
Abstract
Molecular characterization of Bovine leukemia virus (BLV) isolates from Brazil using the env gene sequences revealed a high conservation of this gene. In most cases the substitutions corresponded to silent transitions. In addition, cystein residues, potential glycosylation sites, neutralization domains and other critical residues involved with the envelope structural domains and viral infectivity were conserved. Most of the substitutions found in the aminoacid sequences of the gp51 protein were localized in the G and H epitopes. Using the SIFT software, it was predicted that they should not alter the protein functions. Phylogenetic analyses showed that partial or complete env gene sequences grouped in three or four phylogenetic clusters, respectively. The sequences from the Brazilian isolates had similar mutation rates as compared to samples from other countries, and belonged to at least two phylogenetic clusters.
Similar content being viewed by others


Introduction
Bovine leukemia virus (BLV) is an infectious agent that can cause lymphomas and benign disorders which have direct or indirect financial impacts on the cattle industry. BLV is distributed worldwide, and in Brazil it is mostly found in dairy herds [1–3]. Most of the infected cattle do not show clinical signs of disease and are referred to as aleukemic [AL] [4]. Approximately, 30% of cattle naturally infected with BLV develop persistent lymphocytosis (PL) with non-malignant polyclonal expansion of CD5+ B-cells, the majority of which harbor BLV provirus [5]. After a latency period of 1–8 years, only 1–5% of the infected cattle develop malignant B-cell lymphosarcomas (LS) [6]. Thus, the progression of BLV infection is divided into three stages: AL, PL and LS.
BLV is a type C retrovirus which is genetically and structurally similar to Human and Simian T-cell lymphoma/leukemia viruses (HTLV-1, HTLV-2, STLV-1, STLV-2 and STLV-3). BLV belongs to the Deltaretrovirus genus and these viruses possess a common ancestor. The biological study of the genetic diversity and its implications are not only of interest for veterinary medicine in areas such as diagnosis, pathogenicity, and vaccine development, but also for the possibility of virus transmission to other animal species and/or to humans [7–10].
In contrast to other retroviruses, analysis of the BLV env gene sequences of isolates from different origins have demonstrated a high conservation with predominantly silent substitutions [11, 12]. However, these small alterations may affect infectivity and/or even pathogenicity of the BLV [13–17]. BLV variants were found in different geographic regions [18, 19]. These can also be detected by restriction fragment length polymorphism (RFLP) [20, 21]. In contrast, there seem to be no serologic sub-groups. The high conservation of the nucleotide sequences from isolates from different geographic areas and collected over long periods of time may be of importance in conjunction with epidemiological findings [22].
The purpose of this work was to characterize Brazilian BLV isolates by alignment of partial and complete sequences of the env gene and by phylogenetic analysis.
Materials and methods
Samples
Whole blood was obtained from eight naturally infected bovines (Table 1). At the time of sample collection none of the animals showed clinical signs characteristic for BLV infection.
DNA isolation
DNA was extracted from whole blood of the bovines, the sheep (210 days after inoculation) and the rabbits (425 days after inoculation) using the GFX Genomic Blood DNA Purification Kit® (Amersham Pharmacia Biotech, USA) following the manufacturer’s protocol. The DNA used as a positive control for the PCR was obtained from foetal lamb kidney cells (FLK) persistently infected with the BLV [23].
Amplification of the BLV env gene
DNA samples (200 ng) were amplified with the following sets of primers: BLV1-BLV2, EF-ER, BLV3-BLV4 and BLV5-BLV6 (Invitrogen, USA) (Table 2). The reaction was performed with 10 mM of Tris–HCl, (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 1% glycerol (v/v), 1% DMSO (v/v), 10 ρmoles of each primer set, 1 mM dNTP´s and 0.5 U Taq DNA polymerase (Invitrogen, USA); using the following parameters: initial incubation at 95°C for 3 min., followed by 35 cycles of denaturation at 95°C for 1 min, annealing at the corresponding temperature (Table 2) and extension at 72°C for 1 min each.
