
Abstract
Migraine pain is often preceded, accompanied and followed by dopaminergic symptoms (premonitory yawning and somnolence, accompanying nausea and vomiting, postdromal somnolence, euphoria and polyuria). After reviewing evidence from pharmacological, biochemical, genetic and animal experimental studies on the relationship between dopamine and migraine, and matching these data with patients’ clinical features, we postulate that migraine attacks could be characterized by an ictal dopamine release in a subject with dopamine receptor hypersensitivity due to a chronic dopaminergic deficit synergistic to serotoninergic impairment. Our review suggests that when the attack begins, a low dopamine plasma concentration stimulates hypersensitive central presynaptic dopamine receptors thus causing prodromal symptoms such as yawning and somnolence. Increasing dopamine levels, though still insufficient to stop trigeminovascular activation, stimulate postsynaptic dopamine receptors thus inducing nausea, vomiting and hypotension. Finally, dopamine levels slowly return to baseline, giving rise to somnolence and fatigue, but, in some cases, continue to rise triggering postdromal symptoms such as euphoria and polyuria.
Similar content being viewed by others


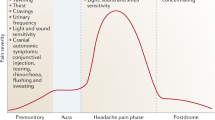
Introduction
Migraine is a common disabling neurovascular disorder characterized by recurrent episodes of moderate or severe head pain associated with vegetative symptoms. The migraine process develops over hours to days across prodromes, aura, headache and postdromes and may progress towards a central sensitization that manifests clinically as cutaneous allodynia [1]. Strong evidence indicates that serotonin plays a major role in migraine probably acting on central pain control pathways and on cranial blood vessels. In 1977, Sicuteri [2] proposed as the second putative protagonist in migraine dopamine (DA); since then, intensive research has investigated DA and migraine. In this review, we outline research advances underlining how DA intervenes in migraine symptoms and mechanisms.
Dopaminergic symptoms in migraine
Head pain is only one among the various symptoms experienced during a migraine attack. Across the different phases, vegetative symptoms develop and often last longer than the IHS criteria suggest [3]. Almost 30 % of migraineurs report experiencing prodromal symptoms hours before the headache begins. They typically feel tired, somnolent and drowsy, yawn excessively, and crave for foods. During the pain phase, nausea, vomiting and sometimes hypotension may induce severe disability. In the postdromal phase, which often needs eliciting, patients are usually tired and weak but sometime feel euphoric [1]. Most, if not all, symptoms preceding, accompanying and following the headache in migraineurs resemble those induced by pharmacologically induced central or peripheral DA receptor (DR) stimulation.
Pharmacological tests
The pharmacological studies we reviewed overall confirm dopaminergic dysfunction in migraine. Pharmacological challenges with DA agonists indicate that dopaminergic hypersensitivity is a specific trait in migraine patients. When given to migraineurs at small (nanomolar) doses, the DRD1-like and DRD2-like agonist apomorphine acts on presynaptic DRs thus reducing DA transmission, whereas at higher doses it acts on postsynaptic DRs thereby stimulating DA transmission. As a pharmacological tool, apomorphine has done much to unmask dopaminergic hypersensitivity in migraine. At the dose of 5 μg/kg sc or 0.25 mg sl it induces a significantly greater number of yawns in migraine sufferers than in controls [4, 5], whereas at the dose of 10 μg/kg sc (by stimulating presynaptic DRs) it causes at the most yawning and drowsiness in controls but (by stimulating postsynaptic DRs) it also causes nausea and vomiting in migraineurs [6]. The DA agonist piribedil at the dose of 0.1 mg/kg iv increases cerebral blood flow and induces nausea, vomiting and hypotension only in migraineurs [7]. In migraineurs but not in healthy controls, bromocriptine (2.5 mg po) provokes an intense hypotensive reaction unrelated to DA beta-hydroxylase plasma levels, suggesting defective central DA control on blood pressure and central DR hypersensitivity [8].
All the aforementioned studies demonstrate that presynaptic and postsynaptic DRs in migraine sufferers have a low activation threshold, in keeping with a chronic dopaminergic hypofunction.
