
Abstract
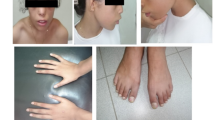
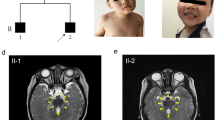
Otofaciocervical syndrome (OFCS) is an autosomal recessively inherited disorder characterized by facial dysmorphism, external ear anomalies with preauricular pits and hearing impairment, branchial cysts or fistulas, anomalies of the vertebrae and the shoulder girdle, and mild intellectual disability. In a large consanguineous family with OFCS from Turkey, we performed whole-exome sequencing (WES) of a single pooled DNA sample of four affected individuals. Filtering for variants with a percentage of alternate reads ≥90 % and a coverage of at least five reads identified only a single novel homozygous variant, c.497G>T, located in PAX1 that co-segregated with the disease in the family. PAX1 encodes a transcription factor with a critical role in pattern formation during embryogenesis in vertebrates. The mutation is predicted to substitute the glycine at position 166 to valine (p.G166V) within the highly conserved paired-box domain of the PAX1 protein. We performed a dual luciferase reporter assay to examine the transactivation of a regulatory sequence in the Nkx3-2 promoter region, which is a direct target of mouse Pax1 transcriptional regulation. We observed a significantly reduced transactivation in HEK293T cells overexpressing Pax1G157V in comparison to Pax1WT expressing cells, indicating a reduced DNA-binding affinity of the mutant protein. Taken together, our results show that the strategy of pooling DNA is a powerful, cost-effective application for WES in consanguineous families and establish PAX1 as a new disease-causing gene for OFCS and as part of the EYA-DACH-SIX-PAX network, important in early embryogenesis.




Similar content being viewed by others

References
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Levi-Acobas F, Cruaud C, Le Merrer M, Mathieu M, Konig R, Vigneron J, Weissenbach J, Petit C, Weil D (1997a) Clustering of mutations responsible for branchio-oto-renal (BOR) syndrome in the eyes absent homologous region (eyaHR) of EYA1. Hum Mol Genet 6:2247–2255
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C (1997b) A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet 15:157–164
Balling R (1994) The undulated mouse and the development of the vertebral column. Is there a human PAX-1 homologue? Clin Dysmorphol 3:185–191
Bannykh SI, Emery SC, Gerber JK, Jones KL, Benirschke K, Masliah E (2003) Aberrant Pax1 and Pax9 expression in Jarcho–Levin syndrome: report of two Caucasian siblings and literature review. Am J Med Genet A 120A:241–246
Bopp D, Burri M, Baumgartner S, Frigerio G, Noll M (1986) Conservation of a large protein domain in the segmentation gene paired and in functionally related genes of Drosophila. Cell 47:1033–1040
Bopp D, Jamet E, Baumgartner S, Burri M, Noll M (1989) Isolation of two tissue-specific Drosophila paired box genes, Pox meso and Pox neuro. EMBO J 8:3447–3457
Burri M, Tromvoukis Y, Bopp D, Frigerio G, Noll M (1989) Conservation of the paired domain in metazoans and its structure in three isolated human genes. EMBO J 8:1183–1190
Chalepakis G, Fritsch R, Fickenscher H, Deutsch U, Goulding M, Gruss P (1991) The molecular basis of the undulated/Pax-1 mutation. Cell 66:873–884
Chi N, Epstein JA (2002) Getting your Pax straight: Pax proteins in development and disease. Trends Genet 18:41–47
Cote S, Preiss A, Haller J, Schuh R, Kienlin A, Seifert E, Jackle H (1987) The gooseberry-zipper region of Drosophila: five genes encode different spatially restricted transcripts in the embryo. EMBO J 6:2793–2801
Dallapiccola B, Mingarelli R (1995) Otofaciocervical syndrome: a sporadic patient supports splitting from the branchio-oto-renal syndrome. J Med Genet 32:816–818
Deutsch U, Dressler GR, Gruss P (1988) Pax 1, a member of a paired box homologous murine gene family, is expressed in segmented structures during development. Cell 53:617–625
