
Abstract
Osteocytes establish an extensive intracellular and extracellular communication system via gap-junction-coupled cell processes and canaliculi throughout bone and the communication system is extended to osteoblasts on the bone surface. The osteocyte network is an ideal mechanosensory system and suitable for mechanotransduction. However, the overall function of the osteocyte network remains to be clarified, since bone resorption is enhanced by osteocyte apoptosis, which is followed by a process of secondary necrosis attributable to the lack of scavengers. The enhanced bone resorption is caused by the release of intracellular content, including immunostimulatory molecules that activate osteoclastogenesis through the canaliculi. Therefore, a mouse model is required in which the osteocyte network is disrupted but in which no bone resorption is induced, in order to evaluate the overall functions of the osteocyte network. One such model is the BCL2 transgenic mouse, in which the osteocyte network, including both intracellular and extracellular networks, is disrupted. Another model is the osteocyte-specific Gja1 knockout mouse, in which intercellular communication through gap junctions is impaired but the canalicular system is intact. Combining the findings from these mouse models with previous histological observations showing the inverse linkage between osteocyte density and bone formation, we conclude that the osteocyte network enhances bone resorption and inhibits bone formation under physiological conditions. Further, studies with BCL2 transgenic mice show that these osteocyte functions are augmented in the unloaded condition. In this condition, Rankl upregulation in osteoblasts and Sost upregulation in osteocytes are, at least in part, responsible for enhanced bone resorption and suppressed bone formation, respectively.
Similar content being viewed by others
Introduction
Osteocytes, which are embedded in the bone matrix, establish an extensive intracellular and extracellular communication system via gap-junction-coupled cell processes and canaliculi through which cell processes pass throughout the bone and the communication system is extended to osteoblasts on the bone surface (Marks and Odgren 2002). Bone tissue is able to adapt its mass and three-dimensional structure to the prevailing mechanical usage to achieve higher load-bearing efficiency (Wolff 1892). The lacunocanalicular network formed by osteocytes is thought to be an ideal mechanosensory system and suitable for mechanotransduction by which mechanical energy is converted into electrical and/or biochemical signals (Burger and Klein-Nulend 1999; Martin 2000; Ehrlich and Lanyon 2002; Knothe Tate 2003; Bonewald and Johnson 2008; Noble 2008). However, the special anatomic features of osteocytes, which are isolated in lacunae but are three-dimensionally connected to each other, make it difficult to study their function. This review focuses on the mechanism of osteocyte apoptosis-induced bone resorption and functions of the osteocyte network in the regulation of bone mass under physiological and unloaded conditions.
Osteocyte death and bone remodeling
Osteocyte death occurs during aging, after menopause, at unloading and under pathological conditions such as microcracks and the death of osteocytes is closely coupled with bone resorption (Noble and Reeve 2000; Cardoso et al. 2009; Emerton et al. 2010). Cells die mainly through one of three pathways, i.e., apoptosis, autophagic cell death and necrosis, under physiological and pathological conditions, although dying cells can share apoptotic and autophagic features (Bursch 2001; Fig. 1). If cells die through apoptosis or autophagy, death is completed with the removal of the cells through engulfment by scavengers. In these cases, the cells are quietly removed without inflammation, because the integrity of the cytoplasmic membranes is maintained when phagocytosis occurs. In contrast, strong insults cause bioenergetic failure and rapid loss of the integrity of the cytoplasmic membrane, both of which are core events of necrosis. Necrosis leads to the rupture of the cytoplasmic membrane and most of the intracellular content is released into the extracellular environment. The released intracellular content encompasses immunostimulatory molecules including the so-called damage-associated molecular pattern molecules, such as S100 family molecules, high-mobility group box 1 protein, purine metabolites, heat-shock proteins and uric acid (Lotze and Tracey 2005; Zong and Thompson 2006). The release of the immunostimulatory molecules actively recruits a defensive or a reparative response to the damaged regions. If cells at the terminal phase of apoptosis or autophagic cell death are not engulfed by phagocytes, a transition to necrosis ensues and the cells are eliminated by cell disruption. This process is called secondary necrosis (Majno and Joris 1995; Silva et al. 2008). As scavengers cannot reach osteocytes, any type of osteocyte death will end in the rupture of the cytoplasmic membrane. After cell rupture, immunostimulatory molecules are released from lacunae through canaliculi to the bone surface and vascular channels and facilitate the recruitment and activation of macrophages, thereby promoting the production of proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-6 and IL-1 (Lotze and Tracey 2005). Further, IL-6 and IL-1 induce receptor activator of nuclear factor κ-B ligand (Rankl) expression, which is essential for osteoclastogenesis (Yasuda et al. 1998; Kong et al. 1999; O’Brien 2010). As TNF-α, IL-6 and IL-1 are the most important proinflammatory cytokines triggering inflammatory bone loss (Firestein 2003), primary and secondary osteocyte necrosis could lead to the enhancement of osteoclastogenesis and bone resorption, which comprise a repair process in bone (Fig. 1).

