
Abstract
To understand the genetic architecture of dyslexia and identify the locations of genes involved, we performed linkage analyses in multigenerational families using a phonological memory phenotype—Nonword Repetition (NWR). A genome scan was first performed on 438 people from 51 families (DS-1) and linkage was assessed using variance components (VC), Bayesian oligogenic (BO), and parametric analyses. For replication, the genome scan and analyses were repeated on 693 people from 93 families (DS-2). For the combined set (DS-C), analyses were performed with all three methods in the regions that were identified in both samples. In DS-1, regions on chromosomes 4p, 6q, 12p, 17q, and 22q exceeded our initial threshold for linkage, with 17q providing a parametric LOD score of 3.2. Analysis with DS-2 confirmed the locations on chromosomes 4p and 12p. The strongest VC and BO signals in both samples were on chromosome 4p in DS-C, with a parametric multipoint LODmax of 2.36 for the 4p locus. Our linkage analyses of NWR in dyslexia provide suggestive and reproducible evidence for linkage to 4p12 and 12p in both samples, and significant evidence for linkage to 17q in one of the samples. These results warrant further studies of phonological memory and chromosomal regions identified here in other datasets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dyslexia or specific reading disability (OMIM 127700) is a common neurocognitive disorder that affects 5–10% of school age children (Shaywitz et al. 1990). The International Dyslexia Association defines dyslexia as a disorder “characterized by difficulties with accurate and/or fluent word recognition and by poor spelling and decoding abilities. These difficulties typically result from a deficit in the phonological component of language that is often unexpected in relation to other cognitive abilities and the provision of effective classroom instructions” (Lyon et al. 2003). Application of these deceptively simple definitions led different investigators to use different criteria to define what constitutes “difficulties” and “unexpectedness”, and different psychometric measures to evaluate dyslexia and “the phonological component of language” that characterizes it. This lack of definitional precision has added to the complexity of conducting rigorous research on dyslexia.
Multiple lines of evidence support the contribution of genetic factors to dyslexia (Fisher and DeFries 2002; Raskind 2001). Twin studies of dyslexia, categorically defined, have found higher concordance rates for monozygotic vs. dyzygotic twins with resulting broad-sense heritability (h2) of 0.6 (DeFries and Alarcon 1996), and family studies have shown a sibling recurrence rate of 40–50% (Gilger et al. 1994; Wolff and Melngailis 1994), which is significantly above the population rate of 5–10% (Shaywitz et al. 1990). Estimates of heritability of dyslexia quantitative traits are also high. Twin studies of component phenotypes of reading estimate h2 of 0.71 for phonological decoding, 0.60 for orthographic choice, and 0.85 for word recognition (Gayan and Olson 2001, 2003). However, in the general population, dyslexia does not show mendelian patterns of inheritance, and is best categorized as a complex genetic disorder. Not surprisingly, targeted and genome-wide linkage analyses have identified numerous localizations for genes that contribute to dyslexia and related cognitive processes, including chromosomes 1p34–36 (DYX8, OMIM 608995) (Grigorenko et al. 2001), 2p16–15 (DYX3, OMIM 604254) (Fagerheim et al. 1999), 3p12-q13 (DYX5, OMIM 606896) (Nopola-Hemmi et al. 2001), 6p22.2 (DYX2, OMIM 600202) (Cardon et al. 1994), 6q13–16.2 (DYX4, OMIM 127700) (Petryshen et al. 2001), 11p15.5 (DYX7, OMIM 127700) (Hsiung et al. 2004), 15q21 (DYX1, OMIM 127700) (Grigorenko et al. 1997), and 18p11 (DYX6 OMIM 606616) (Fisher et al. 2002).
For several of these loci, candidate genes for dyslexia have been proposed. DYX1C1, a candidate gene for the DYX1 region, is disrupted by a chromosomal translocation that co-segregates with intellectual impairment/dyslexia phenotype in a Finnish family (Taipale et al. 2003). Two candidate genes in the DYX2 region, KIAA0319 (Cope et al. 2005) and DCDC2 (Meng et al. 2005) were identified by linkage disequilibrium and association studies. ROBO1, a candidate gene for the DYX5 region, is disrupted by a translocation in one dyslexic individual, and a rare ROBO1 haplotype co-segregates with dyslexia in a large pedigree (Hannula-Jouppi et al. 2005). Causative mutations in these genes have not been identified, making it difficult to elucidate how these genes contribute to dyslexia in the population. In utero RNAi studies in rodents have implicated DYX1C1, KIAA0319 and DCDC2 in neuronal migration (Meng et al. 2005; Paracchini et al. 2006; Rosen et al. 2007), and ROBO1 was implicated in axon guidance and dendritic connections (Kidd et al. 1998). These observations suggest that global brain development disruption might play a role in dyslexia (Galaburda et al. 2006; McGrath et al. 2006).
Learning to read requires a variety of different linguistic and nonlinguistic cognitive abilities (Vellutino et al. 2004), each of which may have some unique genetic components. Phonological awareness, the ability to recognize that words can be decomposed into constituent phonological segments, is thought to be a key skill that is typically impaired in dyslexia. The ability to deal explicitly with the phonemes, the smallest sound units of speech, underlies reading acquisition and conversion of written symbols (orthography) to speech sounds (phonology) (Gathercole et al. 1994; Wagner and Torgesen 1987). Additional phonological skills involved in reading include encoding and retrieving of phonological information from short-term, working, and long-term memory (Kamhi and Catts 1986). To further dissect phonological skills, phonological processing of sounds can be isolated from cognitive skills that rely on the meaning of words by the use of tasks that utilize nonwords. Nonwords can be pronounced but have no associated meaning, and must be processed solely based on sound information they contain. The Nonword Repetition (NWR) task (Wagner et al. 1999) requires orally presented nonwords to be coded into memory while sounds in them are processed and prepared in order to be repeated. Impairments on NWR-like tasks have been found in dyslexia. Children with dyslexia make significantly more word repetition errors and use more phonological processes then normal readers (Kamhi and Catts 1986). Similarly in a group of children and young adults with dyslexia NWR was significantly impaired compared to normal readers (Hulslander et al. 2004). Deficits in NWR have also been described in adults with dyslexia compared to normal readers (Ramus et al. 2003; Szenkovits and Ramus 2005), thus further attesting to the importance of this phenotype in dyslexia. Levels of impairment on the NWR measure have been shown to be stable across development (Bishop et al. 1996), making it additionally suitable for family genetics studies.
