
Abstract
One species of Babesia was identified on the blood smear of 20 different naturally infected sheep in the Northwest of Iran. It was polymorphic, including double pyriform with acute or obtuse angle, single pyriform, and ring form. The size of typical paired pyriforms with acute angle was 2.7 × 0.4 μm (n = 10) and with obtuse angle was 3.5 × 0.6 μm (n = 10). Although the morphological and biometrical parameters resembled the Babesia motasi, the results of seminested polymerase chain reaction (PCR) and PCR-restriction fragment length polymorphism using primers specific for small subunit of 18S rRNA confirmed this species as Babesia ovis. Furthermore, the sequence analysis of hypervariable region of small subunit of 18S rRNA revealed the corresponding sequences for B. ovis as well. Experimental infection of healthy lambs with the morphological larger B. ovis showed a milder clinical signs compared to the small one.
Similar content being viewed by others


Introduction
Babesiosis belongs to the complex of several tick-borne diseases with different etiological agents, such as protozoa, rickettsia, bacteria, and viruses. Babesia causes diseases in the livestock with high morbidity and mortality, thereby resulting in high economical losses worldwide (Barnett 1974a, b; Mehlhorn and Schein 1984; Mehlhorn et al. 1994; Ahmed et al. 2002).
Control and management of livestock health could be understood as the two sides of a gold coin for a successful and healthy economy in stock farming. Here, the control of tick-borne diseases plays a prominent role which is performed based on early diagnosis and therapy.
There are five species of Babesia, which can infect sheep and goats: Babesia ovis (Starcovici 1893), Babesia motasi (Wenyon 1926), Babesia crassa (Hashemi-Fesharki and Uilenberg 1981; Levine 1985; Ray and Raghavachari 1941; Sarwar 1935), Babesia foliata (Ray and Raghavachari 1941), and Babesia taylori (Sarwar 1935).
Diagnosis of Babesia spp. performed traditionally using Giemsa staining of suspicious blood smears and the morphology of the piroplasms in the erythrocytes is decisive for the diagnosis. The Giemsa staining can be accompanied with some technical problems cause false morphological diagnosis, and in some cases needs special diagnostic knowledge. In certain cases, serological methods such as the immune fluorescence antibody test or immunoperoxidase test have also been applied (Jianxus and Hong 1997; Leemans et al. 1997; Shayan and Nabian 2008).
Schnittger et al. (2004) recently described a reverse line blotting method for the simultaneous detection and differentiation of Theileria and Babesia parasites infecting small ruminants. Shayan et al. (2007) have described polymerase chain reaction restriction fragment length polymorphism (PCR-RFLP) and nested PCR for differentiation between B. ovis and B. motasi.
Lian et al. (1997) isolated from the sheep a strain of small Babesia (1.8–2.1 μm in length), which was similar to the B. ovis from Holland, but distinguished significantly from the Dutch B. motasi. Qi Bai et al. (2002) described the large Babesia species infective for sheep and goats with the typical paired pyriforms of 1.8–2.5 to 0.9–1.8 μm, with mean dimensions of 2.21–0.12 μm to 1.17–0.18 μm in size. This species was slightly smaller than that of B. motasi in Northern Europe and was easy to differentiate from B. ovis or Babesia crassa by morphology and size. This study described the large Babesia infective for sheep morphologically comparable to the B. motasi but genetically distinct and identical to the B. ovis.
Materials and methods
Materials
We obtained 20 peripheral blood samples from sheep with suspicion of babesiosis. Blood smears were prepared from the samples and stained with Giemsa. The rest of the samples was used for the DNA extraction. All tissues had been obtained with consent given according to institutional guidelines.
DNA extraction
For more than 200 μl blood, erythrocytes were first lysed in blood samples using Erys-Lysing Buffer. DNA was extracted using a DNA isolation kit (Maleic acid-buffered saline with 0.1% Tween 20 (MBST), Iran) according to the manufacturer’s instructions. Briefly, cells were first lysed in 180 μl lysis buffer and the proteins were degraded with 20 μl proteinase K for 10 min at 55 C. After addition of 360 μl Bindings buffer and incubation for 10 min at 70°C, 270 μl ethanol (100%) was added to the solution and after vortexing, the complete volume was transferred to the MBST column. The MBST column was first centrifuged and then washed twice with 500 μl washing buffer. Finally, DNA was eluted from the carrier with Elution buffer.