Sequencing
PCR fragments were purified from agarose gels using the Wizard™ PCR Preps DNA Purification System Kit™ (Promega, USA). PCR products were directly sequenced. The samples 8513, 89, 135, Sheep, Rabbit-2 and Rabbit-4 were sequenced using only the EF-ER primers. The purified products were sequenced using the same primers and the DNA Sequencing Kit Big Dye Terminator Version III (Applied Biosystems, USA) and the ABI PRISM 377 DNA SEQUENCER ™ (Applied Biosystems, USA).
The program BioEdit Sequence Alignment Editor version 5.0.9 [24] was used for editing, alignment, and translation of the nucleotide sequences. The alignments were constructed using the WWW based program Genomatix (http://www.genomatix.de/software_services/software/Dialign/dialign.html).
Molecular characterization and phylogenetic analysis
The strain AF257515 was used to verify the number of nucleotide and aminoacid substitutions in the Brazilian sequences. The MEGA software version 2.1 [25], implemented with the Kimura 2-parameter substitution model, was used to verify variation among sequences. The on line software SIFT (http://blocks.fhcrc.org/sift/SIFT) [26] was used to check if aminoacid substitutions would affect protein functions. Phylogenetic analyses using the Brazilian sequences and sequences available from GenBank (AF257515-Argentina; D00647-Australia; AF503581, K02251 and M35240-Belgium; AY515273, AY515274, AY515275, AY515276, AY515277, AY515278, AY515279 and AY515280-Chile; M35238-France; U87872-Germany; S83530-Italy; K02120-Japan; AF067081 and AF111171-Poland; AY078387, M35239 and M35242-USA; M92818––HTLV-1) were calculated using the ClustalX software [27], and the PHYLIP software package [28]. The distance matrices were analyzed by the Neighbor-Joining method (Kimura 2-parameter model, transition to transversion rate: 2.0) with 1000 bootstrap replicates.
BLV env sequences (444 nucleotides) from strains 30, 141, 151, 384, 485 e 8513 were submitted to restriction-enzyme site search using the on line software Restriction Mapper, http://www.arabidopsis.org/cgi-bin/patmatch/RestrictionMapper.pl in order to characterize Brazilian strains according to criteria previously used [20, 21]. The results of the restriction-enzyme site search and the phylogenetic analyses were compared.
Results and discussion
Molecular characterization
The env-gene sequences from the Brazilian isolates were submitted to GenBank and received the access numbers listed in Table 1. Using the EF-ER primers the sequences comprised 420 nucleotides (nt), and 381 positions were conserved (90.7%). When aligned with the sequence of the Argentinean isolate B19 (AF257515) the average identities comprised 97.5% among Brazilian isolates and at least 94.8% with all sequences analyzed. The sequences from the isolates obtained from the sheep, rabbit-2 and rabbit-4 were almost identical to the sequence from isolate 485. Most substitutions observed were silent transitions, predominantly G→A and C→T. This was also the case when the BLV sequences were aligned to the reference sequence of the Human immunodeficiency virus [29].
The alignment of the partial BLV env deduced aminoacid (aa) sequences revealed average identities of 92.8% with the B19 isolate (AF257515––Fig. 1). The average substitution rate between the Brazilian and the Argentinean sequences was 2.1%. The evaluated sequences include the second (aa 131–149) and the third (aa 210–225) neutralization domains and three epitopes of the env-gp51 protein: E (175–194), B (228–238) and D (251–270). Of the 32 aa substitutions in the protein of the Brazilian isolates, 12 were found in the second neutralization domain, 3 in the B epitope and 16 in the D epitope. The third neutralization domain and the E epitope were conserved in all Brazilian sequences. The residues 141 and 290 may be considered to be hot spots because of the occurrence of four and three different aa in these positions, respectively.