To examine the genetic architecture of dyslexia, we have adopted a family-based strategy that includes in-depth evaluation of theory-based and empirically validated quantitative phenotypes relevant to dyslexia, followed by aggregation, segregation, and linkage analysis of individual phenotypes (Berninger et al. 2006; Berninger et al. 2001). Our initial familial aggregation (Raskind et al. 2000) and segregation (Wijsman et al. 2000) analyses of dyslexia phenotypes were performed on 102 families, a sample that overlaps with the 144 families presented here. In aggregation studies, amongst all the measures in our test battery, family correlation patterns of performance on NWR and a measure of rate of nonword reading, Pseudoword Decoding Efficiency (PDE) (Torgesen et al. 1999) gave strongest support for a genetic basis. Narrow-sense heritability of NWR, derived from the correlation between parents and offspring and considering additive genetic variance only, was estimated at 0.20. In the same study we found within individual probands pairwise correlations of 0.33 and 0.32, respectively, between NWR and accuracy of word reading—Word Identification (WID) (Woodcock 1987), or rate of word reading—Single Word Efficiency (SWE) (Torgesen et al. 1999), and 0.36 and 0.42, respectively, between accuracy of nonword reading—Word Attack (WA) (Woodcock 1987), or rate of nonword reading—(PDE) (Torgesen et al. 1999). These are moderate correlations compared to individual pairwise correlations of reading measures: WID to SWE, WA to PDA and SWE to PDI of 0.79, 0.6 and 0.71 respectively.
To further evaluate the genetics basis of NWR, estimate the parameters for models of inheritance, and evaluate the suitability of the measure for linkage analysis, we performed segregation analyses. This analysis provided evidence in support of a major-gene mode of inheritance with multiple contributing loci, and in rough agreement with our aggregation analysis, it estimated narrow-sense heritability at ~0.30. Together our familial aggregation and segregation studies show significant evidence for heritability of NWR in our sample.
Although phenotypes associated with dyslexia are multivariate, there are technical difficulties in using multivariate distributions, and multivariate approaches are not well tested. In addition, recent studies using very large numbers of traits in a single sample indicate that multivariate approaches do not appear to provide advantages for linkage detection (Wijsman et al. 2007). A situation in which a multivariate analysis might be warranted, namely evidence for linkage for multiple highly correlated traits at the same location, does not apply to our analyses of NWR because the peaks reported here do not coincide with linkage peaks we have reported for reading measures, and NWR is only moderately correlated with reading measures in our sample. Our approach, which focuses on individual univariate component phenotypes and adjusts for covariate effects, has been shown to be successful in the analysis of simulated complex traits (Wijsman and Amos 1997) and in studies of complex phenotypes in which evidence for linkage was eventually found (Hokanson 1999; Knoblauch et al. 2000; Sviridov and Nestel 2007). Our earlier linkage studies, on a sample of 51 families (DS-1), focused on reading-based phenotypes. For WID, a phenotype based on accuracy of single real word reading, we found supportive evidence for linkage to chromosome 15q21 (Chapman et al. 2004), as well as suggestive evidence for a novel locus on chromosome 12q13 (Igo et al. 2006a). For phenotypes based on speed of single word reading (SWE) and speed of single non-word reading (PDE) we found significant evidence for involvement of loci on chromosomes 13q (Igo et al. 2006a) and 2q (Raskind et al. 2005) respectively. These results provide additional evidence for the genetic complexity of dyslexia with involvement of multiple loci. As a further step in our studies of dyslexia, here we report on genome-wide linkage analysis of NWR, a phonological memory based phenotype. We have performed the analysis in our original sample and followed it with a replication sample and joint analysis of the regions of interest in the combined sample.
Methods and materials
Study subjects
Detailed subject recruitment and evaluation procedures have been described (Berninger et al. 2001; Raskind 2001). Under a protocol approved by the Institutional Review Board of the University of Washington, families were identified through school age children with reading difficulties. Inclusion criteria for probands were a prorated verbal IQ (VIQ) ≥90 (≥25%ile) on the Wechsler Intelligence Scale for Children—3rd edition (Wechsler 1991) and a score below the population mean and at least 1 standard deviation below their VIQ on at least one of the core research measures. These core measures include accuracy and speed of single word or nonword reading, accuracy or rate of oral reading of text, spelling, and automatic writing of alphabet letters. Children with neurological or psychiatric disorders, pervasive developmental disability, or other conditions known to be associated with poor reading were excluded based on a parent questionnaire. Comorbid attention deficit/hyperactivity disorder (ADHD) was not a cause for exclusion. For 209 children from our sample, based on parental rating scale, 10.5% had ratings of ‘often’ on 6 or more symptoms from DSM-IV Inattention or Hyperactivity-Impulsivity criteria (Thomson et al. 2005). This is comparable to 15–35% incidence of ADHD in dyslexia samples reported by others (Willcutt and Pennington 2000; Willcutt et al. 2000). Siblings older than 6.5 years were invited to participate, and nuclear families were extended in a sequential ascertainment procedure through parents who met study criteria of reading or writing impairment. Parental first-degree relatives were recruited, and the process was repeated to the extent possible. The combined set of families used for linkage analysis includes 1131 people in 144 families. The distribution of family sizes and descriptive statistics of probands and their parents are presented in Tables 1 and 2.
Phenotype
The NWR task from the Comprehensive Test of Phonological Processing battery (Wagner et al. 1999) involves storing and analyzing phonological elements for short periods of time before repeating them. Nonwords have been created to represent plausible English language items by randomly combining phonemes to fill slots in syllables and discarding non-pronounceable nonwords. This process should reduce the analogies to real words, and minimize the use of cognitive processes other than phonological memory throughout the task. During the task the nonwords increase in length and phonological complexity. Raw scores are age-normed and converted into percentile ranks and standard scores (mean = 10, SD = 3).
Genotypes
Two genome scans were performed at the Mammalian Genotyping Service (MGS), Marshfield, WI. Screening set 10 (405 STRP markers, 378 autosomal) was used for the first scan on 438 people from 51 families (DS-1). These families were selected for genotyping from a larger set on the basis of the expected value of the family specific maximum LOD score for parametric analysis of NWR, as estimated by simulation with SIMLINK (Boehnke and Ploughman 1997). For the second round of genotyping, screening set 15 (402 STRP markers, 375 autosomal) was used for the set of 693 people in 93 families (DS-2). The DS-2 sample was selected for genotyping based on rank ordering that took all the core ascertainment phenotypes under consideration. The two data sets have a 10-cM average marker spacing and share 68% of the markers. Additional markers, from empirically defined regions of interest (ROIs), on chromosomes 4p (D4S405, D4S3242), 6q (D6S264, D6S1035), 12p (D12S336, D12S1617), 17q (D17S949, D17S1847), and 22q (D22S1170, 22C48788, 22C49177) were typed in our laboratory on DS-1. For a region near the telomere of chromosome 22q no polymorphic markers were available, so 22C48788 and 22C49177 were designed in our laboratory (sequences available on request). Genotypes were checked for mendelian inconsistencies and unlikely genotypes and errors were resolved (Chapman et al. 2004). From screening set 10, two markers were removed because of excessive errors and a third marker was a duplicate. The Marshfield genetic maps were used for analysis (http://research.marshfieldclinic.org/genetics), with all multipoint analysis based on the Haldane map function. Allele frequencies were estimated separately in the two data sets, based on observed counts.
