
Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive fibrotic disease limited to the lung, with high variability in the course of disease from one patient to another. Patients with IPF may experience acute respiratory deteriorations; many of these acute declines are idiopathic and are termed acute exacerbations (AE) of IPF. In these cases, the exclusion of alternative causes of rapid deterioration, including heart failure, bilateral pneumonia or pulmonary embolism, is a challenging goal. AE may occur at any time during the course of IPF, although they are more common in patients with more progressive disease and gastroesophageal reflux. Surgical lung biopsy or even surgical procedures in organs other than the lungs may also trigger AE, mainly in rapidly progressive or advanced IPF. Current diagnostic criteria include the presence of new-onset ground glass opacities or airspace consolidation superimposed on an underlying usual interstitial pneumonia pattern seen on high-resolution computed tomography. The outcome is poor with a short-term mortality in excess of 50 % despite therapy. Currently, there is no treatment with demonstrated efficacy for AE-IPF: empirical high-dose corticosteroid therapy is generally used, with or without immunosuppressive agents, with limited evidence. On the other hand, there is hope that new treatments to slow down progression of IPF will translate into a reduction of AE-IPF’s occurrence. In conclusion, although significant progress in assessing disease severity in IPF has been made, AEs remain unpredictable and are associated with a high risk of death. Improvements in our understanding of the etiology, risk factors, clinical predictors and epidemiology are needed. It is the goal of clinical researchers in the field to provide respiratory physicians with evidence-based guidance to identify patients who may benefit from therapy for preventing or treating AE-IPF.
Similar content being viewed by others

Introduction
Idiopathic pulmonary fibrosis (IPF), the most common form of interstitial pneumonia of unknown cause, is a chronic and progressive fibrotic process limited to the lung, occurring primarily in older adults, and associated with a histopathologic and radiologic pattern of a usual interstitial pneumonia (UIP). The diagnosis of IPF requires the exclusion of other forms of interstitial pneumonia, especially associated with occupational/environmental exposure, medication or systemic disease [1].
Although the median survival is about 2.5–3.5 years from the time of diagnosis, there is high variability in the evolution of IPF from one patient to another, and between different periods in time in a given individual. Therefore, predicting the outcome in IPF is challenging. Major progress has been made in recent years, with particular efforts to identify patients with the highest risk of disease progression who might benefit the most from treatment interventions. Clinical phenotypes with different survival have been identified. Overall, a poorer prognosis is associated with older age (>70 years), smoking history, low body mass index, severe physiological impairment, large radiological extent of disease and the presence of pulmonary hypertension [2].
Patients with IPF generally have a relatively slow, but progressive clinical course and are often referred to a pulmonary specialist months to years after the onset of symptoms [3]. At presentation, lung physiology displays a restrictive pattern, usually with a forced vital capacity (FVC) between 70 and 80 % of predicted values, with or without hypoxemia at rest, worsening during exercise. During the course of the disease, progression is often unpredictable: the decrease in FVC is often not linear from ~75 % (when diagnosis is made) to ~25 % of predicted values (an FVC lower than 25–30 % of predicted is invariably associated with death within weeks or months.) Besides the typical pattern of chronic progressive disease, a subgroup of patients, mainly male cigarette smokers, have a rapidly progressive course associated with shorter survival, known as accelerated IPF, despite similar presentation at the time of diagnosis [4].
Patients with IPF may also experience acute respiratory deteriorations. Many of these acute declines are idiopathic, i.e. without identifiable causes (infections, left heart failure, pulmonary embolism, etc.). These episodes of idiopathic acute deterioration have been termed acute exacerbations (AE) of IPF [5].
Both accelerated IPF and AE-IPF can occur successively in the same patient, suggesting that these different IPF phenotypes may represent an acceleration of the underlying fibroproliferative disorder.
In contrast to AE of asthma or chronic obstructive pulmonary disease, AE-IPF designates idiopathic acute worsening with progression of fibrosis. This article will review the definition, the pathophysiology and the clinical, diagnostic and therapeutic issues related to AE-IPF.