Alignment of the partial aminoacid env sequences from Brazilian BLV strains. A––indicates identity to AF257515 sequence. The numbers above the AF257515 sequence refer to the complete aminoacid env gene sequence
The alignment with the complete deduced env-aa sequences is shown in Fig. 2. Brazilian BLV env protein sequences were highly conserved. When compared to the AF257515 aa sequence, it was found that most substitutions in the nucleotide sequences were silent transitions, thus confirming findings where Argentinean and Japanese isolates were analyzed [22]. Of 1,545 sequenced nucleotides, 1,431 were conserved (92.6% of identity). The average substitution rate in relation to the AF257515 sequence was 3.5%, similar to the rates for env gene sequences from isolates from Australia, Belgium, France, Germany, Italy, Japan and USA (11, 16, 18, 19, 30). The highest variation between Brazilian sequences and the Argentinean sequence AF257515 was 5.3%. There were no deletions and no insertions in the Brazilian env gene sequences. In comparison to the sequence AF257515, the most conserved region was the one that codes for the signal peptide, and the less conserved one was the one that codes for the gp51 protein. This finding is in contrast with previous results [18], that showed that the signal peptide sequence was the most variable and the gp30 transmembrane protein sequence the most conserved.

Alignment of the complete aminoacid env sequences from Brazilian BLV strains. A––indicates identity to the AF257515 sequence. Peptide signal in italic (1–33), gp 51 in bold (34–301); gp30 underlined (302–515); potential glycosylation sites; G epitope: aminoacids 48, 73, 74, 82 and 121, H epitope: aminoacids 56 and 58; F epitope: aminoacid 95; E epitope: aminoacids 175–194; B epitope: aminoacids 228–238; D epitope: aminoacids 251–270; A epitope: aminoacids 282–301
Most nucleotide and aa changes were localized near to the carboxy-termini of the gp51 protein. Of 515 aa, 474 were conserved (92.0%). The average aa substitution rate in relation to the AF257515 sequence was 5.0%, higher than the average rate found between European sequences, which was between 0.5 and 2.7% [18].
The segments between aa 145–253 and 301–432 were highly conserved within Brazilian isolates (only four residues had substitutions: aa 229, 327, 324 and 385). Probably, these conserved regions have important biological functions, related, for example, with the interaction with cell receptors. Other authors reported divergent results, they classified the segments between aa 34–121 and 235–254 as variable, and those between aa 127–234 and aa 255–301 as conserved [14, 18, 30].
None of the aminoacid changes found were located in a region with an important biological function, like cystein residues, potential glycosylation sites, the peptide involved with the fusion ability of BLV, the first and third neutralization domains of gp51 protein, the peptide that induces T-CD8+ response, the P and D residues involved in the proliferative response of T-helper cells in bovines, the WAPE tetrapeptide critical for infection, the cytoplasmatic domains YXXL of the gp30 protein (which are similar to ITAM) and the E epitope. The results suggest that these regions are important to maintain the biological activity of the gp51 and gp30 proteins, thus confirming previous reports [11, 14, 16, 18, 19, 30–35].
In the second neutralization domain (131–149), there were three aa substitutions (aa 134, 141 and 144). This region may be critical for the interaction with the cell receptor and for infection [36]. There was only one aa substitution (385, P→S) in the highly immunogenic gp30 protein epitope GD21 (aa 351–398). Three (aa 48, 74 and 82) of the five aa that constitute the G epitope were changed in the isolate 151 [18]. These changes may alter this epitope, although none of the Brazilian isolates has lost the H and F epitopes on the env protein [18]. In addition, one aa of the B epitope (aa 228–238) on the protein from isolate 141 and one aa from isolate 384 were changed. Four residues of the D epitope (251–270) were substituted. One residue of the A epitope (282–301) was altered in the protein of the Brazilian isolate 151. Twenty-two residues of the BLV gp51 protein were modified in the Brazilian isolates, and 17 of these residues are part of epitopes. This is in concert with the finding that most aa substitutions in the gp51 protein were not distributed at random, but occurred predominantly in epitopes [14]. Of 14 tyrosine residues, just one, residue 229, was changed in the protein of the Brazilian isolates.