Statistical analyses
General approach
Initial genome scans were performed in DS-1 using both variance component (VC) and Bayesian oligogenic (BO) Markov chain Monte Carlo (MCMC) linkage analysis methods. For follow-up analysis, ROIs were empirically defined as VC-LOD > 1.0 and MCMC IR > 5 (log10(IR) > 0.7), where the intensity ratio (IR) is the ratio of the acceptance rate for a QTL position and the rate expected given the uniform distribution of QTLs in the model (Wijsman and Yu 2004). The IR is an easily-computed, excellent approximation to the Bayes Factor (Igo Jr and Wijsman 2008). For ROIs we added markers and computed both single marker and multipoint parametric LOD scores, using a model estimated by complex segregation analysis. For replication in DS-2, a preliminary genome scan with VC and BO methods was performed, and ROIs determined by the same criteria. In regions where DS-2 confirmed the DS-1 findings, the data sets were combined (DS-C) and reanalyzed.
We designed our approach to maximize the chance that our analysis would be robust to potential errors and false positive results. To guard against false positives due to map inaccuracy or genotype misspecifications, we checked key signals with single-marker and multipoint analysis. We used VC, BO, and parametric linkage analyses because they have complementary strengths and weaknesses. The VC approach has the advantages that it is commonly used, well characterized, and it does not require QTL model assumptions. Disadvantages include poor QTL localization relative to parametric modeling (Amos and de Andrade 2001; Atwood and Heard-Costa 2003), sensitivity to skewness or kurtosis (Blangero et al. 2000), sensitivity to pedigree ascertainment (Forrest and Feingold 2000), and less efficient use of mapping information compared to a parametric approach. These disadvantages are balanced by the BO approach that provides relatively accurate QTL localization (Daw et al. 1999), characterizes the underlying trait models, and is robust to pedigree ascertainment and underlying distributional assumptions (Wijsman and Yu 2004). Disadvantages of the BO approach include the less familiar Bayesian framework and the need for assumptions of prior distributions for model parameters. Finally, the parametric approach is most thoroughly tested. It is a full likelihood method, and as such, statistical theory predicts that it is the most efficient method. The disadvantage is that to achieve this high efficiency requires the underlying model to be well specified. To specify the trait model for NWR in our sample, we performed both maximum-likelihood complex segregation analysis and BO segregation analysis. In the final parametric linkage analysis we used the transmission model that was best supported with both methods because of previous results indicating that this approach provides robust models (Igo et al. 2006b). Although use of the parametric method increases the power of analysis, potential trait misspecification typically reduces the evidence for linkage and as a consequence is unlikely to give false positive results (Ott 1999). All three approaches are summarized in Table 3.
In all analyses, NWR was adjusted for VIQ, age with separate linear terms for children and adults, and sex. For VC analyses, the covariates were explicitly modeled, while for all other analyses a pre-adjusted NWR measure was used.
Variance components linkage analyses
The VC approach involves partitioning the variance of the trait of interest into components reflecting the contribution of covariates, a major gene, and a polygenic component (Almasy and Blangero 1998; Amos 1994). Estimation of variance components requires the use of genotyping data in the form of estimated pairwise identity-by-descent (IBD) sharing between relatives. Exact single-marker and multipoint IBD scores were calculated at each marker locus with Merlin (Abecasis et al. 2002) or with Loki (Heath 1997) for larger pedigrees, and used for variance components linkage analysis with SOLAR 2.1 (http://www.sfbr.org/sfbr/public/software/solar/). In addition to handling larger pedigrees than other alternatives such as Merlin, SOLAR allows for incorporation of a dominance variance component in the model that has shown to be important in our data set. Four models were fit at each locus: M0-a, the null polygenic model allowing additive variance; M0-d, the null polygenic model allowing additive and dominance variance; M1-a, an additive model, where the additive variance is estimated, and the dominance variance is assumed to be zero in the QTL and polygenic components; and M1-d, a dominance model, where the additive and dominance variance are estimated in the QTL and polygenic components. LOD scores were obtained by comparing the log-likelihoods of either M1-a to M0-a or M1-d to M0-d. We use the term VC-LOD for results from VC analyses, and LOD or LODmax for LOD scores or maximum LOD scores from parametric analyses. A VC-LOD > 1.0 was set as a threshold to define a region of interest.
Bayesian oligogenic joint segregation and linkage analyses
BO segregation analyses and joint segregation and linkage analyses were carried out using Loki version 2.4.5 (http://www.stat.washington.edu/thompson/Genepi/Loki.shtml). This approach allows analysis of a quantitative phenotype, using simultaneous modeling of multiple trait loci and multiple marker loci. Loki estimates posterior distributions of unknown parameters using a Bayesian reversible-jump MCMC sampler (Heath 1997), conditional on the data and assumptions of prior distributions on several parameters (Wijsman 2002). A Poisson distribution with mean 2 was used for the prior distribution of the number of QTLs. Genotype effects were sampled from a normal distribution with mean 0 and variance τβ. Optimal values of τβ were chosen by maximizing the mean total genetic variance in a segregation analysis. A uniform distribution was assumed for the location of QTLs. Adequacy of assumed prior distributions and of the resulting MCMC samples were evaluated with methods described in detail elsewhere (Wijsman and Yu 2004). An analysis with Loki yields posterior distributions for the total number of QTLs and QTLs with significant effect, the allele frequencies and genotype effects for each QTL, and the locations of linked QTLs. We report results as intensity ratios (IRs), the ratios of the number of QTLs accepted in a region relative to the number expected if the distribution of QTLs was uniform, given the posterior mean of the total number of QTLs per iteration. For the current work, 50,000 MCMC iterations were performed with every 2nd iteration saved. IRs were calculated over 2-cM intervals with an IR cutoff of 5 used to define a region of interest.
Segregation analyses
Complex segregation analyses (CSA) were performed using DS-1 and DS-C. We used DS-C to assess the potential bias in the DS-1 selection that was based on power to detect linkage to NWR. We estimated parameters of the mendelian model using the class D of the logistic regression models of Bonney (Bonney 1986) with the REGC program in S.A.G.E. (S.A.G.E. 1997). We matched mendelian models with and without polygenic components and compared them to one another and to an environmental model. Adequacy of the resulting models was assessed by fitting vs. fixing the value of the heterozygous transmission parameter. BO segregation analyses were performed using Loki with 50,000 iterations, in the same manner as linkage analyses but without including marker data. Each iteration yielded values for the total genetic variance over all QTLs, an effect size (defined as the square root of the genetic variance) for each QTL, and the residual (environmental) variance. QTL models from both analysis methods were compared to verify model similarity, and the point estimates from the CSA were used in the linkage analyses. Although trait model inaccuracy is to be expected, linkage detection with parametric approaches is robust to parameter misspecification, with only modest reduction in power to detect linkage (Blangero et al. 2000; Ott 1999).