Estefania E, Ramirez-Camacho R, Gomar M, Trinidad A, Arellano B, Garcia-Berrocal JR, Verdaguer JM, Vilches C (2005) Point mutation of an EYA1-gene splice site in a patient with oto-facio-cervical syndrome. Ann Hum Genet 70:140–144
Fara M, Chlupackova V, Hrivnakova J (1967) Familial oto-facio-cervical dysmorphia. Acta Chir Orthop Traumatol Cech 34:511–520
Fischer S, Ludecke HJ, Wieczorek D, Bohringer S, Gillessen-Kaesbach G, Horsthemke B (2006) Histone acetylation dependent allelic expression imbalance of BAPX1 in patients with the oculo-auriculo-vertebral spectrum. Hum Mol Genet 15:581–587
Hol FA, Geurds MP, Chatkupt S, Shugart YY, Balling R, Schrander-Stumpel CT, Johnson WG, Hamel BC, Mariman EC (1996) PAX genes and human neural tube defects: an amino acid substitution in PAX1 in a patient with spina bifida. J Med Genet 33:655–660
Kaiser R, Posteguillo EG, Muller D, Just W (2007) Exclusion of genes from the EYA-DACH-SIX-PAX pathway as candidates for Branchio-Oculo-Facial syndrome (BOFS). Am J Med Genet A 143A:2185–2188
Lang D, Powell SK, Plummer RS, Young KP, Ruggeri BA (2007) PAX genes: roles in development, pathophysiology, and cancer. Biochem Pharmacol 73:1–14
McGaughran JM, Oates A, Donnai D, Read AP, Tassabehji M (2003) Mutations in PAX1 may be associated with Klippel-Feil syndrome. Eur J Hum Genet 11:468–474
Noll M (1993) Evolution and role of Pax genes. Curr Opin Genet Dev 3:595–605
Read AP, van Heyningen V (1994) PAX genes in human developmental anomalies. Semin Dev Biol 5:323–332
Rickard S, Boxer M, Trompeter R, Bitner-Glindzicz M (2000) Importance of clinical evaluation and molecular testing in branchio-oto-renal (BOR) syndrome and overlapping phenotypes. J Med Genet 37:623–627
Rickard S, Parker M, van‘t Hoff W, Barnicoat A, Russell-Eggitt I, Winter RM, Bitner-Glindzicz M (2001) Oto-facio-cervical (OFC) syndrome is a contiguous gene deletion syndrome involving EYA1: molecular analysis confirms allelism with BOR syndrome and further narrows the Duane syndrome critical region to 1 cM. Hum Genet 108:398–403
Rodrigo I, Hill RE, Balling R, Munsterberg A, Imai K (2003) Pax1 and Pax9 activate Bapx1 to induce chondrogenic differentiation in the sclerotome. Development 130:473–482
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM Jr, Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ, Stratakis CA, Weil D, Petit C, Hildebrandt F (2004) SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA 101:8090–8095
Schnittger S, Rao VV, Deutsch U, Gruss P, Balling R, Hansmann I (1992) Pax1, a member of the paired box-containing class of developmental control genes, is mapped to human chromosome 20p11.2 by in situ hybridization (ISH and FISH). Genomics 14:740–744
Treisman J, Harris E, Desplan C (1991) The paired box encodes a second DNA-binding domain in the paired homeo domain protein. Genes Dev 5:594–604
Wallin J, Wilting J, Koseki H, Fritsch R, Christ B, Balling R (1994) The role of Pax-1 in axial skeleton development. Development 120:1109–1121
Wallin J, Eibel H, Neubuser A, Wilting J, Koseki H, Balling R (1996) Pax1 is expressed during development of the thymus epithelium and is required for normal T-cell maturation. Development 122:23–30
Wilm B, Dahl E, Peters H, Balling R, Imai K (1998) Targeted disruption of Pax1 defines its null phenotype and proves haploinsufficiency. Proc Natl Acad Sci USA 95:8692–8697
Acknowledgments
We are grateful to all family members who participated in this study, to Esther Milz for excellent technical assistance, to Andrea Pannes for her technical advice in slow-down PCR, to Simon von Ameln for cochlea preparation, and to Karin Boss for critically reading the manuscript. This work was supported by the German Federal Ministry of Education and Research (BMBF) by grant number 01GM1211A (E-RARE network CRANIRARE-2) to B.W.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pohl, E., Aykut, A., Beleggia, F. et al. A hypofunctional PAX1 mutation causes autosomal recessively inherited otofaciocervical syndrome. Hum Genet 132, 1311–1320 (2013). https://doi.org/10.1007/s00439-013-1337-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-013-1337-9