Twenty-four aa substitutions were considered to be tolerant and 17 to be intolerant to the protein function, as evaluated using the SIFT software. This software had an accuracy, which varied from 63 to 81%, and may be useful to select regions to study altered phenotypes [26]. Although the change in aa 74 was considered intolerant, this change was evaluated in vitro and found to be tolerant [18]. Exposed proteins on the viral surface exhibited fast evolutionary changes under host immune system selection pressure. Considering this, the high conservation of the env gene of BLV and HTLV [37] may be an evolutionary constraint or a good host adaptation [38]. Other reasons for the low variation rates of BLV may be the less number of replication cycles, the replication as a provirus, a Deltaretrovirus reverse transcriptase less prone to errors and the minimum expression of structural genes in vivo [39, 40]. A low number of mutations on the env gene of Retroviruses may affect viral replication, the capacity to infect new hosts, the ability to form syncitia, the processing of the precursor glycoprotein, and alter glycoprotein expression on the cell surface [37–43]. Previous reports showed a low level of intrastrain variation in the env and ltr genes of BLV in the asymptomatic and symptomatic stages of the infection [44, 45] and among sequences of different isolates [10–12, 19–12, 30], in agreement with this work.
Restriction-enzyme site search and phylogenetic analysis of partial (5099–5542) nucleotide sequences of BLV env gene
Table 3 shows the results of the restriction-enzyme sites found in the partial nucleotide sequences (444) of the env gene of the Brazilian isolates. Four of six Brazilian isolates were classified in the Australian genotype [20] and two were not classified. Ninety percent of the 309 sequences analyzed by RFLP were classified in Belgian, Australian or Japanese genotypes [20]. Using another classification model [21], four Brazilian sequences would be classified in Genotype 1 and two in Genotype 6. Japanese samples and isolates from bovines with lymphosarcomas predominate in Genotype 1, as well as samples from cell lines infected with BLV [21]. The samples 141, 384 and 485 originated from the same farm, where the herd consisted of animals bought from different places, were classified in at least two genotypes. The presence of more than one genotype in the same herd was noticed in open herds and in herds that used public pastures [20, 21]. However it was observed that the presence of more than one genotype in a closed herd and the introduction of a second genotype might have been a consequence of transmission of the virus by insect vectors from neighbur farms, or might be due to the repeated use of needles in mass vaccination campaigns [38]. In our work the presence of sites for the restriction enzymes BamHI and Bgl1 did not contribute to genotype differentiation because they were present in all sequences [46].
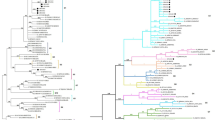
The typing of Brazilian BLV env sequences by phylogenetic analysis correlates well to the typing by the presence of restriction enzyme sites. Just one sequence in this work showed divergence between these two methods of typing. Other authors found divergences [20, 47], or a high agreement between RFLP and phylogenetic analysis [38]. The phylogenetic analysis with 444 nucleotide sequences of the BLV env gene demonstrated the presence of four phylogenetic clusters, but with low bootstrap values (Fig. 3a): Cluster 1, with sequences from Argentina and Brazil; Cluster 2, with sequences from Australia, Brazil, Japan and USA; Cluster 3, with sequences from Chile and Europe; and Cluster 4, with sequences from Brazil, Chile and Italy. Other authors analyzing the same region of the env gene found four clusters: the first with sequences from Argentina and Brazil; the second with European and Brazilian sequences; the third with sequences from Germany and Japan; and the fourth with sequences from Australia, USA and other countries [38]. A different distribution of the isolates in four clusters was also reported [47]: the first with sequences from Chile, Europe and USA; the second with sequences from Argentina, Brazil and Japan; the third with sequences from Argentina, Australia, Brazil, Japan and USA; and the fourth with sequences from Chile and Italy.