Biochemistry
Our review also identified several studies describing DA changes in blood, urine and cerebrospinal fluid (CSF) in migraine. DA plasma levels measured during the menses are higher in women with migraine without aura than in controls [9]. The DA metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) increases in CSF during the migraine attack and increased levels correlate with pain severity [10]. Increased DA urinary levels have been described in female but not in male migraineurs during the headache-free period [11]. DA levels in platelets are higher in migraine patients than in controls but only in migraine without aura [12]. Biochemical change could reflect reduced activity of the enzyme DA beta-hydroxylase which catalyzes DA conversion into norepinephrine. In their study, D’Andrea et al. also suggest that during the migraine attack, impaired energy metabolism shifts tyrosine metabolism from hydroxylation, which physiologically gives rise to a metabolic cascade leading to catecholamine synthesis, toward decarboxylation, resulting in increased synthesis of trace amines (tyrosine, octopamine, synephrine). The imbalance between neurotransmitters (DA, norepinephrine) and neuromodulators (trace amines) activates the trigeminovascular system [13]. Receptor binding studies show that peripheral blood lymphocytes from migraineurs contain higher DRD3, DRD4 and DRD5 densities than control lymphocytes, with no differences in radioligand affinity. This finding indicates central or peripheral DR hypersensitivity or both due to dopaminergic system hypofunction [14, 15].
Genetics
Several studies have explored the correlation between specific DA-related genotypes and susceptibility, comorbidity, clinical expression and pharmacological responsiveness in migraine. The migraine risk seems increased in ANKK1 DRD2 loci missense polymorphism or DBH rs7239728 polymorphism carriers and lowered in DRD2 rs6275 polymorphism carriers [16]. Migraine with aura is associated with polymorphisms in DBH (rs2097629), DRD2 (rs7131056) and the dopamine transporter gene SLC6A3 (rs40184) [17]. The incidence of migraine with aura, major depression, generalized anxiety disorder, panic attacks and phobia is higher in individuals with the DRD2 NcoI C/C genotype than in those with a DRD2 NcoI T allele [18]. In their genetic studies, Stochino et al. [19, 20] reported that the DRD2/NcoI C allele could be a susceptibility factor for good rizatriptan responsiveness, but did not confirm an association between dopaminergic genes and the migraine-panic phenotype. Using a family-based association method, others have reported that the allelic distribution at the DRD2 locus differs significantly in the dopaminergic migraineurs subgroup [21], although a more recent study argued against a role for the DRD2 gene in the migraine clinical phenotype [22]. Genetic variability in the DRD4 gene is involved in the predisposition to episodic migraine without aura, whereas susceptibility to chronic daily headache seems to involve genetic variability in the DAT gene [23, 24].
Brain dopaminergic pathways and migraine
Research over the past 15 years both in humans and animals advanced our knowledge on the involvement of the central dopaminergic circuitry in migraine [25]. The substantia nigra (SN), a brain area that actively controls the cerebral cortical microcirculation and neuronal activity, contains a high density of 5HT1B receptors, targeted by triptans [25]. Objective evidence confirming SN involvement in migraine comes from a functional magnetic resonance imaging study showing blood oxygen level-dependent SN activation during visually triggered migraine [26]. Parkinson’s disease (PD), the paradigm for DA system impairment, seems to shorten the clinical course of migraine. In a study on 237 PD patients aimed to assess the prevalence of lifetime and current migraine, approximately two-thirds of patients reported that migraine improved or remitted after PD onset [27]. SN degeneration, the PD hallmark, could be responsible for the migraine decline in parkinsonian patients, reinforcing the view that migraine needs an efficient DA system. Again underlining dopaminergic system involvement in migraine, patients with PD and current or prior migraine respond better to dopaminergic medications and experience less severe disabilities than patients without migraine, further supporting dopaminergic hypersensitivity in migraine [28].
