briones ML, Garcia-Saavedra A, Rajas O, Blanquer J, Caballero-Hidalgo A, Marcos-Ramos JA, Hernandez-Lopez J, Rodriguez-Gallego C (2008) Mannose-binding lectin and mannose-binding lectin-associated serine protease 2 in susceptibility, severity, and outcome of pneumonia in adults. J Allergy Clin Immunol 122:368–374, 374 e1-2
Garred P, Larsen F, Madsen HO, Koch C (2003) Mannose-binding lectin deficiency—revisited. Mol Immunol 40:73–84
Garred P, Larsen F, Seyfarth J, Fujita R, Madsen HO (2006) Mannose-binding lectin and its genetic variants. Genes Immun 7:85–94
Haerynck F, Van Steen K, Cattaert T, Loeys B, Van Daele S, Schelstraete P, Claes K, Van Thielen M, De Canck I, Mahachie John JM, De Baets F (2012) Polymorphisms in the lectin pathway genes as a possible cause of early chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients. Hum Immunol 73:1175–1183
Henckaerts L, Nielsen KR, Steffensen R, Van Steen K, Mathieu C, Giulietti A, Wouters PJ, Milants I, Vanhorebeek I, Langouche L, Vermeire S, Rutgeerts P, Thiel S, Wilmer A, Hansen TK, Van den Berghe G (2009) Polymorphisms in innate immunity genes predispose to bacteremia and death in the medical intensive care unit. Crit Care Med 37(192-201):e1–e3
Henriksen ML, Brandt J, Andrieu JP, Nielsen C, Jensen PH, Holmskov U, Jorgensen TJ, Palarasah Y, Thielens NM, Hansen S (2013) Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol 191:6117–6127
Herpers BL, Immink MM, de Jong BA, van Velzen-Blad H, de Jongh BM, van Hannen EJ (2006) Coding and non-coding polymorphisms in the lectin pathway activator L-ficolin gene in 188 Dutch blood bank donors. Mol Immunol 43:851–855
Honoré C, Hummelshoj T, Hansen BE, Madsen HO, Eggleton P, Garred P (2007) The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum 56:1598–1607
Hummelshoj T, Fog LM, Madsen HO, Sim RB, Garred P (2008) Comparative study of the human ficolins reveals unique features of Ficolin-3 (Hakata antigen). Mol Immunol 45:1623–1632
Hummelshoj T, Munthe-Fog L, Madsen HO, Fujita T, Matsushita M, Garred P (2005) Polymorphisms in the FCN2 gene determine serum variation and function of Ficolin-2. Hum Mol Genet 14:1651–1658
Hummelshøj T, Ma YJ, Munthe-Fog L, Bjarnsholt T, Moser C, Skjoedt MO, Romani L, Fujita T, Endo Y, Garred P (2012) The interaction pattern of murine serum ficolin-A with microorganisms. PLoS ONE 7:e38196
Héja D, Kocsis A, Dobó J, Szilágyi K, Szász R, Závodszky P, Pál G, Gál P (2012) Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc Natl Acad Sci U S A 109:10498–10503
Ishii M, Ohsawa I, Inoshita H, Kusaba G, Onda K, Wakabayashi M, Ohi H, Horikoshi S, Matsushita M, Tomino Y (2011) Serum concentration of complement components of the lectin pathway in maintenance hemodialysis patients, and relatively higher levels of L-Ficolin and MASP-2 in Mannose-binding lectin deficiency. Ther Apher Dial 15:441–447
Jensen ML, Honoré C, Hummelshøj T, Hansen BE, Madsen HO, Garred P (2007) Ficolin-2 recognizes DNA and participates in the clearance of dying host cells. Mol Immunol 44:856–865
Kilpatrick DC (2002) Mannan-binding lectin: clinical significance and applications. Biochim Biophys Acta 1572:401–413
Kilpatrick DC, Chalmers JD (2012) Human L-ficolin (ficolin-2) and its clinical significance. J Biomed Biotechnol 2012:138797
Kilpatrick DC, St Swierzko A, Matsushita M, Domzalska-Popadiuk I, Borkowska-Klos M, Szczapa J, Cedzynski M (2013) The relationship between FCN2 genotypes and serum ficolin-2 (L-ficolin) protein concentrations from a large cohort of neonates. Hum Immunol 74:867–871
Kjaer TR, Thiel S, Andersen GR (2013) Toward a structure-based comprehension of the lectin pathway of complement. Mol Immunol 56:413–422
Krarup A, Sørensen UB, Matsushita M, Jensenius JC, Thiel S (2005) Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan-binding lectin, L-ficolin, and H-ficolin. Infect Immun 73:1052–1060