DNA extraction from blood smears
Each blood smear was cleaned in separate vessels by a short passage in acetone and ethanol. Approximately, half the blood smear after Giemsa staining was dissolved in 100 μl Lysis buffer of the MBST kit. DNA was extracted as described above and dissolved in 50 μl TE buffer.
Polymerase chain reaction and seminested PCR
Approximately 100 to 500 ng DNA, or 5–10 μl DNA solution in the case of extraction from blood smear, was used for the PCR analysis. The PCR was performed on 100 μl total volume including one time PCR buffer, 2.5 U Taq Polymerase (Cina gene, Iran), 2 μl of each primer (20 mM, MWG, Germany), 200 μM of each dATP, dTTP, dCTP, and dGTP (Fermenta), and 1.5 mM MgCl2 in automated Thermocycler (MWG, Germany) with the following program: 5 min incubation at 95°C to denature double strand DNA, 35–38 cycles of 45 s at 54–58°C (annealing step), 45 s, at 72°C (extension step), and 45 s at 94°C (denaturing step). Finally, PCR was completed with the additional extension step for 10 min. The PCR products were analyzed on 1.8% Agarose gel in 0.5 times Tris–Borate–ethylenediaminetetraacetic acid (EDTA) buffer and visualized using Ethidium bromide and UV eluminator.
To control the specificity of the PCR products from the 18s rRNA, seminested PCR technique was used in which the additional primer is designated within the hyper variable region of the V4 region of 18s rRNA gene. The primers are listed in the Table 1. Seminested PCR was performed with the PCR product isolated from agarose gel using the MBST kit according to the manufacturer’s instructions. Briefly, the DNA bands were cut from the gel under UV control and dissolved in the binding buffer at 60°C. The dissolved agarose was transferred into the MBST column. After washing, the bound DNA was eluted using 30 μl TE buffer. A 1–5-μl eluted DNA was amplified with the primers P3–P2 and P4–P2 separately. Seminested PCR was performed directly with the 1 μl PCR product as well.
PCR product purification
PCR product was purified from the salts and proteins using PCR purification kit (MBST, Iran). Briefly, 200 μl binding buffer was added to 100 μl PCR product solution. After adding of 150 μl ethanol (96%) to the sample, the mixture was applied into the column. The column was washed twice with washing buffer, and PCR product was eluted from the column using 100 μl elution buffer.
PCR-RFLP with Nde I and Mbo II
DNA extracted from blood or blood smears was amplified using forward and reverse primer, resulting a PCR product of 389 bp for B. ovis. This product should be for B. motasi 395 bp. The PCR products were purified from enzyme and salts using PCR product purification kit (MBST). Twenty microliters of purified PCR product was, then, cut with 2 μl Nde I or Bsr D I (Fermenta, 10 U/μl) in 3 μl 10 × corresponding buffer and 5 μl H2O for 16 h by 37°C. As control, 20 μl PCR product was treated with 3 μl 10× corresponding buffer and 7 μl H2O without adding of enzyme.
Experimental infection of sheep with morphological diagnosed as B. ovis or B. motasi
Fifteen male lambs aged 4–6 months local breed sheep prepared from Animal Breed Research Institute of Tehran University were used. A week prior to initiation of the study, the blood samples from all animals were negative for Babesia spp. using Giemsa staining and PCR analysis with Babesia specific primers derived from 18S rRNA. The lambs were divided in three groups. The lambs in two groups received 6 × 106 of small B. ovis or large B. ovis infected erythrocytes which was isolated from west Azarbayejan of Iran. The third group received saline buffer and served as control group. To obtain hematological parameters, blood samples were drawn from the jugular vein into EDTA of all sheep prior to inoculation and every 2 days on post inoculation day. Daily physical examination was performed on each animal. The parasitemia and clinical signs such as the rectal temperature, color of the mucous membranes (conjunctival, oral, and vaginal), pulse, respiratory rate, anorexia, and death time were considered.