Experimental evidence shows that DRs exist in the trigeminal ganglion and spinal nucleus and that DA exerts a potent inhibitory effect on trigeminocervical complex neurons by stimulating DRD2; central DRD2 are involved in trigeminovascular nociceptive processing whereas peripheral DRD1 might play a role in peripheral sensitization [29]. More help in understanding trigeminal DA mechanisms in migraine comes from a study conducted in recent years showing that DRD2 and 5-HT1B/1D receptors are colocalized in the trigeminocervical complex, suggesting that DA and serotonin act simultaneously and synergistically at that level to reduce neuronal firing and to reverse trigeminal sensitization [30].
DA agonists and DA antagonists in migraine treatment
In current practice, migraine therapy includes drugs acting on the DA system.
In preventive treatment, DA agonists have proven efficacy. In patients who are resistant to migraine prophylaxis and have a high attack frequency, continuous sc apomorphine infusion effectively reduces the frequency and severity of migraine attacks [31]. Lisuride, an 8-α-amino-ergoline derivative with a high affinity for DRD1 and DRD2, at the dose 0.05–0.075 mg significantly reduces attack frequency and severity [32]. α-Dihydroergocryptine, a semisynthetic ergot derivative, is a DR agonist which at the dose of 10 mg bid is as effective as flunarizine in migraine prevention [33]. Evidence that DR stimulation is meaningful for migraine prevention is apparently weakened by the potential neuroleptic-like profile of flunarizine, an established antimigraine prophylactic agent characterized by the risk of inducing parkinsonism in the elderly. Its therapeutic action in migraine could, however, be independent from antidopaminergic mechanisms given that single photon-emission computer tomography with [123I]iodobenzamide, a radioligand with high affinity and specificity for DRD2, failed to show a correlation between efficacy and degree of the DRD2 blockade [34]. After treatment for 1 month, flunarizine left the pretherapy inhibitory prolactin response to an acute bromocriptine challenge observed before therapy unchanged, contrasting with the view that flunarizine acts as a direct antagonist at DRD2, at least in the tuberoinfundibular system [35].
In acute migraine treatment, DR antagonists have recognized efficacy. Domperidone, a peripheral DRD1–DRD2 antagonist, taken at doses ranging from 20 to 40 mg during prodromes, may stop progression to the headache phase [36]. Metoclopramide (10–20 mg), taken alone or combined with other agents, is an effective acute migraine treatment not only for nausea and vomiting but also for headache. Other neuroleptics, i.e., prochlorperazine (5–10 mg), chlorpromazine (25–100 mg), droperidol (1.25–2.5 mg) and haloperidol (2–5 mg), regardless of the delivery route, provide effective acute migraine treatment and in some comparative studies outperform NSAIDs, valproate, opioids and sumatriptan [37].
Dopaminergic symptoms in migraine: a unifying hypothesis
Even though the literature on DA and migraine remains to some extent controversial, more recent experimental studies have provided new insights into DA involvement in migraine pathophysiology demonstrating that DA acts as a brake on trigeminocervical neurons. Current consensus agrees that migraineurs suffer from a chronic dopaminergic hypofunction due to defective DA levels, defective DA-related genotypes or both. Dopaminergic dysfunction would lead to a DR upregulation, rendering the patient more sensitive to DA stimulation, and to a reduced inhibitory control on trigeminocervical neurons that increases their firing susceptibility in response to internal or external migraine triggers. When an attack begins, during the prodromal phase, DA is probably released (as demonstrated by increased DOPAC cerebrospinal fluid levels during the attack) and, at a relatively low plasma concentration, DA stimulates hypersensitive central presynaptic DRs causing yawning and somnolence. Thereafter, increasing DA levels, too late to stop incipient trigeminovascular activation, would then also stimulate central and peripheral postsynaptic DRs inducing nausea, vomiting and hypotension. In the postdromal phase, DA levels would slowly return to baseline, giving rise to somnolence and fatigue, but, in some cases, could continue to rise thus precipitating postdromal symptoms such as euphoria and polyuria. In this view, antidopaminergic agents would work by acutely blocking peripheral DRD1- and central DRD2-mediated sensitization, whereas chronic dopaminergic stimulation would help prevent migraine by increasing DA negative control on trigeminocervical neurons.