Kronborg G, Weis N, Madsen HO, Pedersen SS, Wejse C, Nielsen H, Skinhøj P, Garred P (2002) Variant mannose-binding lectin alleles are not associated with susceptibility to or outcome of invasive pneumococcal infection in randomly included patients. J Infect Dis 185:1517–1520
Kuraya M, Ming Z, Liu X, Matsushita M, Fujita T (2005) Specific binding of L-ficolin and H-ficolin to apoptotic cells leads to complement activation. Immunobiology 209:689–697
Laursen TL, Sandahl TD, Støy S, Schiødt FV, Lee WM, Vilstrup H, Thiel S, Grønbaek H, Group UALFS (2015) Circulating mannan-binding lectin, M-, L-, H-ficolin and collectin-liver-1 levels in patients with acute liver failure. Liver Int 35:756–763
Lee YH, Witte T, Momot T, Schmidt RE, Kaufman KM, Harley JB, Sestak AL (2005) The mannose-binding lectin gene polymorphisms and systemic lupus erythematosus: two case-control studies and a meta-analysis. Arthritis Rheum 52:3966–3974
Lipscombe RJ, Sumiya M, Summerfield JA, Turner MW (1995) Distinct physicochemical characteristics of human mannose binding protein expressed by individuals of differing genotype. Immunology 85:660–667
Madsen HO, Garred P, Kurtzhals JA, Lamm LU, Ryder LP, Thiel S, Svejgaard A (1994) A new frequent allele is the missing link in the structural polymorphism of the human mannan-binding protein. Immunogenetics 40:37–44
Madsen HO, Garred P, Thiel S, Kurtzhals JA, Lamm LU, Ryder LP, Svejgaard A (1995) Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol 155:3013–3020
Madsen HO, Satz ML, Hogh B, Svejgaard A, Garred P (1998) Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J Immunol 161:3169–3175
Mead R, Jack D, Pembrey M, Tyfield L, Turner M (1997) Mannose-binding lectin alleles in a prospectively recruited UK population. Lancet 349:1669–1670
Metzger ML, Michelfelder I, Goldacker S, Melkaoui K, Litzman J, Guzman D, Grimbacher B, Salzer U (2015) Low ficolin-2 levels in common variable immunodeficiency patients with bronchiectasis. Clin Exp Immunol 179:256–264
Michalski M, St Swierzko A, Lukasiewicz J, Man-Kupisinska A, Karwaciak I, Przygodzka P, Cedzynski M (2015a) Ficolin-3 activity towards the opportunistic pathogen, Hafnia alvei. Immunobiology 220:117–123
Michalski M, Szala A, St Swierzko A, Lukasiewicz J, Maciejewska A, Kilpatrick DC, Matsushita M, Domzalska-Popadiuk I, Borkowska-Klos M, Sokolowska A, Szczapa J, Lugowski C, Cedzynski M (2011) H-ficolin (ficolin-3) concentrations and FCN3 gene polymorphism in neonates. Immunobiology 217:730–737
Michalski M, Świerzko AS, Pągowska-Klimek I, Niemir ZI, Mazerant K, Domżalska-Popadiuk I, Moll M, Cedzyński M (2015b) Primary Ficolin-3 deficiency—is it associated with increased susceptibility to infections? Immunobiology 220:711–713
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Munthe-Fog L, Hummelshoj T, Hansen BE, Koch C, Madsen HO, Skjodt K, Garred P (2007) The impact of FCN2 polymorphisms and haplotypes on the ficolin-2 serum levels. Scand J Immunol 65:383–392
Munthe-Fog L, Hummelshoj T, Honore C, Madsen HO, Permin H, Garred P (2009) Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N Engl J Med 360:2637–2644
Munthe-Fog L, Hummelshoj T, Ma YJ, Hansen BE, Koch C, Madsen HO, Skjodt K, Garred P (2008) Characterization of a polymorphism in the coding sequence of FCN3 resulting in a ficolin-3 (Hakata antigen) deficiency state. Mol Immunol 45:2660–2666
Ojurongbe O, Abou Ouf E, Van Tong H, Toan NL, Song LH, Luz PR, Messias-Reason IJ, Nurjadi D, Zanger P, Kun JF, Kremsner PG, Velavan TP (2012) Reliable and rapid characterization of functional FCN2 gene variants reveals diverse geographical patterns. BMC Med Genet 13:37
Olesen HV, Jensenius JC, Steffensen R, Thiel S, Schiotz PO (2006) The mannan-binding lectin pathway and lung disease in cystic fibrosis—disfunction of mannan-binding lectin-associated serine protease 2 (MASP-2) may be a major modifier. Clin Immunol 121:324–331