Osteocyte death and bone resorption. Cells die mainly through one of three pathways: apoptosis, autophagic cell death and necrosis. As osteocytes at the terminal phase of apoptosis or autophagic cell death are not engulfed by phagocytes, the process of secondary necrosis ensues. After cell rupture, immunostimulatory molecules including high-mobility group box 1 (HMGB1), S100, heat shock protein (HSP), adenosine triphosphate (ATP) and uric acid are released from lacunae through canaliculi to the bone surface and vascular channels, where they facilitate the recruitment and activation of macrophages, thereby promoting the production of proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6) and interleukin 1 (IL-1), which induce osteoclastogenesis and bone resorption. Apoptotic osteocytes release osteoclastogenesis-promoting factors, including soluble receptor activator of nuclear factor κ-B ligand (sRankl), IL-6 and soluble IL-6 receptor (sIL-6R) and induce TNF-α expression in osteoclast precursors in vitro
Apoptotic osteocytes show enhancement of osteoclastogenesis in vitro (Kogianni et al. 2008; Al-Dujaili et al. 2011; Cheung et al. 2012; Shandala et al. 2012). Addition of apoptotic bodies from osteocytes but not osteoblasts induces osteoclastogenesis through the induction of TNF-α production by mononuclear osteoclast precursors (Kogianni et al. 2008). Conditioned medium from apoptotic osteocytic cell line MLO-Y4 contains more soluble Rankl than that from non-apoptotic MLO-Y4 cells and induces osteoclastogenesis (Al-Dujaili et al. 2011). Methotrexate (MTX)-induced apoptosis of MLO-Y4 cells increases IL-6 and IL-11 expression and the conditioned media promote osteoclastogenesis (Shandala et al. 2012). Further, apoptotic MLO-Y4-conditioned media promote osteoclast precursor adhesion to endothelial cells via intercellular adhesion molecule 1 (ICAM-1), probably through the induction of IL-6 and soluble IL-6 receptor (Cheung et al. 2012). Therefore, apoptotic osteocytes can upregulate the expression of osteoclastogenesis-promoting factors (Fig. 1).
Another special feature of osteocyte death is the sustained detection of degraded DNA by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL; Weinstein et al. 2000; Moriishi et al. 2011, 2012). This indicates that a part of the DNA degraded during the process of apoptosis is retained in the lacunae, even after secondary necrosis. Even during primary necrosis, randomly degraded DNA retained in the lacunae can be detected by TUNEL. The reactivity of TUNEL in the lacunae is retained until the bone, including the lacunae, is resorbed.
Functions of osteocytes in bone resorption
As the death of osteocytes induces bone resorption, the physiological function of osteocytes has been considered to be the inhibition of bone resorption (Gu et al. 2005; Tatsumi et al. 2007). A typical example is osteocyte ablation by diphtheria toxin (Tatsumi et al. 2007). Transgenic mice expressing diphtheria toxin receptor under the control of the dentin matrix protein 1 (Dmp1) promoter show enhanced bone resorption resulting in severe osteoporosis after the injection of diphtheria toxin. Dmp1 expression begins in osteoblasts when they are about to be embedded into the bone matrix and Dmp1 expression is high in immature osteocytes but decreases during osteocyte maturation (Toyosawa et al. 2001; Xiong et al. 2011). After primary or secondary necrosis caused by diphtheria toxin, the intracellular content of osteocytes is released from the lacunae onto the bone surface and vascular channels and promotes osteoclastogenesis as a process of repair (Fig. 1). This seems to be the case, because Rankl is highly induced after the injection of diphtheria toxin (Tatsumi et al. 2007). Although osteocyte death causes bone resorption, whether live osteocytes inhibit bone resorption remains unclear.