Parametric LOD score analyses
FASTLINK (Cottingham et al. 1993) was used to calculate exact single-marker parametric LOD scores. Because of computational constraints of multipoint analysis, whole-chromosome multipoint parametric LOD scores were calculated using the MCMC-based program lm_markers from the MORGAN suite of programs (http://www.stat.washington.edu/Thompson/Genepi/MORGAN/Morgan.shtml). FASTLINK and lm_markers are the only available programs that can compute parametric LOD scores for a quantitative trait model. The transmission models for these analyses were obtained from CSA using DS-1.
Analysis of DS-C
Genotyping in DS-1 and DS-2 was performed using two different MGS marker panels. It was important to allow for different marker allele frequencies in DS-1 and DS-2 because there are differences in allele scoring for markers shared between the two panels. For the VC analyses, the IBD scores were calculated separately, and in the MCMC analysis, an option was used that allowed for different allele frequencies in two data sets. For the parametric LOD-score analysis, DS-1 and DS-2 were analyzed separately, and the resulting LOD scores were added.
Empirical significance levels
We carried out simulations to obtain estimates of empirical significance levels in the DS-C analysis. The large data set, relatively large pedigrees, and the need to accommodate two different sets of genome-scan marker panels created certain complications with obtaining such simulations with existing software. The existence of some large pedigrees in the sample that required multipoint marker IBD estimation with MCMC-based methods prevented use of the marker simulation approach implemented in Merlin. To achieve practical computation, we used trait simulation provided by the SimSuite program (http://faculty.washington.edu/wijsman/software.shtml), retaining the original-data marker IBD estimates. We used samples from the posterior distribution of oligogenic models obtained from a 100,000-iteration run with Loki as the source of generating models for unlinked trait distributions (Igo Jr and Wijsman 2008), while retaining the original marker data. This trait simulation-based approach yields slightly more conservative empirical estimates of P-values than does marker simulation (Igo and Wijsman 2008), and is more computationally tractable than is marker simulation. To allow computations within the constraints of the software, markers used in both marker panels were treated as separate pairs of markers that were spaced by 0.01 cM on the marker map because of prohibitions in the MCMC approach against use of multiple markers at identical positions. To further limit the total computation time, we restricted analysis to markers in the ~100 cM region of chr 4 with the strongest evidence of linkage. Even so, the 10,000 replicates needed for reasonably accurate estimation of the empirical P-value required >7 days of CPU time.
Results
Segregation analyses for NWR were performed in both DS-1 and DS-C, and yielded similar models. We explored the use of the DS-C based models for the parametric analyses, but the results were not substantially different from those using DS-1 (data not shown), and the CSA major gene (CSA-mg) model from DS-1 was used for parametric linkage detection. This model describes a population where 5% of individuals homozygous for the risk allele are in the lowest performing group (genotype mean −1.74), the 59% homozygous for the protective allele are in the highest performing group (genotype mean +0.27), and the remaining 36% (heterozygous) have a genotype mean of −0.16. In the context of a single-locus, two-allele model, this pattern describes a mendelian co-dominant model with additive and dominance variances of 0.16 and 0.04, respectively. The residual (within genotype) variance is estimated at 0.46. This QTL accounts for approximately 31% of the variation of NWR in the population. Oligogenic analysis estimated an average of 3.2 QTLs contributing to NWR in DS-1.
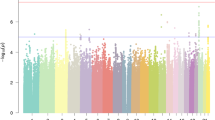
The DS-1 genome scan identified five ROIs. These regions, defined as VC-LOD > 1.0 and MCMC IR > 5 (log10(IR) > 0.7), were on chromosomes 4p, 6q, 12p, 17q, and 22q (Fig. 1). For each of the ROIs, additional markers were genotyped and VC, BO, and parametric linkage analyses were performed. With the additional markers for chromosomes 6 and 22, only modest VC-LOD and log10(IR) scores remained, and parametric multipoint LOD scores were under 1.5 (data not shown). Evidence for linkage to chromosome 17q was statistically significant with a parametric single-marker LOD score of 3.2 for D17S2193 at 98 cM. However the addition of markers resulted in reduction of the multipoint parametric LODmax to 1.99 at D17S784 at 126.8 cM, a location that did not coincide with the maximum multipoint VC-LOD and log10(IR) at 102.9 cM (D17S949) (Table 4). On chromosomes 4 and 12 the multipoint parametric LOD score results remained strong. For chromosome 4 a multipoint parametric LODmax = 2.47 was obtained at 81.5 cM, and for chromosome 12 LODmax = 2.12 was obtained at 35.2 cM. These locations approximately coincided with the locations of the maximum multipoint VC-LOD and log10(IR) (Table 4, Fig. 3).

DS-1 whole genome scan for nonword repetition using variance component linkage and Bayesian oligogenic MCMC joint segregation and linkage analysis. The dotted lines mark the thresholds for regions of interest for further analysis
The second genome scan using DS-2 identified seven ROIs, including 4p and 12p that were also identified in analysis of DS-1, (Fig. 2). For five regions identified only in DS-2 (5q, 7q, 8p, 14p and 20p), the regions with the strongest evidence for linkage were 8p, with VC-LODmax = 2.91 at 74.5 cM, 20p, with VC-LODmax = 2.57 at 23.4 cM and 7q, with a VC-LODmax = 2.42 at 190.9 cM. The 8p and 20p regions did not show evidence for linkage in DS-1, but 7q almost achieved the status as a ROI in the DS-1 sample. Detailed DS-2 analyses of regions on chromosomes 4p and 12p are shown in Table 5 and Fig. 3. On chromosome 4, the multipoint VC-LOD scores maximized close to the location implicated in the DS-1 analysis, the log10(IR) shows a broad region of linkage support, and multipoint parametric LOD scores were weakly positive in the same area. On chromosome 12, the multipoint VC-LOD scores support linkage in the same region as in DS-1, the BO analysis supports a broad region with weak evidence for linkage, and multipoint parametric LOD score analysis shows weak results outside the region of interest.

DS-2 whole genome scan for nonword repetition using variance component linkage and Bayesian oligogenic MCMC joint segregation and linkage analysis. The dotted lines mark the thresholds for regions of interest for further analysis

Multipoint variance component linkage, Bayesian oligogenic MCMC segregation and linkage and parametric linkage analysis for chromosomes 4 and 12 for DS-1, DS-2, and DS-C
Analysis of DS-C was performed with all available markers for chromosomes 4, 7, and 12. For chromosome 7q, the positions with evidence for linkage differed in the two data sets, and the combined analysis of DS-C weakened the overall evidence for linkage (results not shown). On chromosome 12, combined analysis confirmed moderately positive results over a broad region of about 20 cM (Fig. 3). The VC-LOD maximized at 1.93 (33.61 cM) and exceeded 1.5 in the interval of 21–39 cM. The MCMC log10(IR) maximized at 1.11 (27 cM) and exceeded 0.7 between 23 and 35 cM. However the strength of the parametric LOD score decreased as compared to DS-1 alone. On chromosome 4, DS-C analysis improved the evidence for linkage, with substantially increased VC-LOD and MCMC IR scores and more precise localization with LOD score analysis (Fig. 3). Multipoint parametric linkage analysis yielded a LODmax of 2.36 at 72.3 cM. The position of the maximum parametric LOD score is in good agreement with those of the maximum VC-LOD of 2.15 at D4S3242 (68.89 cM) and the maximum log10(IR) of 1.33 at 79 cM.