(a) Phylogenetic analysis with partial sequences of BLV env gene (Corresponds to base 5099–5542). Neighbor-joining method, 1,000 bootstrap replications. The same set of sequences were subjected to a restriction enzyme site search. (b) Phylogenetic analysis with complete sequences of BLV env gene. Neighbor-joining method, 1,000 bootstrap replications
The distribution of Brazilian isolates in three phylogenetic clusters and the presence of isolates from a single herd in distinct phylogenetic clusters (141, 384 and 485) may be due to the introduction of bovines from different countries and/or to the transit of animals within Brazil.
Phylogenetic analysis of complete nucleotide sequences of BLV env gene
The phylogenetic analysis of the complete sequences of the BLV env gene showed the presence of three phylogenetic clusters (Fig. 3b): Cluster 1, with sequences from Argentina and Brazil; Cluster 2, with sequences from Brazil, Japan and USA; and Cluster 3, with European sequences.
Although the three phylogenetic analyses employed different groups of nucleotide sequences, some similarities were found between them. Each one of the three phylogenetic analyses showed at least two clusters with Brazilian isolates, one cluster with sequences from Brazil, Japan and the USA, one cluster with sequences from Argentina and Brazil, and finally one cluster with sequences from Europe. Isolate 151-BRA was not included in any cluster; as up to date no complete nucleotide sequences are available in GenBank that allow grouping of this isolate in comparison to the use of the partial env nucleotide sequences (Fig. 3b). Contrary to what was previously described [47], there was a tendency of the isolates to be grouped by geographical origin. Neither breed nor age interfered with the results of phylogenetic analysis. It is possible that there was a relation between animal importation and distribution in the different phylogenetic clusters.
In some of the nodes, the bootstrap values were low, as observed previously [38], even when the complete sequences of the env gene were used. This may be due to the small number of available sequences and/or limited genetic variation. Some authors performed phylogenetic analyses with sequences of the BLV pol gene and HTLV-1 p21e gene, which proved inadequate to distinguish between viral strains [48, 49]. The genetic variation of Primate T Leukemia Virus 1 (PTLV-1) in the env gene was greater than that of HTLV-II and BLV [50]. As the variation among viruses of the same genus is not expected to differ much, probably a more extensive analysis with bovine samples would reveal a more distinct phylogenetic tree [50]. These analyses should consider other regions of the BLV genome and use a greater number of samples from all over the world.
It was not possible to conclude if the observed changes in the sequences of the Brazilian BLV isolates had functional consequences. This gene, however, is highly conserved and immunodominant. Small alterations in its structure may change viral pathogenesis and cell and host tropism. Further studies are necessary in the area of virus evolution, variability, and BLV biological characteristics, such as cell tropism, replication kinetic, antigenicity and pathogeneticity. The results of this work provide some supplementary information to studies involving structure, diagnosis, vaccine development and phylogenetic analysis of BLV.