In conclusion, as research in animal models now confirms, dopaminergic symptoms in migraine are probably the clinical expression of ictal DA release in subjects who have a dopaminergic dysfunction associated with and synergistic to serotoninergic impairment.
References
Blau JN (2005) Feeling and seeing headaches. J Headache Pain 6:10–19
Sicuteri F (1977) Dopamine the second putative protagonist in headache. Headache 17:129–131
Headache Classification Subcommittee of the International Headache Society (2004) The International Classification of Headache Disorders. 2nd edn. Cephalalgia 24(1):1–160
Blin O, Azulay JP, Masson G, Aubrespy G, Serratrice G (1991) Apomorphine-induced yawning in migraine patients: enhanced responsiveness. Clin Neuropharmacol 14:91–95
Del Bene E, Poggioni M, De Tommasi F (1994) Video assessment of yawning induced by sublingual apomorphine in migraine. Headache 34:536–538
Cerbo R, Barbanti P, Buzzi MG, Fabbrini G, Brusa L, Roberti C et al (1997) Dopamine hypersensitivity in migraine: role of the apomorphine test. Clin Neuropharmacol 20:36–41
Bussone G, Boiardi A, La Mantia L, Frediani F, Lamperti E, Peccarisi C et al (1986) Clinical usefulness of a dopaminergic agonist in headache diagnosis. Int J Clin Pharmacol Res 6:23–26
Fanciullacci M, Michelacci S, Curradi C, Sicuteri F (1980) Hyperresponsiveness of migraine patients to the hypotensive action of bromocriptine. Headache 20:99–102
Nagel-Leiby S, Welch KM, D’Andrea G, Grunfeld S, Brown E (1990) Event-related slow potentials and associated catecholamine function in migraine. Cephalalgia 10:317–329
Castillo J, Martinez F, Suarez C, Naveiro J, Lema M, Noya M (1996) Cerebrospinal fluid tyrosine and 3,4-dihydroxyphenylacetic acid levels in migraine patients. Cephalalgia 16:56–61
Gruber HJ, Bernecker C, Pailer S, Lechner A, Horejsi R, Möller R et al (2010) Increased dopamine is associated with the cGMP and homocysteine pathway in female migraineurs. Headache 50:109–116
D’Andrea G, Granella F, Perini F, Farruggio A, Leone M, Bussone G (2006) Platelet levels of dopamine are increased in migraine and cluster headache. Headache 46:585–591
D’Andrea G, D’Arrigo A, Dalle Caerbonare M, Leon A (2012) Pathogenesis of migraine: role of neuromodulators. Headache 52:1155–1163
Barbanti P, Bronzetti E, Ricci A, Cerbo R, Fabbrini G, Buzzi MG et al (1996) Increased density of dopamine D5 receptor in peripheral blood lymphocytes of migraineurs: a marker for migraine? Neurosci Lett 29(207):73–76
Barbanti P, Fabbrini G, Ricci A, Pascali MP, Bronzetti E, Amenta F et al (2000) Migraine patients show an increased density of dopamine D3 and D4 receptors on lymphocytes. Cephalalgia 20:15–19
Ghosh J, Pradhan S, Mittal B (2013) Identification of a novel ANKK1 and other dopaminergic (DRD2 and DBH) gene variants in migraine susceptibility. Neuromolecular Med 15:61–73
Todt U, Netzer C, Toliat M, Heinze A, Goebel I, Nürnberg P et al (2009) New genetic evidence for involvement of the dopamine system in migraine with aura. Hum Genet 125:265–279
Peroutka SJ, Price SC, Wilhoit TL, Jones KW (1998) Comorbid migraine with aura, anxiety, and depression is associated with dopamine D2 receptor (DRD2) NcoI alleles. Mol Med 4:14–21
Asuni C, Cherchi A, Congiu D, Piccardi MP, Del Zompo M, Stochino ME (2007) Association study between clinical response to rizatriptan and some candidate genes. J Headache Pain 8:185–189