We obtained approximate significance levels for the linkage results presented here, uncorrected for multiple testing, under both asymptotic assumptions and with an empirical approach. LOD scores can be converted to likelihood ratio statistics, which follow a 50:50 distribution of 0 and a χ2 1 distribution under the null hypothesis of no linkage. This gives p-values under asymptotic assumptions of 0.00006, 0.0004, 0.0005, and 0.0009 for parametric LOD scores of 3.2 (on chromosome 17 in DS-1), 2.47 (on chromosome 4 in DS-1), 2.36 (on chromosome 4 in DS-C), and 2.12 (on chromosome 12 in DS-1), respectively. The VC LOD score of 2.15 that included dominance variance on chr 4 at 68.9 cM in DS-C had an asymptotic p-value of 0.0008 and an empirical p-value of 0.0051 (95% CI: 0.0037–0.0065). In the MCMC analysis of DS-C, the region of chromosome 4 between 50 and 100 cM has a posterior probability for linkage of 0.437 (0.513 for the whole chromosome).
Discussion
We performed linkage analyses using a NWR measure in multigenerational families ascertained for dyslexia. A genome scan was initially performed on 51 families with a follow-up genome scan in a replication sample of 93 families as well as targeted analysis of the combined data set. Our analyses of this extensive data set have provided suggestive and reproducible evidence in both samples for linkage of NWR to chromosomes 4p12 and 12p, and significant evidence for linkage in one of the samples to chromosome 17q.
Reading is a cognitively complex process that involves numerous functions, including perception, language, memory, and executive function as they apply to conversion of written words to speech sounds. Nonword repetition, a measure of phonological memory for reproduction of word-like speech sounds is impaired in dyslexic individuals, heritable, and stable across development. NWR may have a less complex genetic structure than the actual process of reading that draws on a wider range of neuronal-cognitive abilities. Behavioral phenotype research shows NWR is strongly associated with dyslexia (Wagner and Torgesen 1987). For these reasons, we chose to study NWR as a component phenotype for genetic linkage analysis in dyslexia.
Impairments on NWR-related tasks characterize not only dyslexia but also specific language impairment (SLI, OMIM 602081) and speech sound disorder (SSD, OMIM 608445) which overlap phenotypically. SLI is characterized by failure to acquire age-appropriate language despite normal non-verbal intelligence. SSD is characterized by developmentally inappropriate errors in speech production. All three disorders are associated with phonological impairments. In dyslexia, evidence for association of NWR to alleles of DYX1C1, a candidate gene on chromosome 15q21, was found by quantitative transmission disequilibrium testing in family trios (Wigg et al. 2004), while in a Dutch sib-pair study, evidence was also found for linkage of NWR to chromosome 15q21, as well as to 11p (de Kovel et al. 2008). In a quantitative genome-wide linkage analysis of NWR in sib pairs with SLI, loci on chromosomes 16q and 19q were identified (Newbury et al. 2002; Newbury et al. 2004). In sib pairs with SSD, a regression-based analysis of regions previously linked to dyslexia found evidence for linkage of NWR to chromosomes 6p22 and 15q21 (Smith et al. 2005).
Our detailed genome-wide analyses with three methods (VC, BO, and parametric) in multigenerational families with dyslexia did not find evidence for linkage of NWR to the previously identified loci in studies of dyslexia, SLI or SSD. Instead, we have identified significant evidence for linkage to 17q and suggestive but reproducible evidence for linkage to loci on chromosomes 4p and 12p. For chromosome 4p, addition of a replication sample and analysis of the combined samples resulted in strengthened evidence for linkage with VC and BO linkage methods. Whereas parametric LOD score analysis of the DS-2 sample alone did not show evidence for or against the linkage in the region, analysis of the combined DS-C sample allowed for better localization of the linkage peak to a region on 4p12. For chromosome 12, analysis of replication and combined samples with VC and BO methods confirmed positive results over a broad region of 20 cM.
There are several considerations to be taken into account when interpreting strength of evidence and locations of our linkage signals. Our results on chromosome 17q were statistically significant in the first sample, but did not replicate in the second sample. This lack of replication may be explained by an initial false-positive result, by use of an ill-suited analysis model, or by differences in ascertainment of pedigrees for genotyping in the two samples, which resulted in different fractions of pedigrees segregating a QTL at this position in the genome. The possibility of a poor model for parametric analysis, even for DS-1, is likely because of the positive multipoint LOD scores across a large interval and the sensitivity of the position of the maximum LOD score to multipoint vs. two-point analysis. Our results for chromosomes 4 and 12 are in the suggestive range, which raises possibility that these findings could be spurious. However, there are several reasons why we consider that to be unlikely. First is the replication of the findings with VC and BO analytic approaches. The lack of evidence for linkage in the replication sample with parametric analysis may reflect inaccuracies in the model used, which was based on a relatively simplistic segregation analysis in which only a single diallelic susceptibility locus is modeled. The BO method, which has shown evidence for the linkage in the replication sample, may more accurately approximate more complex situations that exist in this data set, such as more than one QTL in the region of interest or in the genome. Second, the identification of more than one QTL location might indicate genetic heterogeneity that could further decrease the power of the study and depress the LOD scores. Although NWR is conceptualized as cognitively and genetically less complex then dyslexia, it is still likely to be influenced by more than one QTL, and our prior segregation study of NWR provided evidence in support of an oligogenic major-gene mode of inheritance with several contributing QTLs (Wijsman et al. 2000). Third, although NWR is correlated with other phenotypes of dyslexia in our sample, the correlations between NWR and measures of accuracy and rate of real word and nonword reading are weaker (0.33–0.42) than are pairwise correlations between these reading measures (0.54–0.79) (Raskind et al. 2000). This finding supports the notion that distinct QTL’s influence different phenotypes.
The lack of the replication of the loci identified by others also warrants consideration. First, the power of our sample to detect linkage for NWR may not have been sufficient to detect the loci identified in other studies, especially if these loci were of relative small effect in our sample. Assuming four equally frequent alleles and a recombination fraction of 0.05 we estimate 0.59 and 0.32 power to obtain a LOD score of 2 or higher in parametric analysis in DS-1 and DS-2, respectively. Second, incorporation of an adjustment for VIQ may be influential because use of an IQ discrepancy criterion results in increased heritability in dyslexia (Francks et al. 2004; Olson et al. 1999), but has little effect in SLI (Bishop et al. 1995). Because linkage studies of NWR in SLI and SSD were performed without IQ-adjustment, and removing VIQ could result in removal of genetic variance and increase in error variance in residual NWR scores, we reanalyzed DS-1 without IQ as a covariate. This analysis did not find evidence for linkage of NWR to SLI or SSD regions reported by others (data not shown). Finally, we hypothesize that discrepancies in the findings are most likely due to different inclusion criteria and ascertainment strategies between the studies that might have led to identification of different biological-genetic bases.