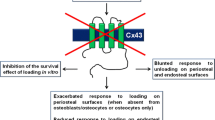
Gja1 (connexin 43) is the major gap junction protein and plays important roles in osteogenesis and osteoblast function (Lecanda et al. 2000; Chung et al. 2006; Watkins et al. 2011). Targeted deletion of Gja1 in osteocytes results in the disruption of the intercellular communication of osteocytes through gap junctions, while retaining communication through the canalicular network (Bivi et al. 2012). In this mouse model, osteocyte apoptosis is increased, the osteoclast number and surface are increased at the endocortical surface and the marrow cavity is enlarged. This indicates that osteocyte apoptosis in conditional Gja1 knockout mice could induce bone resorption, probably because the intracellular content of dead osteocytes could be released through the intact canalicular network and trigger osteoclastogenesis and bone resorption (Fig. 1). The abundance of osteoclast precursor cells and immune cells in bone marrow should facilitate bone resorption at the endocortical surface after osteocyte apoptosis.
Another model of dysfunction of the osteocyte network is the osteoblast-specific BCL2 transgenic mouse. BCL2 transgenic mice have a reduced number of osteocyte processes probably because of the function of BCL2 to alter cytoskeletal organization; the osteocytes gradually die by apoptosis and the frequency of TUNEL-positive lacunae reaches 80% at 4 months of age (Moriishi et al. 2011, 2012). However, bone resorption is not induced by osteocyte apoptosis in BCL2 transgenic mice and both the intracellular and extracellular osteocyte networks are disrupted at 4 months of age by the reduced number of canaliculi and the accumulation of TUNEL-positive lacunae. The absence of enhanced bone resorption in BCL2 transgenic mice seems to be attributable to the failure of canalicular (extracellular) network formation, which interrupts the release of the intracellular content of dead osteocytes onto the bone surface and into vascular channels. Contrary to many previous observations that osteocyte apoptosis enhances bone resorption, the number of osteoclasts and bone resorption are reduced in BCL2 transgenic mice. Interestingly, the number of osteoclasts is reduced on the endocortical surface but not on the periosteal surface in cortical bone, suggesting that the osteocyte network plays a major role in regulating osteoclastogenesis on the endocortical surface of cortical bone. These findings indicate that the disruption of both the intercellular and extracellular communication systems of osteocytes suppresses osteoclastogenesis and bone resorption. As BCL2 overexpression in osteoblasts has no effect on osteoclastogenesis and bone resorption (Moriishi et al. 2011, 2012), we conclude that the osteocyte network enhances osteoclastogenesis and bone resorption under physiological conditions (Fig. 2).

Regulation of bone mass by the osteocyte network. a Regulation of bone mass under physiological conditions. The osteocyte network mildly stimulates osteoclastogenesis and bone resorption and mildly inhibits osteoblast function and bone formation. Rankl expression in osteoblasts and sclerostin (Sost) expression in osteocytes are, at least in part, responsible for the enhanced bone resorption and suppressed bone formation, respectively. Both Rankl and osteoprotegerin (Opg) are highly expressed in osteocytes and Opg seems to be trapped by Rankl on the surface of osteocytes. sRankl secreted by osteocytes might be involved in the regulation of osteoclastogenesis (dashed arrow). b Regulation of bone mass in the unloaded condition. The osteocyte network strongly stimulates osteoclastogenesis and bone resorption and strongly inhibits osteoblast function and bone formation. The osteocyte network enhances Rankl expression in osteoblasts and Sost expression in osteocytes in the unloaded condition. The signals from osteocytes that stimulate Rankl expression in osteoblasts remain to be identified. Upregulation of Rankl in osteocytes in the unloaded condition is controversial. In a, b, the thickness of the lines and arrows reflects the strength of the effects and the height of the osteoblasts reflect their activities