Results and discussion
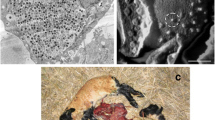
Babesiosis is an important disease in the livestock with high morbidity and mortality, thereby, resulting in high economical losses worldwide (Barnett 1974a, b; Mehlhorn and Schein 1984; Mehlhorn et al. 1994; Ahmed et al. 2002). B. ovis and B. motasi were described as the more common causative agent of babesiosis in Iran (Maghani 1977; Niak and Huttner 1977). Identification and characterization of Babesia spp. is routinely performed using Giemsa staining of blood smears in Iran. A large Babesia were identified on the blood smear of 20 naturally infected sheep in the Northwest of Iran, which were polymorphic, including double pyriform with acute or obtuse angle, single pyriform, and ring form. The size of typical paired pyriforms with acute angle was with mean of 2.7 × 0.4 μm (n = 10) and with obtuse angle was with mean of 3.5 × 0.6 μm (n = 10). The round forms were also in different size detectable. Babesia spp in small ruminant can be differentiated on the basis of morphometrical data. Soulsby (1982) described B. ovis as small Babesia being 1–2.5 μm in length (<2.5 μm), round or comparatively rare pyriform with obtuse angle occurring at the margin of the red cell. He described B. motasi as a large Babesia measuring 2.5–4 × 2 μm (>2.5 μm) with acute angle in pyriform. According to the biometrical and morphological data, the large Babesia was routinely diagnosed as a B. motasi in Iran. In contrast to our results, Lewis et al. (1981) have described a small B. motasi in Wales as double pyriform with the mean length of one side being 2.23 μm, which appeared to be morphometrically and serologically closed to the other North European B. motasi strains. Bai et al. (2002) also reported about a large Babesia, which was polymorphic, including double pyriform, single pyriform, ring form, rod-like, three leafed, and budding forms. The size of its typical paired pyriforms was 1.8–2.5 × 0.9–1.8 μm, with mean dimensions of 2.21 ± 0.12 × 1.17 ± 0.18 μm. This parasite causes a severe course of infection associated with a parasitemia as high as 23.7% and with a high mortality rate in both sheep and goats. It seems that morphometrical parameters could not be a gold standard method in the differential diagnosis of Babesia spp. This consideration was supported by the studies of Thomford et al. (1993) and Persing et al. (1995). They have detected a Babesia-like organism (WA1) and have characterized it as morphologically identical to Babesia microti but biologically and genetically distinct from it. To determine the species of large Babesia, DNA was isolated from the large and small Babesia from infected blood cells of sheep (Fig. 1 a–d). The DNA was, then, analyzed in PCR using common specific primers for Babesia spp. and Theileria spp. derived from flanking part of hypervariable region of 18S rRNA (Shayan and Rahbari 2005 and Shayan et al. 2007). The application of the mentioned primer pair (P1 and P2) could easily and simultaneously differentiate between Theileria spp. and Babeia spp. The PCR products of Theilera spp. and Babesia spp. are 426–430 and 389–402 bp, respectively. The difference of approximately 30 bp in the nucleotide sequence of the PCR products is easily detectable in 1.8% agarose gel (Shayan and Rahbari 2005). The specificity of the PCR products for B. ovis or B. motasi was performed twice, first with the seminested PCR using forward primers specific for B. ovis or B. motasi and common reverse primer and second using RFLP method. Our results showed that the DNA from all samples (small Babesia and larger Babesia) had the expected PCR product of approximately 389 bp for Babesia species (Fig. 1c). One microliter of the purified PCR product or 5 μl of isolated PCR product from agarose gel was amplified with the specific primers derived from V4 region of 18S rRNA for each species of Babesia (P3, P4), their sequence was within the first forward (P1) and reverse primer (P2). The amplification could only be detected by primers specific for B. ovis resulted in the expected 184 bp PCR product (Fig. 1d). The PCR product could not be amplified with the primers specific for B. motasi. In the second way to show that the first PCR product is specific for B. ovis, the PCR products were first purified and 20 μl of them was, then, cut with the restriction endonucleases Nde I and Mbo II. The restriction endonucleasis Nde I can recognize the sequence (CATATG) in corresponding PCR product of B. motasi and cut it at the nucleotide in the position 38, whereas