A recent genome wide VC linkage analysis in an epidemiological sample of twins unselected for reading ability found support for seven of the eleven previously identified dyslexia locations, and evidence for two novel regions at 4p15.33 and 17p13.3 (Bates et al. 2006). The chromosome 4 multipoint VC-LODmax of 2.08 was the highest among the reading measures, and it maximized at D4S403 (29.91 cM), approximately 50 cM distal from our highest signal. As one disadvantage of the VC in comparison to parametric methods is poor QTL localization, it is possible that these two signals represent the same locus. It is also of interest that the sample in that study was representative of the population in terms of intellectual ability and not ascertained with regard to reading, although results in this study support most of the reported dyslexia loci. Our families were ascertained through probands with dyslexia, and the VIQ selection criterion resulted in an average VIQ 2/3 SD above the mean. In a genome wide scan of dyslexia-related phenotypes, in the US sib pair sample evidence was found for linkage of the marker D4S392 (78.97 cM) with a phoneme awareness task with nominal P-value of 0.0002 (Fisher et al. 2002). This marker is very close to our strongest signal on chromosome 4. Interestingly, the measure for which linkage was found, phoneme awareness, also requires use of phonological memory for storing the incoming speech sounds while they are analyzed and prepared for reproduction. However, it is a more complex measure than NWR, because in addition to repeating the nonwords, subjects are required to delete a phoneme and then only repeat the part of the word remaining after the deletion (Gayan et al. 1999). Furthermore, an additional related cognitive measure, verbal working memory, was mapped within the same broadly defined chromosome 4 linkage signal in families ascertained through a proband with schizophrenia (Paunio et al. 2004). These findings suggest that NWR may assess the storage and processing mechanism for phonological word forms and their parts in a verbal working memory architecture (Berninger et al. 2006). Further molecular genetics studies of the chromosome 4 and 12 regions with suggestive evidence for linkage to NWR in our families are needed to investigate the significance of the findings, refine locations and evaluate evidence for candidate genes.
References
Abecasis G, Cherny S, Cookson W, Cardon L (2002) Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101. doi:10.1038/ng786
Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211. doi:10.1086/301844
Amos CI (1994) Robust variance-components approach for assessing genetic linkage in pedigrees. Am J Hum Genet 54:535–543
Amos C, de Andrade M (2001) Genetic linkage methods for quantitative traits. Stat Methods Med Res 10:3–25. doi:10.1191/096228001677031143
Atwood LD, Heard-Costa NL (2003) Limits of fine-mapping a quantitative trait. Genet Epidemiol 24:99–106. doi:10.1002/gepi.10225
Bates TC, Luciano M, Castles A, Coltheart M, Wright MJ, Martin NG (2006) Replication of reported linkages for dyslexia and spelling and suggestive evidence for novel regions on chromosomes 4 and 17. Eur J Hum Genet 15(2):194–203
Berninger VW, Abbott RD, Thomson JB, Raskind WH (2001) Language phenotype for reading and writing disability: a family approach. Sci Stud Read 5:59–106. doi:10.1207/S1532799XSSR0501_3
Berninger V, Abbott R, Thomson J, Wagner R, Swanson HL, Wijsman E et al (2006) Modeling developmental phonological core deficits within a working memory architecture in children and adults with developmental dyslexia. Sci Stud Read 10:165–198. doi:10.1207/s1532799xssr1002_3
Bishop DVM, North T, Donlan C (1995) Genetic-basis of specific language impairment—evidence from a twin study. Dev Med Child Neurol 37:56–71
Bishop D, North T, Donlan C (1996) Nonword repetition as a behavioural marker for inherited language impairment: evidence from a twin study. J Child Psychol Psychiatry 37:391–403. doi:10.1111/j.1469-7610.1996.tb01420.x
Blangero J, Williams J, Almasy L (2000) Robust LOD scores for variance component-based linkage analysis. Genet Epidemiol 19:S8–S14. doi :10.1002/1098-2272(2000)19:1+<::AID-GEPI2>3.0.CO;2-Y
Boehnke M, Ploughman LM (1997) SIMLINK: a program for estimating the power of a proposed linkage study by computer simulation. Version 4.12
Bonney G (1986) Regressive logistic models for familial disease and other binary traits. Biometrics 42:611–625. doi:10.2307/2531211
Cardon LR, Smith SD, Fulker DW, Kimberling WJ, Pennington BF, DeFries JC (1994) Quantitative trait locus for reading disability on chromosome 6. Science 266:276–279. doi:10.1126/science.7939663
Chapman NH, Igo RP, Thomson JB, Matsushita M, Brkanac Z, Holzman T et al (2004) Linkage analyses of four regions previously implicated in dyslexia: confirmation of a locus on chromosome 15q. Am J Med Genet B Neuropsychiatr Genet 131:67–75
Cope N, Harold D, Hill G, Moskvina V, Stevenson J, Holmans P et al (2005) Strong evidence that KIA0319 on chromosome 6p is a susceptibility gene for developmental dyslexia. Am J Hum Genet 76:581–591. doi:10.1086/429131
Cottingham RW Jr, Idury RM, Schaffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263
Daw EW, Heath SC, Wijsman EM (1999) Multipoint oligogenic analysis of age-at-onset data with applications to Alzheimer disease pedigrees. Am J Hum Genet 64:839–851. doi:10.1086/302276
DeFries JC, Alarcon M (1996) Genetics of specific reading disability. Ment Ret Devel Dis Res Rev 2:39–47
de Kovel CG, Franke B, Hol FA, Lebrec JJ, Maassen B, Brunner H et al (2008) Confirmation of dyslexia susceptibility loci on chromosomes 1p and 2p, but not 6p in a Dutch sib-pair collection. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics 147:294–300. doi:10.1002/ajmg.b.30598
Fagerheim T, Raeymaekers P, Tonnessen FE, Pedersen M, Tranebjaerg L, Lubs HA (1999) A new gene (DYX3) for dyslexia is located on chromosome 2. J Med Genet 36:664–669
Fisher SE, DeFries JC (2002) Developmental dyslexia: genetic dissection of a complex cognitive trait. Nat Rev Neurosci 3:767–780. doi:10.1038/nrn936
Fisher SE, Francks C, Marlow AJ, MacPhie IL, Newbury DF, Cardon LR et al (2002) Independent genome-wide scans identify a chromosome 18 quantitative-trait locus influencing dyslexia. Nat Genet 30:86–91. doi:10.1038/ng792