Stochino ME, Asuni C, Congiu D, Del Zompo M, Severino G (2003) Association study between the phenotype migraine without aura-panic disorder and dopaminergic receptor genes. Pharmacol Res 48:531–534
Del Zompo M, Cherchi A, Palmas MA, Ponti M, Bocchetta A, Gessa GL, Piccardi MP (1998) Association between dopamine receptor genes and migraine without aura in a Sardinian sample. Neurology 51:781–786
Rebaudengo N, Rainero I, Parziale A, Rosina F, Pavanelli E, Rubino E et al (2004) Lack of interaction between a polymorphism in the dopamine D2 receptor gene and the clinical features of migraine. Cephalalgia 24:503–507
Mochi M, Cevoli S, Cortelli P, Pierangeli G, Soriani S, Scapoli C, Montagna P (2003) A genetic association study of migraine with dopamine receptor 4, dopamine transporter and dopamine-beta-hydroxylase genes. Neurol Sci 23:301–305
Cevoli S, Mochi M, Scapoli C, Marzocchi N, Pierangeli G, Pini LA et al (2006) Genetic association study of dopamine metabolism-related genes and chronic headache with drug abuse. Eur J Neurol 13:1009–1013
Barbanti P, Fabbrini G (2002) Migraine and the extrapyramidal system. Cephalalgia 22:2–11
Cao Y, Aurora SK, Nagesh V, Patel SC, Welch KM (2002) Functional MRI-BOLD of brainstem structures during visually triggered migraine. Neurology 59:72–78
Barbanti P, Fabbrini G, Vanacore N, Rum A, Lenzi GL, Meco G, Cerbo R (2000) Dopamine and migraine: does Parkinson’s disease modify migraine course? Cephalalgia 20:720–723
Cubo E, Kompoliti K, Leurgans SE, Raman R (2004) Dopaminergic hypersensitivity in patients with Parkinson disease and migraine. Clin Neuropharmacol 27:30–32
Charbit AR, Akerman S, Goadsby PJ (2009) Comparison of the effects of central and peripheral dopamine receptor activation on evoked firing in the trigeminocervical complex. J Pharmacol Exp Ther 331:752–763
Charbit AR, Akerman S, Goadsby PJ (2011) Trigeminocervical complex responses after lesioning dopaminergic A11 nucleus are modified by dopamine and serotonin mechanisms. Pain 152:2365–2376
Lai M, Loi V, Pisano MR, Del Zompo M (1997) Therapy of migraine by modulating dopamine hypersensitivity: its effect on mood and pain. Int J Clin Pharmacol Res 17:101–103
Somerville BW, Herrmann WM (1978) Migraine prophylaxis with Lisuride hydrogen maleate—a double blind study of Lisuride versus placebo. Headache 18:75–79
Bussone G, Cerbo R, Martucci N, Micieli G, Zanferrari C, Grazzi L et al (1999) Alpha-dihydroergocryptine in the prophylaxis of migraine: a multicenter double-blind study versus flunarizine. Headache 39:426–431
Wöber C, Brücke T, Wöber-Bingöl C, Asenbaum S, Wessely P, Podreka I (1994) Dopamine D2 receptor blockade and antimigraine action of flunarizine. Cephalalgia 14:235–240
Cupini LM, Troisi E, Placidi F, Diomedi M, Silvestrini M, Argiro G, Bernardi G (1999) Does the antimigraine action of flunarizine involve the dopaminergic system? A clinical-neuroendocrinological study. Cephalalgia 19:27–31
Waelkens J (1984) Dopamine blockade with domperidone: bridge between prophylactic and abortive treatment of migraine? A dose-finding study. Cephalalgia 4:85–90
Marmura J (2012) Use of dopamine antagonists in treatment of migraine. Curr Treat Options Neurol 14:27–35
Conflict of interest
We certify that there is no actual or potential conflict of interest in relation to this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barbanti, P., Fofi, L., Aurilia, C. et al. Dopaminergic symptoms in migraine. Neurol Sci 34 (Suppl 1), 67–70 (2013). https://doi.org/10.1007/s10072-013-1415-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-013-1415-8