References
R.S. Castro, R.C. Leite, J.J. Abreu, A.P. Lage, I.B. Ferraz, Z.I. Lobato, S.L. Balsamao, Trop. Anim. Hlth. Prod. 24, 173–176 (1992)
J.L. D´Angelino, M. Garcia, E.H. Birgel, Trop. Anim. Hlth. Prod. 30, 13–158 (1998)
M.F. Camargos, C.B. Melo, R.C. Leite, D. Stancek, Z.I.P. Lobato, M.A. Rocha, G.N. Souza, J.K.P. Reis, Ciênc. Vet. Tróp. 5, 20–26 (2002)
A. Burny, Y. Cleuter, R. Kettmann, M. Mammerickx, G. Marbaix, D. Portetelle, A. vanden Broeke, L. Willems, R. Thomas, Vet. Microbiol. 17, 197–218 (1988)
M.L. Mirsky, C.A. Olmstead, Y. Da, H.A. Lewin, J. Virol. 70, 2178–2183 (1996)
I. Schwartz, D. Levy, Vet. Res. 25, 521–36 (1994)
R. Johnson, J.B. Kaneene, Vet. Bull. 62, 287–312 (1992)
C.R. Wyatt, D. Wingett, J.S. White, C.D. Buck, D. Knowles, R. Reeves, N.S. Magnuson, J. Virol. 63, 4498–4506 (1989)
R.B. Brandon, H. Naif, R.C. Daniel, M.F. Lavin, Res. Vet. Sci. 50, 89–94 (1991)
N. Sagata, T. Yasunaga, J. Tsuzuku-Kawamura, K. Ohishi, Y. Ogawa, Y. Ikawa, Proc. Natl. Acad. Sci. USA 82, 677–681 (1985)
S. Dube, G. Dolcini, L. Abbot, S. Mehta, D. Dube, S. Gutierrez, C. Ceriani, E. Esteban, J. Ferrer, B.J. Poiesz, Virology 277, 379–386 (2000)
M.F. Camargos, D. Stancek, M.A. Rocha, L.M. Lessa, J.K.P. Reis, R.C. Leite, J. Vet. Med. B 49, 325–331 (2002)
N.R. Rice, R.M. Stephens, D. Couez, J. Deschamps, R. Kettmann, A. Burny, R.V. Gilden, Virology 138, 82–93 (1984)
D. Portetelle, D. Couez, C. Bruck, R. Kettmann, M. Mammerickx, M. Vander Maaten, R. Brasseur, A. Burny, Virology 169, 27–33 (1989)
L. Willems, J.S. Gatot, M. Mammerickx, D. Portetelle, A. Burny, P. Kerkhofs, R. Kettmann, J. Virol. 69, 4137–4141 (1995)
H. Fechner, P. Blankenstein, A.C. Looman, J. Elwert, L. Geue, C. Albrecht, A. Kurg, D. Beier, O. Marquardt, D. Ebner, Virology 237, 261–269 (1997)
L. Willems, A. Burny, D. Collete, O. Dangoisse, F. Dequiedt, J.S. Gatot, P. Kerkhofs, L. Lefebvre, C. Merezak, T. Peremans, D. Portetelle, J.C. Twizere, R. Kettmann, Aids. Res. Hum. Retroviruses 16, 1787–1795 (2000)
R.Z. Mamoun, M. Morisson, N. Rebeyrotte, B. Busetta, D. Couez, R. Kettmann, M. Hospital, B. Guillemain, J. Virol. 64, 4180–4188 (1990)
E. Molteni, A. Agresti, R. Meneveri, A. Marozzi, M. Malcovati, L. Bonizzi, G. Poli, E. Ginelli, J. Vet. Med. B 43, 201–211 (1996)
D. Beier, P. Blankenstein, O. Marquardt, J. Kuzmak, Berl Munch Tierartztl Wochensch 114, 252–256 (2001)
M. Licursi, Y. Inoshima, D. Wu, T. Yokoyama, E.T. Gonzalez, H. Sentsui, Virus. Res. 86, 101–110 (2002)
M. Licursi, Y. Inoshima, D. Wu, T. Yokoyama, E.T. Gonzalez, H. Sentsui, Vet. Microbiol. 96, 17–23 (2003)
M.J. Van der Maaten, J.M. Miller, Bibl. Haematol. 43, 360–362 (1976)
T.A. Hall, Nucl. Acids. Symp. Ser. 41, 95–98 (1999)
S. Kumar, K. Tamura, I.B. Jakobsen, M. Nei, Bioinformatics 17, 1244–1245 (2001)
P.C. Ng, S. Henikoff, Genome Res. 11, 863–874 (2001)
J.D. Thompson, D.G. Higgins, T.J. Gibson, Nucleic Acid. Res. 22, 4673–4680 (1994)