Olszowski T, Poziomkowska-Gęsicka I, Jensenius JC, Adler G (2014) Lectin pathway of complement activation in a Polish woman with MASP-2 deficiency. Immunobiology 219:261–262
Pan Q, Chen H, Wang F, Jeza VT, Hou W, Zhao Y, Xiang T, Zhu Y, Endo Y, Fujita T, Zhang XL (2012) L-ficolin binds to the glycoproteins hemagglutinin and neuraminidase and inhibits influenza A virus infection both in vitro and in vivo. J Innate Immun 4:312–324
Roy S, Knox K, Segal S, Griffiths D, Moore CE, Welsh KI, Smarason A, Day NP, McPheat WL, Crook DW, Hill AV (2002) MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet 359:1569–1573
Sahagún-Ruiz A, Breda LC, Valencia MM, Elias WP, Munthe-Fog L, Garred P, Barbosa AS, Isaac L (2015) Studies of the binding of ficolin-2 and ficolin-3 from the complement lectin pathway to Leptospira biflexa, Pasteurella pneumotropica and Diarrheagenic Escherichia coli. Immunobiology 220:1177–1185
Sallenbach S, Thiel S, Aebi C, Otth M, Bigler S, Jensenius JC, Schlapbach LJ, Ammann RA (2011) Serum concentrations of lectin-pathway components in healthy neonates, children and adults: mannan-binding lectin (MBL), M-, L-, and H-ficolin, and MBL-associated serine protease-2 (MASP-2). Pediatr Allergy Immunol 22:424–430
Sato T, Endo Y, Matsushita M, Fujita T (1994) Molecular characterization of a novel serine protease involved in activation of the complement system by mannose-binding protein. Int Immunol 6:665–669
Schlapbach LJ, Thiel S, Kessler U, Ammann RA, Aebi C, Jensenius JC (2011) Congenital H-ficolin deficiency in premature infants with severe necrotising enterocolitis. Gut 60:1438–1439
Segat L, Fabris A, Padovan L, Milanese M, Pirulli D, Lupo F, Salizzoni M, Amoroso A, Crovella S (2008) MBL2 and MASP2 gene polymorphisms in patients with hepatocellular carcinoma. J Viral Hepat 15:387–391
Skjoedt MO, Hummelshoj T, Palarasah Y, Hein E, Munthe-Fog L, Koch C, Skjodt K, Garred P (2011) Serum concentration and interaction properties of MBL/ficolin associated protein-1. Immunobiology 216:625–632
Sokolowska A, Szala A, St Swierzko A, Kozinska M, Niemiec T, Blachnio M, Augustynowicz-Kopec E, Dziadek J, Cedzynski M (2015) Mannan-binding lectin-associated serine protease-2 (MASP-2) deficiency in two patients with pulmonary tuberculosis and one healthy control. Cell Mol Immunol 12:119–121
Steffensen R, Hoffmann K, Varming K (2003) Rapid genotyping of MBL2 gene mutations using real-time PCR with fluorescent hybridisation probes. J Immunol Methods 278:191–199
Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC (2000) Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods 241:33–42
Stengaard-Pedersen K, Thiel S, Gadjeva M, Moller-Kristensen M, Sorensen R, Jensen LT, Sjoholm AG, Fugger L, Jensenius JC (2003) Inherited deficiency of mannan-binding lectin-associated serine protease 2. N Engl J Med 349:554–560
Stover C, Barrett S, Lynch NJ, Barker JN, Burden D, Trembath R, Schwaeble W, Veal C (2005) Functional MASP2 single nucleotide polymorphism plays no role in psoriasis. Br J Dermatol 152:1313–1315
Stover CM, Thiel S, Thelen M, Lynch NJ, Vorup-Jensen T, Jensenius JC, Schwaeble WJ (1999) Two constituents of the initiation complex of the mannan-binding lectin activation pathway of complement are encoded by a single structural gene. J Immunol 162:3481–3490
Sugimoto R, Yae Y, Akaiwa M, Kitajima S, Shibata Y, Sato H, Hirata J, Okochi K, Izuhara K, Hamasaki N (1998) Cloning and characterization of the Hakata antigen, a member of the ficolin/opsonin p35 lectin family. J Biol Chem 273:20721–20727
Sumiya M, Super M, Tabona P, Levinsky RJ, Arai T, Turner MW, Summerfield JA (1991) Molecular basis of opsonic defect in immunodeficient children. Lancet 337:1569–1570