Osteocytes highly express Rankl compared with osteoblasts (Nakashima et al. 2011; Xiong et al. 2011; Moriishi et al. 2012). Conditional Rankl deletion by using Dmp1 Cre transgenic mice results in increased bone mass (Nakashima et al. 2011; Xiong et al. 2011). Rankl is produced as a membrane-bound or soluble form, although the membrane-bound form is much more efficient in terms of osteoclast formation than the soluble form (Lum et al. 1999; Nakashima et al. 2000; Ikeda et al. 2001; Hikita et al. 2006). The production of soluble Rankl (sRankl) in MLO-Y4 cells is controversial; one study has shown that the conditioned media from MLO-Y4 cells support osteoclastogenesis, whereas another has demonstrated that conditioned media fail to support osteoclastogenesis (Zhao et al. 2002; Al-Dujaili et al. 2011). As sRankl is detected in the circulatory system of humans (Schett et al. 2004; Trofimov et al. 2004; Rogers and Eastell 2005; Ziegler et al. 2005), the physiological importance of sRankl produced by osteocytes needs to be investigated, because the direct interaction of osteocytes with osteoclasts and their precursors will be limited (Fig. 2). The secreted factor osteoprotegerin (Opg), which is produced in osteocytes, might also be released onto the bone surface and vascular channel through canaliculi without being trapped by the Rankl on the surface of osteocytes in the Rankl conditional knockout mice, because Opg is also highly expressed in osteocytes (Moriishi et al. 2012). Under physiological conditions, most of the Opg produced by osteocytes seem to be trapped by Rankl on the surface of osteocytes, because the osteocyte network enhances osteoclastogenesis under physiological conditions (Moriishi et al. 2012; Fig. 2).
Functions of osteocytes in bone formation
The function of osteocytes in bone formation under physiological conditions has been controversial. Ablation of osteocytes by diphtheria toxin severely reduces bone formation, indicating that osteocyte death also affects bone formation. Reduced bone formation also seems to be caused by secondary necrosis-induced inflammation, because osteoblast maturation is severely inhibited in diphtheria toxin receptor transgenic mice (Tatsumi et al. 2007). In contrast, osteocyte density is negatively correlated with bone formation and both the empty lacunar density and periosteal bone apposition increase with age, suggesting a linkage between the two phenomena (Lazenby 1990; Qiu et al. 2002; Metz et al. 2003; Russo et al. 2006; Hedgecock et al. 2007). Mice carrying a targeted mutation of Col1a1, encoding a collagenase-resistant form of type I collagen, exhibit osteocyte apoptosis and increased bone formation (Zhao et al. 2000). Further, enhanced bone formation at the periosteal and endocortical surfaces is localized in the area in which viable osteocytes are lost in osteocyte-specific Gja1 knockout mice (Bivi et al. 2012). In a study of BCL2 transgenic mice at 4 months of age, bone formation in both trabecular and cortical bones was enhanced when the osteocyte network was disrupted, although the contribution of BCL2 overexpression in osteoblasts to bone formation could not be completely excluded (Moriishi et al. 2012). Combining the findings from these mouse models with the histological observations showing the inverse linkage between osteocyte density and bone formation, we conclude that the osteocyte network suppresses bone formation under physiological conditions (Fig. 2).
Sost (sclerostin) is specifically expressed in osteocytes and inhibits osteoblast function and bone formation by antagonizing canonical Wnt signaling through binding to Wnt co-receptor low density lipoprotein receptor-related protein (LRP) 5 and LRP6, and bone formation is enhanced in trabecular bone and on the endocortical and periosteal surfaces of cortical bone in Sost-deficient mice (Winkler et al. 2003; van Bezooijen et al. 2004, 2007; Bellido et al. 2005; Li et al. 2005, 2008; Poole et al. 2005; Semenov et al. 2005). Further, osteocyte-specific deletion of the parathyroid hormone (PTH) receptor increases Sost expression and the specific expression of constitutively active PTH receptor in osteocytes reduces it, indicating the involvement of Sost in the anabolic action of PTH (Powell et al. 2011; Rhee et al. 2011). Therefore, Sost is a major negative regulator of osteoblast differentiation and function produced by osteocytes. As the dissemination of Sost through canaliculi is impaired in BCL2 transgenic mice, impaired secretion to the bone surface should greatly contribute to enhanced bone formation (Moriishi et al. 2012). However, bone formation is enhanced in trabecular bone and on the periosteal surface but not on the endocortical surface of cortical bone in BCL2 transgenic mice (Moriishi et al. 2012). Thus, the osteocyte network might send some positive signals to osteoblasts on the endocortical surface, in addition to the negative signal by Sost.