Mbo II can recognize the sequence (GAAGA) in the corresponding PCR product of B. ovis and cut it at the nucleotide in the position 297. These restriction sites are specific for each mentioned Babesia species. Our results revealed that all PCR products could be cut only with the restriction endonucleasis Mbo II (Fig. 1e), which explained the specificity of the PCR product for B. ovis and confirmed the results of seminested PCR. It is widely accepted that B. motasi is a valid species and many large Babesia described in the world are considered to be this species. According to some published data, B. motasi is not pathogenic to intact sheep (Alani and Herbert 1980a, b; Uilengberg et al. 1980) and not infective to goats (Madhava 1966; Uilengberg et al. 1980). However, Lewis et al. (1981) reported successful infection of goats by inoculation with parasitized blood, and Jagannath et al. (1974) showed that B. motasi could cause disease and even death in sheep. Because we could detect a large Babesia as B. ovis, which considered wrongly as B. motasi in Iran, it was interesting to know if both forms differ in their pathogenicity. Therefore, we inoculated two groups of lambs separately with 6 × 106 small (SB group) or large (LB group) B. ovis infected erythrocytes, respectively. All animals developed fever concurrent with parasitemia that occurred at day 3 and 7 post inoculation for small Babesia and large Babesia, respectively. Maximum of parasitemia was by small Babesia 5.5% and by large Babesia 7%. Animals in both groups showed anemia. Lambs in SB group showed higher hemoglobinuria than in LB group. The count of whole white blood cells and lymphocytes was comparable in both groups. Hematocrit was higher in SB group (14%) than in LB group (10%). The lethality was significantly higher in SB group (80%) than in LB group (20%) and occurred mostly 6 to 10 days after the first sign of fever. In the control group, no clinical sign of disease was observed. Our preliminary results showed that the pathogenicity of small B. ovis was higher than large one. But it is very soon to speculate about the difference in pathogenicity of the two forms according to the limited amount of experimental materials. Therefore, the experiments with more probes from each form of B. ovis are required to confirm the above-mentioned statements. Currently, we showed B. ovis in the salivary gland of Rhipicephalus bursa, Rhipicephalus turanicus, and Rhipicephalus sanguineous using Seminested PCR, PCR-RFLP, and sequencing. B. ovis was detected in salivary gland of 18.5% Rh. bursa, 9.1% Rh.turanicus, and 8.1% Rh. sanguineus from 269 ticks (108 Rh. bursa, 87 Rh. turanicus, 74 Rh. sanguineus), respectively (Shayan et al. 2007). Interestingly, there was no B. motasi detectable in any examined ticks. In addition, all examined blood smears from infected sheep were previously diagnosed as B. motasi by Giemsa staining in our department, were characterized as B. ovis using PCR in this study. It can be concluded that despite the limited amount of examined blood samples in this study, the causative organism of many cases of Babesiosis in previous studies could be B. ovis instead of B. motasi. We believe that under this circumstances, the biometrical polymorphisms could exist within B. ovis in Iran. This polymorphism could be a main problem in differentiation between B. ovis and B. motasi by Giemsa staining, which could be dissolved by specific PCR analysis.

Blood smear from Babesia-infected sheep was stained with Giemsa. Giemsa-stained blood smear from large Babesia (a and b) and from small Babesia (c and d). DNA was isolated from the blood smear of Babesia-infected animals and analyzed by PCR. e PCR analysis with primers P1–P2 specific for 18S rRNA gene of Theileria and Babesia (lane 1 DNA from small Babesia, lane 2 DNA from large Babesia). f DNA from small Babesia was analyzed by PCR with primers P1–P2 (lane 1), the corresponding PCR product was analyzed by seminested PCR using Babesia ovis specific primers P3–P2 (lane 2). DNA from large Babesia was analyzed by PCR with primers P1–P2 (lane 3), the corresponding PCR product was analyzed by seminested PCR using B. ovis specific primers P3–P2 (lane 4). The PCR product with primers P1–P2 was cut with restriction endonucleasis Mbo II from small B. ovis (lane 1) and large B. ovis (lanes 2 and 3). Lane 4 uncut PCR product. M is 100 bp marker
References
Ahmed J, Yin H, Schnittger L, Jongejan F (2002) Ticks and tick-borne diseases in Asia with special emphasis on China. Parasitol Res 88:S51–S55