Swierzko AS, Atkinson AP, Cedzynski M, Macdonald SL, Szala A, Domzalska-Popadiuk I, Borkowska-Klos M, Jopek A, Szczapa J, Matsushita M, Szemraj J, Turner ML, Kilpatrick DC (2009) Two factors of the lectin pathway of complement, l-ficolin and mannan-binding lectin, and their associations with prematurity, low birthweight and infections in a large cohort of Polish neonates. Mol Immunol 46:551–558
Szala A, St Swierzko A, Cedzynski M (2013) Cost-effective procedures for genotyping of human FCN2 gene single nucleotide polymorphisms. Immunogenetics 65:439–446
Takahashi M, Endo Y, Fujita T, Matsushita M (1999) A truncated form of mannose-binding lectin-associated serine protease (MASP)-2 expressed by alternative polyadenylation is a component of the lectin complement pathway. Int Immunol 11:859–863
Takahashi M, Iwaki D, Kanno K, Ishida Y, Xiong J, Matsushita M, Endo Y, Miura S, Ishii N, Sugamura K, Fujita T (2008) Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J Immunol 180:6132–6138
Thiel S, Frederiksen PD, Jensenius JC (2006) Clinical manifestations of mannan-binding lectin deficiency. Mol Immunol 43:86–96
Thiel S, Kolev M, Degn S, Steffensen R, Hansen AG, Ruseva M, Jensenius JC (2009) Polymorphisms in mannan-binding lectin (MBL)-associated serine protease 2 affect stability, binding to MBL, and enzymatic activity. J Immunol 182:2939–2947
Thiel S, Steffensen R, Christensen IJ, Ip WK, Lau YL, Reason IJ, Eiberg H, Gadjeva M, Ruseva M, Jensenius JC (2007) Deficiency of mannan-binding lectin associated serine protease-2 due to missense polymorphisms. Genes Immun 8:154–163
Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, Willis AC, Eggleton P, Hansen S, Holmskov U, Reid KB, Jensenius JC (1997) A second serine protease associated with mannan-binding lectin that activates complement. Nature 386:506–510
Troldborg A, Thiel S, Jensen L, Hansen S, Laska MJ, Deleuran B, Jensenius JC, Stengaard-Pedersen K (2015a) Collectin liver 1 and collectin kidney 1 and other complement-associated pattern recognition molecules in systemic lupus erythematosus. Clin Exp Immunol 182:132–138
Troldborg A, Thiel S, Laska MJ, Deleuran B, Jensenius JC, Stengaard-Pedersen K (2015b) Levels in plasma of the serine proteases and associated proteins of the lectin pathway are altered in patients with systemic lupus erythematosus. J Rheumatol 42:948–951
Wang CC, Yim KW, Poon TC, Choy KW, Chu CY, Lui WT, Lau TK, Rogers MS, Leung TN (2007) Innate immune response by ficolin binding in apoptotic placenta is associated with the clinical syndrome of preeclampsia. Clin Chem 53:42–52
Wang Y, Kasper LH (2014) The role of microbiome in central nervous system disorders. Brain Behav Immun 38:1–12
Worthley DL, Bardy PG, Mullighan CG (2005) Mannose-binding lectin: biology and clinical implications. Intern Med J 35:548–555
Acknowledgments
The authors would like to thank Dr. Sveinn Gudmundsson at the Icelandic Blood Bank for his help and valuable support of this project. This study was supported by the Icelandic Research Fund, the Unversity of Iceland Research Fund and the Landspitali University Hospital Research Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No financial support or benefits come from commercial sources. The authors declare that they have no competing interests.
Ethics approval
Informed consent was obtained from participants and only ethnic origin, gender and age was recorded. The study was approved by the Bioethics Committee of Iceland and the Data Protection Committee of Iceland.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bjarnadottir, H., Arnardottir, M. & Ludviksson, B.R. Frequency and distribution of FCN2 and FCN3 functional variants among MBL2 genotypes. Immunogenetics 68, 315–325 (2016). https://doi.org/10.1007/s00251-016-0903-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-016-0903-4