Regulation of bone mass by mechanical stress
Application of mechanical stimuli to osteocytes in vitro induces the release of anabolic factors, such as prostaglandin E2, prostaglandin I2, nitric oxide, cyclooxygenase-2 and endothelial nitric oxide synthase (Burger and Klein-Nulend 1999). Further, it is widely accepted that mechanical stress increases bone mass by enhancing bone formation and inhibiting bone resorption, whereas unloading reduces bone mass by inhibiting bone formation and enhancing bone resorption and that the osteocyte network is an appropriate system for the sensing apparatus (Burger and Klein-Nulend 1999; Martin 2000; Ehrlich and Lanyon 2002; Knothe Tate 2003; Noble 2008; Bonewald and Johnson 2008). However, difficulties have been experienced in establishing that the osteocyte network is responsible for the regulation of bone mass by mechanical stress, because osteocyte death provokes bone resorption.
Bone-specific Gja1 conditional knockout mice, in which intercellular communication through gap junctions is disrupted, represent a potential mouse model for evaluating whether the osteocyte network acts as the mechanosensing apparatus. However, the responses to mechanical stress in the Gja1 conditional knockout mice are variable. In the unloaded condition following hind limb muscle paralysis, bone resorption is enhanced on the endocortical surface of tibiae in wild-type mice but not in the conditional Gja1 knockout mice, in which Gja1 is deleted in osteoblasts and osteocytes using 2.3-kb Col1a1 promoter Cre transgenic mice (Grimston et al. 2011). However, the finding that bone resorption on the endocortical surface of the conditional Gja1 knockout mice is enhanced under physiological conditions makes the evaluation difficult. Two groups have reported the response to mechanical stress in Gja1 conditional knockout mice, in which Gja1 is deleted in mature osteoblasts and osteocytes using human osteocalcin promoter Cre transgenic mice. Zhang et al. (2011) have shown that periosteal bone formation is enhanced by mechanical loading in the conditional Gja1 knockout mice but not in wild-type mice, whereas Lloyd et al. (2012) have demonstrated that bone formation on both endocortical and periosteal surfaces is decreased in wild-type mice but not in Gja1 conditional knockout mice at unloading. As hemichannels and gap junctions, both of which are formed by Gja1, play important roles in the response to mechanical stress in osteocytes in vitro (Cherian et al. 2005; Batra et al. 2012), we need to evaluate the physiological importance of Gja1 in the response to mechanical stress by using osteocyte-specific Gja1 conditional knockout mice.
In the unloaded condition, bone resorption is enhanced and bone formation is mildly suppressed in trabecular bone of wild-type mice but not of BCL2 transgenic mice at 4 months of age, indicating that the osteocyte network is responsible for bone loss at unloading (Moriishi et al. 2012). Rankl is upregulated in osteoblasts of wild-type mice but not of BCL2 transgenic mice at unloading (Moriishi et al. 2012). Therefore, the osteocyte network is required for the induction of Rankl in osteoblasts at unloading. Although Rankl upregulation in osteocytes in the unloaded condition might increase bone resorption by reducing Opg release from osteocytes to the bone surface, the upregulation of Rankl in osteocytes by unloading is inconsistently observed (Xiong et al. 2011; Moriishi et al. 2012).
The reduction in bone formation in response to unloading is abrogated in Sost -/- mice and the level of Sost mRNA but not Sost-positive osteocytes is increased after unloading (Robling et al. 2008; Lin et al. 2009). Further, the downregulation of Sost after loading is dependent on the site in the tibiae (Moustafa et al. 2011). In hind limb unloading by tail suspension, Sost-positive cells are regionally increased in wild-type mice but not in BCL2 transgenic mice at 4 months of age (Moriishi et al. 2012). Therefore, the osteocyte network is required for the upregulation of Sost expression at unloading, which is, at least in part, responsible for the reduction of bone formation in the unloaded condition.