Forrest W, Feingold E (2000) Composite statistics for QTL mapping with moderately discordant sibling pairs. Am J Hum Genet 66:1642–1660. doi:10.1086/302897
Francks C, Paracchini S, Smith S, Richardson A, Scerri T, Cardon L et al (2004) A 77-kilobase region of chromosome 6p22.2 is associated with dyslexia in families from the United Kingdom and from the United States. Am J Hum Genet 75:1046–1058. doi:10.1086/426404
Galaburda AM, LoTurco J, Ramus F, Fitch RH, Rosen GD (2006) From genes to behavior in developmental dyslexia. Nat Neurosci 9:1213–1217. doi:10.1038/nn1772
Gathercole SE, Willis CS, Baddeley AD, Emslie H (1994) The Children’s Test of Nonword Repetition: a test of phonological working memory. Memory (Hove, England) 2:103–127. doi:10.1080/09658219408258940
Gayan J, Olson RK (2001) Genetic and environmental influences on orthographic and phonological skills in children with reading disabilities. Dev Neuropsychol 20:483–507. doi:10.1207/S15326942DN2002_3
Gayan J, Olson RK (2003) Genetic and environmental influences on individual differences in printed word recognition. J Exp Child Psychol 84:97–123. doi:10.1016/S0022-0965(02)00181-9
Gayan J, Smith SD, Cherny SS, Cardon LR, Fulker DW, Brower AM et al (1999) Quantitative-trait locus for specific language and reading deficits on chromosome 6p. Am J Hum Genet 64:157–164. doi:10.1086/302191
Gilger JW, Borecki IB, DeFries JC, Pennington BF (1994) Commingling and segregation analysis of reading performance in families of normal reading probands. Behav Genet 24:345–355. doi:10.1007/BF01067536
Grigorenko EL, Wood FB, Meyer MS, Hart LA, Speed WC, Shuster A et al (1997) Susceptibility loci for distinct components of developmental dyslexia on chromosomes 6 and 15. Am J Hum Genet 60:27–39
Grigorenko EL, Wood FB, Meyer MS, Pauls JE, Hart LA, Pauls DL (2001) Linkage studies suggest a possible locus for developmental dyslexia on chromosome 1p. Am J Med Genet 105:120–129. doi :10.1002/1096-8628(20010108)105:1<120::AID-AJMG1075>3.0.CO;2-T
Hannula-Jouppi K, Kaminen-Ahola N, Taipale M, Eklund R, Nopola-Hemmi J, Kaariainen H et al (2005) The axon guidance receptor gene ROBO1 is a candidate gene for developmental dyslexia. PLOS Genetics 1:467–474. doi:10.1371/journal.pgen.0010050
Heath SC (1997) Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet 61:748–760. doi:10.1086/515506
Hokanson JE (1999) Functional variants in the lipoprotein lipase gene and risk cardiovascular disease. Curr Opin Lipidol 10:393–399. doi:10.1097/00041433-199910000-00003
Hsiung GY, Kaplan BJ, Petryshen TL, Lu S, Field LL (2004) A dyslexia susceptibility locus (DYX7) linked to dopamine D4 receptor (DRD4) region on chromosome 11p15.5. Am J Med Genet 125B:112–119. doi:10.1002/ajmg.b.20082
Hulslander J, Talcott J, Witton C, DeFries J, Pennington B, Wadsworth S et al (2004) Sensory processing, reading, IQ, and attention. J Exp Child Psychol 88:274–295. doi:10.1016/j.jecp. 2004.03.006
Igo RP Jr, Wijsman EM (2008) Empirical significance values for linkage analysis: trait simulation using posterior model distributions from MCMC oligogenic segregation analysis. Genet Epidemiol 32:119–131
Igo RP Jr, Chapman NH, Berninger VW, Matsushita M, Brkanac Z, Rothstein JH et al (2006a) Genomewide scan for real-word reading subphenotypes of dyslexia: novel chromosome 13 locus and genetic complexity. Am J Med Genet B Neuropsychiatr Genet 141:15–27. doi:10.1002/ajmg.b.30245
Igo RP Jr, Chapman NH, Wijsman EM (2006b) Segregation analysis of a complex quantitative trait: approaches for identifying influential data points. Hum Hered 61:80–86. doi:10.1159/000093085
Kamhi A, Catts H (1986) Toward an understanding of developmental language and reading disorders. J Speech Hear Disord 51:337–347
Kidd T, Brose K, Mitchell KJ, Fetter RD, Tessier-Lavigne M, Goodman CS et al (1998) Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell 92:205–215. doi:10.1016/S0092-8674(00)80915-0
Knoblauch H, Schuster H, Luft FC, Reich J (2000) A pathway model of lipid metabolism to predict the effect of genetic variability on lipid levels. J Mol Med 78:507–515. doi:10.1007/s001090000156
Lyon GR, Shaywitz SE, Shaywitz B (2003) A definition of dyslexia. Ann Dyslexia 53:1–14. doi:10.1007/s11881-003-0001-9
McGrath LM, Smith SD, Pennington BF (2006) Breakthroughs in the search for dyslexia candidate genes. Trends Mol Med 12:333–341. doi:10.1016/j.molmed.2006.05.007
Meng H, Smith SD, Hager K, Held M, Liu J, Olson RK et al (2005) DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc Nat Acad Sci USA 102:17053–17058. doi:10.1073/pnas.0508591102
Newbury DF, Bonora E, Lamb JA, Fisher SE, Lai CSL, Baird G et al (2002) FOXP2 is not a major susceptibility gene for autism or specific language impairment. Am J Hum Genet 70:1318–1327. doi:10.1086/339931
Newbury DF, Cleak JD, Banfield E, Marlow AJ, Fisher SE, Monaco AP et al (2004) Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected by specific language impairment. Am J Hum Genet 74:1225–1238. doi:10.1086/421529
Nopola-Hemmi J, Myllyluoma B, Haltia T, Taipale M, Ollikainen V, Ahonen T et al (2001) A dominant gene for developmental dyslexia on chromosome 3. J Med Genet 38:658–664. doi:10.1136/jmg.38.10.658
Olson R, Datta H, Gagan J, DeFries J (1999) A behavioral genetic analysis of reading disabilities and component processes. In: Klein R, McMullen P (eds) Converging methods for understanding reading and dyslexia. MIT Press, Cambridge, pp 133–151
Ott J (1999) Analysis of human genetic linkage, 3rd edn. Johns Hopkins University Press, Baltimore
Paracchini S, Thomas A, Castro S, Lai C, Paramasivam M, Wang Y et al (2006) The chromosome 6p22 haplotype associated with dyslexia reduces the expression of KIAA0319, a novel gene involved in neuronal migration. Hum Mol Genet 15:1659–1666. doi:10.1093/hmg/ddl089
Paunio T, Tuulio-Henriksson A, Hiekkalinna T, Perola M, Varilo T, Partonen T et al (2004) Search for cognitive trait components of schizophrenia reveals a locus for verbal learning and memory on 4q and for visual working memory on 2q. Hum Mol Genet 13:1693–1702. doi:10.1093/hmg/ddh184
Petryshen TL, Kaplan BJ, Fu Liu M, de French NS, Tobias R, Hughes ML et al (2001) Evidence for a susceptibility locus on chromosome 6q influencing phonological coding dyslexia. Am J Med Genet 105:507–517. doi:10.1002/ajmg.1475