J. Felsenstein, Cladistics 5, 164–166 (1989)
J.E. Fitzgibbon, S. Mazar, D.T. Dubin, AIDS Res. Hum. Retroviruses 9, 833–838 (1993)
J. Coulston, H. Naif, R. Brandon, S. Kumar, S. Khan, R.C.W. Daniel, M.F. Lavin, J. Gen. Virol. 71, 1737–1746 (1990)
C. Bruck, D. Portetelle, A. Burny, J. Zavada, Virology 122, 353–362 (1982)
C. Bruck, N. Rensonnet, D. Portetelle, Y. Cleuter, M. Mammerickx, A. Burny, R. Mamoun, B. Guillemain, M.J. Van der Maaten, J. Ghysdael, Virology 136, 20–31 (1984)
I. Callebaut, A. Burny, V. Krchnak, H. Gras-Masse, B. Wathelet, D. Portetelle, Virology 185, 48–55 (1991)
J.S. Gatot, I. Callebaut, J.P. Mornon, D. Portetelle, A. Burny, P. Kerkhofs, R. Kettmann, L. Willems, J. Biol. Chem. 273, 12870–12880 (1998)
E.R. Johnston, K. Radke, J. Virol. 74, 2930–2935 (2000)
J.S. Gatot, I. Callebaut, C. Van Lint, D. Demonte, P. Kerkhofs, D. Portetelle, A. Burny, L. Willems, R. Kettmann, J. Virol. 76, 7956–7967 (2002)
C. Pique, T. Tursz, M.C. Dokhelar, Embo J. 9, 4243–4248 (1990)
G. Monti, R. Schrijver, D. Beier, Arch. Virol. 150, 443–458 (2005)
L. Haas, T. Divers, J.W. Casey, J. Virol. 66, 6223–6225 (1992)
L.M. Mansky, H.M. Temin, J. Virol. 68, 494–499 (1994)
C. Cheng-Mayer, T. Shioda, J.A. Levy, J. Virol. 65, 6931–6941 (1991)
V. Voneche, D. Portetelle, R. Kettmann, L. Willems, K. Limbach, E. Paoletti, J.M. Ruysschaert, A. Burny, R. Brasseur, Proc. Natl. Acad. Sci. USA 89, 3810–3814 (1992)
L. Willems, R. Kettmann, F. Dequiedt, D. Portetelle, V. Voneche, I. Cornil, P. Kerkhofs, A. Burny, M. Mammerickx, J. Virol. 67, 4078–4085 (1993)
L. Willems, E. Thienpont, P. Kerkhofs, A. Burny, M. Mammerickx, R. Kettmann, J. Virol. 67, 1086–1089 (1993)
L. Willems, P. Kerkhofs, A. Burny, M. Mammerickx, R. Kettmann, Virology 206, 769–772 (1995)
Y. Asfaw, S. Tsuduku, M. Konishi, K. Murakami, T. Tsuboi, D. Wu, H. Sentsui, Arch. Virol. 150, 493–505 (2005)
R. Felmer, G. Munoz, J. Zuniga, M. Recabal, Vet. Microbiol. 108, 39–47 (2005)
G.D. Ehrlich, J. Andrews, M.P. Sherman, S.J. Greenberg, B.J. Poiesz, Virology, 186, 619–627 (1992)
S. Dube, S. Bachman, T. Spicer, J. Love, D. Choi, E. Esteban, J.F. Ferrer, B.J. Poiesz, J. Gen. Virol. 78, 1389–1398 (1997)
R.N. Perzova, T.P. Loughran, S. Dube, J. Ferrer, E. Esteban, B.J. Poiesz, Br. J. Haematol. 109, 64–70 (2000)
Acknowledgements
We thank Leandro Moreira Lessa for the technical help and Mr. Maurício Baltazar de Carvalho Filho for critical reading of the manuscript. This study was funded by the Fundação de Amparo a Pesquisa de Minas Gerais (FAPEMIG).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Camargos, M.F., Pereda, A., Stancek, D. et al. Molecular characterization of the env gene from Brazilian field isolates of Bovine Leukemia Virus. Virus Genes 34, 343–350 (2007). https://doi.org/10.1007/s11262-006-0011-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-006-0011-x