Many other molecules, including PTH, estrogen receptor α, androgen receptor, leptin, periostin, β1 integrin and IL-11, are also involved in the response to mechanical loading (Price et al. 2011). Loading above physiological conditions activates anabolic pathways and suppresses anti-anabolic pathways for bone formation and makes the overall function of osteocytes more anabolic. Recently, we have shown that the mRNA expression of pyruvate dehydrogenase kinase 4 (Pdk4), which inactivates the pyruvate dehydrogenase complex, is upregulated in osteoblasts and osteocytes in the unloaded condition in wild-type mice but not in BCL2 transgenic mice at 4 months of age and that bone resorption is not enhanced in the unloaded condition in Pdk4-deficient mice. Thus, Pdk4 is involved in a catabolic pathway in the unloaded condition (Wang et al. 2012).
Concluding remarks
Gene targeting experiments, especially the generation of osteocyte-specific conditional knockout mice, are revealing the importance of osteocytes in bone modeling and remodeling. Although these experiments have uncovered the function of individual genes in osteocytes, the overall function of the osteocyte network remains to be clarified, because osteocyte death causes apoptosis- or necrosis-induced bone resorption. Indeed, an ideal mouse model for the evaluation of the overall function of the osteocyte network seems to be an impossibility. Taking into consideration recent findings from osteocyte-network-disrupted mouse models with previous histological observations, however, the overall functions of the osteocyte network are now evident. The osteocyte network enhances bone resorption by activating osteoclastogenesis but inhibits bone formation by suppressing osteoblast function under physiological conditions. In the unloaded condition, the functions of the osteocyte network are augmented and bone resorption is further enhanced and bone formation is further inhibited. These functions of the osteocyte network also explain the increases in bone mass seen after exercise in many clinical studies (Schwab and Klein 2008), because the osteocyte network decreases the inhibitory effects on bone mass by reducing the stimulatory effect on osteoclastogenesis and the inhibitory effect on osteoblast function in the loaded condition (under physiological conditions) compared with the unloaded condition.
References
Al-Dujaili SA, Lau E, Al-Dujaili H, Tsang K, Guenther A, You L (2011) Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem 112:2412–2423
Batra N, Burra S, Siller-Jackson AJ, Gu S, Xia X, Weber GF, DeSimone D, Bonewald LF, Lafer EM, Sprague E, Schwartz MA, Jiang JX (2012) Mechanical stress-activated integrin α5β1 induces opening of connexin 43 hemichannels. Proc Natl Acad Sci USA 109:3359–3364
Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146:4577–4583
Bivi N, Condon KW, Allen MR, Farlow N, Passeri G, Brun LR, Rhee Y, Bellido T, Plotkin LI (2012) Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res 27:374–389
Bonewald LF, Johnson ML (2008) Osteocytes, mechanosensing and Wnt signaling. Bone 42:606–615
Burger EH, Klein-Nulend J (1999) Mechanotransduction in bone—role of the lacuno-canalicular network. FASEB J 13:S101–S112
Bursch W (2001) The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ 8:569–581
Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB (2009) Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res 24:597–605
Cherian PP, Siller-Jackson AJ, Gu S, Wang X, Bonewald LF, Sprague E, Jiang JX (2005) Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol Biol Cell 16:3100–3106
Cheung WY, Simmons CA, You L (2012) Osteocyte apoptosis regulates osteoclast precursor adhesion via osteocytic IL-6 secretion and endothelial ICAM-1 expression. Bone 50:104–110
Chung DJ, Castro CH, Watkins M, Stains JP, Chung MY, Szejnfeld VL, Willecke K, Theis M, Civitelli R (2006) Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin 43. J Cell Sci 119:4187–4198
Ehrlich PJ, Lanyon LE (2002) Mechanical strain and bone cell function: a review. Osteoporos Int 13:688–700
Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, Jepsen KJ, Schaffler MB (2010) Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone 46:577–583
Firestein GS (2003) Evolving concepts of rheumatoid arthritis. Nature (Lond) 423:356–361
Grimston SK, Goldberg DB, Watkins M, Brodt MD, Silva MJ, Civitelli R (2011) Connexin 43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J Bone Miner Res 26:2151–2160