Alani AJ, Herbert IV (1980a) The pathogenesis of Babesia motasi (Wales) infection in sheep. Vet Parasitol 27:209–220
Alani AJ, Herbert IV (1980b) The morphometrics of Babesia motasi (Wales) and its transmission by Haemaphysalis punctata (Canestrni and Fanzago, 1877) to sheep. Vet Parasitol 30:87–95
Bai Q, Liu G, Liu D, Ren J, Li X (2002) Isolation and preliminary characterization of a large Babesia sp from sheep and goats in the eastern part of Gansu Province, China. Parasitol Res 88(Supplement 1):16–21
Barnett SF (1974a) Economical aspects of protozoal tick-borne diseases in livestock in parts of the world other than Britain. Bull Off Int Epiz 81(1–2):183–196
Barnett SF (1974b) Economical aspects of tick-borne disease control in Britain. Bull Off Int Epiz 81(1–2):167–182
Hashemi-Fesharki R, Uilenberg G (1981) Babesia crassa n.sp. (Sporozoa, Babesiidae) of domestic sheep in Iran. Vet Q 2:3–14
Jagannath MS, Hedge KS, Shivaram K, Nagaraja KV (1974) An outbreak of babesiosis in sheep and goats and its control. Mysor J Agric Sci 8:441–443
Jianxus L, Hong Y (1997) Theileriosis of sheep and goats in china. Trop Anim Health Peod 29(Suppl 9):8S–10S
Leemans I, Hooshmand-Rad P, Uggla A (1997) The indirect fluorescent antibody test based on schizont antigen for study of the sheep parasite Theileria lestoquardi. Vet Parasitol 69:9–18
Levine ND (1985) Veterinary protozoology. Iowa State University Press, Ames
Lewis D, Holman MR, Purnell RE, Young ER (1981) Investigations on Babesia motasi isolated from Wales. Res Vet Sci 31:239–243
Lian C, Bai Q, He H, Han G, Liu G (1997) A large new Babesia sp. found from infected sheep in China (in Chinese). Chin J Vet Sci 17:116–119
Madhava AKR (1966) Some observations on Babesia motasi infection in sheep in Andhra Pradesh. Ind Vet J 43:785–789
Maghani G (1977) The present situation of ticks and tick-borne diseases in Iran. The first Mediterranean conference on parasitology. 5–10 October, Izmir, Turkey. Summaries, pp 97–98
Mehlhorn H, Schein E (1984) The piroplasms: life cycle and sexual stages. Adv Parasitol 23:37–103
Mehlhorn H, Schein E, Ahmed JS (1994) Theileria. In: Kreier JP (ed) Parasitic protozoa, vol 7. Academic, San Diego, pp 217–304
Niak A and Huttner SH (1977)Ruminant babesiosis in Iran. The fifth International Congress Of Protozoology, New York, 26 June–2 July 1977, 417 pp.
Persing DH, Herwaldt BL, Glaser C, Lane RS, Thomford JW, Mathiesen D, Krause PJ, Phillip DF, Conrad PA (1995) Infection with a Babesia-like organism in northern California. N Engl J Med 2 332(5):298–303
Ray HN, Raghavachari K (1941) Observations on Babesia foliata n.sp. from a sheep. Ind J Vet Sci 11:239–242
Sarwar SM (1935) A hitherto undescribed piroplasm of goats (Piroplasma taylori). Ind J Vet Sci 5:171–176
Schnittger L, Yin H, Qi B, Gubbels MJ, Beyer D, Niemann S, Jongejan F, Ahmed JS (2004) Simultaneous detection and differentiation of Theileria and Babesia parasites infecting small ruminants by reverse line blotting. Parasitol Res 92(3):189–96
Shayan P, Nabian S (2008) The use of immunostaining for determination of Babesia and Theileria and gene expression of proliferation associated with nuclear protein in Theileria infected cells. Iran Vet J 3(3):21–29
Shayan P, Rahbari S (2005) Simultaneous differentiation between Theileria spp. and Babesia spp. on stained blood smear using PCR. Parasitol Res 97(4):281–286
Shayan P, Hooshmand E, Rahbari S, Nabian S (2007) Determination of Rhipicephalus spp. As vectors for Babesia ovis in Iran. Parasitol Res 101:1029–1033
Soulsby EJL (1982) Helminths, arthropods and protozoa of domesticated animals, 7th edn. Tindall, London, pp 706–728
Thomford JW, Conrad PA, Boyce WM, Holman PJ (1993) Isolation and in vitro cultivation of Babesia parasites from free-ranging desert bighorn sheep (Ovis canadensis nelsoni) and mule deer (Odocoileus hemionus) in California. J Parasitol 79(1):77–84 Feb
Uilengberg G, Rombach MC, Perie NM, Zwart D (1980) Blood parasites of sheep in the Netherlands. II. Babesia motasi (Sporozoa, Babesiidae). Vet Q 2:3–14
Wenyon CM (1926) Protozoology, vol. 3. Hafner, New York
Acknowledgment
This work was supported by Grand no. 76/9203 of University of Tehran and by the Investigating Unit Molecular Biological System Transfer (Iran).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shayan, P., Hooshmand, E., Nabian, S. et al. Biometrical and genetical characterization of large Babesia Ovis in Iran. Parasitol Res 103, 217–221 (2008). https://doi.org/10.1007/s00436-008-0960-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-0960-1