Ramus F, Rosen S, Dakin SC, Day BL, Castellote JM, White S et al (2003) Theories of developmental dyslexia: insights from a multiple case study of dyslexic adults. Brain 126:841–865. doi:10.1093/brain/awg076
Raskind W (2001) Current understanding of the genetic basis of reading and spelling disability. Learn Disabil Q 24:141–157. doi:10.2307/1511240
Raskind WH, Hsu L, Berninger VW, Thomson JB, Wijsman EM (2000) Familial aggregation of dyslexia phenotypes. Behav Genet 30:385–396. doi:10.1023/A:1002700605187
Raskind WH, Igo RP, Chapman NH, Berninger VW, Thomson JB, Matsushita M et al (2005) A genome scan in multigenerational families with dyslexia: identification of a novel locus on chromosome 2q that contributes to phonological decoding efficiency. Mol Psychiatry 10:699–711. doi:10.1038/sj.mp.4001657
Rosen GD, Bai J, Wang Y, Fiondella CG, Threlkeld SW, Loturco JJ et al (2007) Disruption of neuronal migration by RNAi of Dyx1c1 results in neocortical and hippocampal malformations. Cereb Cortex 17(11):2562–2572
SAGE (1997) Statistical analysis for genetic epidemiology, Computer program package available from the Department of Epidemiology and Biostatistics, 3.1 edn. Cleveland: Rammelkamp Center for Education and Research, MetroHealth Campus, Case Western Reserve University
Shaywitz SE, Shaywitz BA, Fletcher JM, Escobar MD (1990) Prevalence of reading disability in boys and girls. Results of the Connecticut Longitudinal Study. J Am Med Assoc 264:998–1002. doi:10.1001/jama.264.8.998
Smith SD, Pennington BF, Boada R, Shriberg LD, Tunick RA, Raitano NA (2005) Linkage of speech sound disorder to reading disability loci. J Child Psychol Psychiatry 46:1057–1066. doi:10.1111/j.1469-7610.2005.01534.x
Sviridov D, Nestel PJ (2007) Genetic factors affecting HDL levels, structure, metabolism and function. Curr Opin Lipidol 18:157–163. doi:10.1097/MOL.0b013e32803dbdd6
Szenkovits G, Ramus F (2005) Exploring dyslexics’ phonological deficit I: lexical vs sub-lexical and input vs output processes. Dyslexia (Chichester, England) 11:253–268. doi:10.1002/dys.308
Taipale M, Kaminen N, Nopola-Hemmi J, Haltia T, Myllyluoma B, Lyytinen H et al (2003) A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc Natl Acad Sci USA 100:11553–11558. doi:10.1073/pnas.1833911100
Thomson JB, Chenault B, Abbott RD, Raskind WH, Richards T, Aylward E et al (2005) Converging evidence for attentional influences on the orthographic word form in child dyslexics. J Neurolinguistics 18:93–126. doi:10.1016/j.jneuroling.2004.11.005
Torgesen J, Wagner R, Reshotte C (1999) Test of word reading efficiency (TOWRE). Pro-Ed, Austin
Vellutino FR, Fletcher JM, Snowling MJ, Scanlon DM (2004) Specific reading disability (dyslexia): what have we learned in the past four decades? J Child Psychol Psychiatry 45:2–40. doi:10.1046/j.0021-9630.2003.00305.x
Wagner R, Torgesen J (1987) The nature of phonological processing and its causal role in the acquisition of reading skills. Psychol Bull 101:192–212. doi:10.1037/0033-2909.101.2.192
Wagner R, Torgesen J, Rashotte C (1999) Comprehensive test of phonological processing (CTOPP). PRO-ED, Austin, TX
Wechsler D (1991) Wechsler intelligence scale for children—third edition (WISC-III). The Psychological Corporation, San Antonio
Wigg K, Couto J, Feng Y, Anderson B, Cate-Carter T, Macciardi F et al (2004) Support for EKN1 as the susceptibility locus for dyslexia on 15q21. Mol Psychiatry 9:1111–1121. doi:10.1038/sj.mp.4001543
Willcutt EG, Pennington BF (2000) Psychiatric comorbidity in children and adolescents with reading disability. J Child Psychol Psychiatry 41:1039–1048
Willcutt EG, Pennington BF, DeFries JC (2000) Twin study of the etiology of comorbidity between reading disability and attention-deficit/hyperactivity disorder. Am J Med Genet 96:293–301
Wijsman EM (2002) Joint linkage and segregation analysis using Markov chain Monte Carlo methods. In: Camp N, Cox A (eds) Quantitative trait loci: methods and protocols, vol 125. Humana Press, Totowa, pp 139–161
Wijsman EM, Amos CI (1997) Genetic analysis of simulated oligogenic traits in nuclear and extended pedigrees: summary of GAW10 contributions. Genet Epidemiol 14:719–735. doi :10.1002/(SICI)1098-2272(1997)14:6<719::AID-GEPI28>3.0.CO;2-S
Wijsman E, Yu D (2004) Joint oligogenic segregation and linkage analysis using Bayesian Markov chain Monte Carlo methods. Mol Biotechnol 28:205–226. doi:10.1385/MB:28:3:205
Wijsman EM, Peterson D, Leutenegger AL, Thomson JB, Goddard KA, Hsu L et al (2000) Segregation analysis of phenotypic components of learning disabilities I. Nonword memory and digit span. Am J Hum Genet 67:631–646. doi:10.1086/303044
Wijsman EM, Sung YJ, Buil A, Atkinson E, Bastone L, Christensen GB et al (2007) Summary of genetic analysis workshop 15: group 9 linkage analysis of the CEPH expression data. Genet Epidemiol 31(Suppl 1):S75–S85. doi:10.1002/gepi.20283
Wolff PH, Melngailis I (1994) Family patterns of developmental dyslexia: clinical findings. J Med Genet 54:122–131. Neuropsych Genet. doi:10.1002/ajmg.1320540207
Woodcock R (1987) Woodcock reading master tests—revised (WRMT-R). American Guidance Service, Circle Pines, MN
Acknowledgments
We thank School Psychology graduate students for administering the tests, Jennifer Thompson for contributions in working with families and supervising testers, Patricia Stock and Joan Waiss for assistance in working with families, John Wolff for sample processing and Hiep Nguyen and Ted Holzman for computer and data base support. We thank the families that have made this study meaningful and possible. Some of the analyses were performed using S.A.G.E. supported by U.S. PHS 1P41 RR03655 from NCRR. Genotyping at Marshfield MGS was supported by NO1-HV-48141 from NHLBI. P50 HD33812, K08 HD049342, R01 HD054562, and R01 GM46255 from NIH, and funds from Children’s Hospital and Regional Medical Center, Seattle, WA also supported this research. Authors report no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Stacey Cherny.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Brkanac, Z., Chapman, N.H., Igo, R.P. et al. Genome Scan of a Nonword Repetition Phenotype in Families with Dyslexia: Evidence for Multiple Loci. Behav Genet 38, 462–475 (2008). https://doi.org/10.1007/s10519-008-9215-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-008-9215-2