Gu G, Mulari M, Peng Z, Hentunen TA, Vaananen HK (2005) Death of osteocytes turns off the inhibition of osteoclasts and triggers local bone resorption. Biochem Biophys Res Commun 335:1095–1101
Hedgecock NL, Hadi T, Chen AA, Curtiss SB, Martin RB, Hazelwood SJ (2007) Quantitative regional associations between remodeling, modeling, and osteocyte apoptosis and density in rabbit tibial midshafts. Bone 40:627–637
Hikita A, Yana I, Wakeyama H, Nakamura M, Kadono Y, Oshima Y, Nakamura K, Seiki M, Tanaka S (2006) Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-κB ligand. J Biol Chem 281:36846–36855
Ikeda T, Kasai M, Utsuyama M, Hirokawa K (2001) Determination of three isoforms of the receptor activator of nuclear factor-κB ligand and their differential expression in bone and thymus. Endocrinology 142:1419–1426
Knothe Tate ML (2003) “Whither flows the fluid in bone?” An osteocyte’s perspective. J Biomech 36:1409–1424
Kogianni G, Mann V, Noble BS (2008) Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res 23:915–927
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature (Lond) 397:315–323
Lazenby RA (1990) Continuing periosteal apposition. II. The significance of peak bone mass, strain equilibrium, and age-related activity differentials for mechanical compensation in human tubular bones. Am J Phys Anthropol 82:473–484
Lecanda F, Warlow PM, Sheikh S, Furlan F, Steinberg TH, Civitelli R (2000) Connexin 43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J Cell Biol 151:931–944
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 23:860–869
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D (2005) Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 280:19883–19887
Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, Li Y, Feng G, Gao X, He L (2009) Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/β-catenin signaling. J Bone Miner Res 24:1651–1661
Lloyd SA, Lewis GS, Zhang Y, Paul EM, Donahue HJ (2012) Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. J Bone Miner Res 27:2359–2372
Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5:331–342
Lum L, Wong BR, Josien R, Becherer JD, Erdjument-Bromage H, Schlondorff J, Tempst P, Choi Y, Blobel CP (1999) Evidence for a role of a tumor necrosis factor-α (TNF-α)-converting enzyme-like protease in shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dendritic cell survival. J Biol Chem 274:13613–13618
Majno G, Joris I (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146:3–15
Marks SC Jr, Odgren PR (2002) The structure and development of the skeleton. In: Bilezikian JP, Raisz LG, Rodan GA (eds) Principles of bone biology. Academic Press, New York, pp 3–15
Martin RB (2000) Does osteocyte formation cause the nonlinear refilling of osteons? Bone 26:71–78
Metz LN, Martin RB, Turner AS (2003) Histomorphometric analysis of the effects of osteocyte density on osteonal morphology and remodeling. Bone 33:753–759
Moriishi T, Fukuyama R, Ito M, Miyazaki T, Maeno T, Kawai Y, Komori H, Komori T (2012) Osteocyte network; a negative regulatory system for bone mass augmented by the induction of rankl in osteoblasts and sost in osteocytes at unloading. PLoS One 7:e40143
Moriishi T, Maruyama Z, Fukuyama R, Ito M, Miyazaki T, Kitaura H, Ohnishi H, Furuichi T, Kawai Y, Masuyama R, Komori H, Takada K, Kawaguchi H, Komori T (2011) Overexpression of Bcl2 in osteoblasts inhibits osteoblast differentiation and induces osteocyte apoptosis. PLoS One 6:e27487
Moustafa A, Sugiyama T, Prasad J, Zaman G, Gross TS, Lanyon LE, Price JS (2011) Mechanical loading-related changes in osteocyte sclerostin expression in mice are more closely associated with the subsequent osteogenic response than the peak strains engendered. Osteoporos Int 23:1225–1234
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17:1231–1234
Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H (2000) Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-κB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun 275:768–775
Noble B (2008) The osteocyte lineage. Arch Biochem Biophys 473:106–111
Noble BS, Reeve J (2000) Osteocyte function, osteocyte death and bone fracture resistance. Mol Cell Endocrinol 159:7–13
O’Brien CA (2010) Control of RANKL gene expression. Bone 46:911–919
Poole KE, Bezooijen RL van, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19:1842–1844
Powell WF Jr, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, Pajevic PD (2011) Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol 209:21–32
Price JS, Sugiyama T, Galea GL, Meakin LB, Sunters A, Lanyon LE (2011) Role of endocrine and paracrine factors in the adaptation of bone to mechanical loading. Curr Osteoporos Rep 9:76–82
Qiu S, Rao DS, Palnitkar S, Parfitt AM (2002) Relationships between osteocyte density and bone formation rate in human cancellous bone. Bone 31:709–711
Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, Olivos N, Passeri G, O’Brien CA, Bivi N, Plotkin LI, Bellido T (2011) PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J Bone Miner Res 26:1035–1046
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283:5866–5875
Rogers A, Eastell R (2005) Circulating osteoprotegerin and receptor activator for nuclear factor κB ligand: clinical utility in metabolic bone disease assessment. J Clin Endocrinol Metab 90:6323–6331
Russo C, Lauretani F, Seeman E, Bartali B, Bandinelli S, Di lorio A, Guralnik J, Ferrucci L (2006) Structural adaptations to bone loss in aging men and women. Bone 38:112–118
Schett G, Kiechl S, Redlich K, Oberhollenzer F, Weger S, Egger G, Mayr A, Jocher J, Xu Q, Pietschmann P, Teitelbaum S, Smolen J, Willeit J (2004) Soluble RANKL and risk of nontraumatic fracture. JAMA 291:1108–1113
Schwab P, Klein RF (2008) Nonpharmacological approaches to improve bone health and reduce osteoporosis. Curr Opin Rheumatol 20:213–217
Semenov M, Tamai K, He X (2005) SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem 280:26770–26775
Shandala T, Shen Ng Y, Hopwood B, Yip YC, Foster BK, Xian CJ (2012) The role of osteocyte apoptosis in cancer chemotherapy-induced bone loss. J Cell Physiol 227:2889–2897
Silva MT, do Vale A, dos Santos NM (2008) Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis 13:463–482
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K (2007) Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab 5:464–475
Toyosawa S, Shintani S, Fujiwara T, Ooshima T, Sato A, Ijuhin N, Komori T (2001) Dentin matrix protein 1 is predominantly expressed in chicken and rat osteocytes but not in osteoblasts. J Bone Miner Res 16:2017–2026
Trofimov S, Pantsulaia I, Kobyliansky E, Livshits G (2004) Circulating levels of receptor activator of nuclear factor-κB ligand/osteoprotegerin/macrophage-colony stimulating factor in a presumably healthy human population. Eur J Endocrinol 150:305–311
van Bezooijen RL, Roelen BA, Visser A, Wee-Pals L van der, Wilt E de, Karperien M, Hamersma H, Papapoulos SE, Dijke P ten, Lowik CW (2004) Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199:805–814
van Bezooijen RL, Svensson JP, Eefting D, Visser A, Horst G van der, Karperien M, Quax PH, Vrieling H, Papapoulos SE, Dijke P ten, Lowik CW (2007) Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res 22:19–28
Wang Y, Liu W, Masuyama R, Fukuyama R, Ito M, Zhang Q, Komori H, Murakami T, Moriishi T, Miyazaki T, Kitazawa R, Yoshida CA, Kawai Y, Izumi S, Komori T (2012) Pyruvate dehydrogenase kinase 4 induces bone loss at unloading by promoting osteoclastogenesis. Bone 50:409–419
Watkins M, Grimston SK, Norris JY, Guillotin B, Shaw A, Beniash E, Civitelli R (2011) Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol Biol Cell 22:1240–1251
Weinstein RS, Nicholas RW, Manolagas SC (2000) Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab 85:2907–2912
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22:6267–6276
Wolff J (1892) Das Gesetz der Transformation der Knochen. Hirschwald, Berlin
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation. Nat Med 17:1235–1241
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA 95:3597–3602
Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, Srinivasan S, Gross TS, Donahue HJ (2011) Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One 6:e23516
Zhao S, Zhang YK, Harris S, Ahuja SS, Bonewald LF (2002) MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J Bone Miner Res 17:2068–2079
Zhao W, Byrne MH, Wang Y, Krane SM (2000) Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest 106:941–949
Ziegler S, Niessner A, Richter B, Wirth S, Billensteiner E, Woloszczuk W, Slany J, Geyer G (2005) Endurance running acutely raises plasma osteoprotegerin and lowers plasma receptor activator of nuclear factor κB ligand. Metabolism 54:935–938
Zong WX, Thompson CB (2006) Necrotic death as a cell fate. Genes Dev 20:1–15
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Komori, T. Functions of the osteocyte network in the regulation of bone mass. Cell Tissue Res 352, 191–198 (2013). https://doi.org/10.1007/s00441-012-1546-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-012-1546-x