Definition
In 1993, Kondoh et al. [6] described an acute clinical deterioration in three IPF patients in whom acute influenza-like symptoms, cough, fever, leukocytosis and progressive hypoxia developed in the absence of an identified infection. Histologic findings from open-lung biopsy showed both UIP and an organizing acute lung injury pattern.
More recently, Collard et al. [5]—in a consensus statement aimed at giving a standard definition of AE-IPF—describe this event as an acute, clinically significant deterioration of unidentifiable cause in a patient with underlying IPF and distinct from acute respiratory worsening related to a known cause. Criteria for the consensus definition of AE-IPF are listed in Table 1. As criteria may be lacking [in patients with a clinical worsening for more than 30 days, or in patients in whom HRCT or bronchoalveolar lavage (BAL) has not been performed], IPF patients with clinical worsening of unknown cause but not fulfilling all criteria should be considered as “suspected AE-IPF,” an event that nevertheless is relevant in practice and possibly in clinical trials [5].
Acute exacerbations are not restricted to IPF, but may also complicate the course of other interstitial lung diseases, including nonspecific interstitial pneumonia [7], connective tissue diseases-related interstitial lung disease (mainly rheumatoid arthritis) [8] and hypersensitivity pneumonitis [9]. Acute worsening of sarcoidosis has been described [10], but does not seem to be comparable to AE-IPF.
Pathophysiology
Triggers
Although AE-IPF is considered an idiopathic event, the finding of diffuse alveolar damage (DAD) suggests an acute injurious insult and a histological similarity with fibroproliferative ARDS. However, the presentation of some patients with fever, “flu-like” symptoms and neutrophilia in BAL fluid specimens plausibly implicates an unrecognized infectious etiology. Wootton et al. [11] observed in 9 % of the patients with AE-IPF the presence of common respiratory viral infection (parainfluenza, rhinovirus, coronavirus); conversely, no viruses were detected in the BAL from stable patients. Furthermore, pan-viral microarrays reveal additional evidence of viral infection [herpes simplex virus, Epstein–Barr virus and torque teno virus (TTV)], in patients with AE-IPF. TTV infections are significantly more common in patients with acute exacerbation than stable controls, but present in a similar percentage of disease controls with acute lung injury. Deep sequencing of a subset of acute exacerbation cases confirms the presence of TTV, but does not identify additional viruses.
Furthermore, the detection of elevated pepsin levels in the BAL of AE-IPF patients suggests that aspiration could be responsible for the occurrence of AE-IPF in a subgroup of patients [12]. Consistent with these results, treatment with proton pump inhibitor significantly reduces the incidence of AE-IPF [13].
In another subgroup of patients, the occurrence of AE-IPF may be caused by the increased mechanical stress of the lungs that may be secondary to surgery (lung biopsies, lung resection or nonrespiratory surgery) or BAL. Prolonged mechanical ventilation, high tidal volume and high concentration of supplemental oxygen during surgery have been suggested as potential causes [14]. A recent study indicates that ambient air pollution may also cause AE-IPF, particularly by ozone and nitrogen dioxide [15]. Finally, some drugs may be responsible for the occurrence of AE-IPF, including biologic (anakinra, etanercept and infliximab) and nonbiologic (ambrisentan) agents and immunomodulatory (everolimus, interferon alpha and beta) and antineoplastic agents (both conventional chemotherapy and new drugs as tyrosine kinase inhibitors). All these potential causative drugs suggest that different causes or pathways may be responsible for the occurrence of AE-IPF in different patients.
Biological research
The molecular mechanisms related to this condition are also poorly known. Recent studies emphasize the role of immunologic regulation. It has been in fact shown that annexin 1 is an autoantigen able to induce both a humoral and cellular immunologic response in patients with AE-IPF, and that the N-terminal portion of annexin plays a role in the pathogenesis of this condition [16].
Reduced expression of heat shock protein 47 (HSP47)—a collagen-specific molecular chaperone, essential for the synthesis and secretion of collagen molecules—correlates with reduced collagen production and fibrosis progression. Serum HSP47 levels are significantly higher in AE-IPF than in stable IPF, suggesting a role for this molecule in AE-IPF pathogenesis. Furthermore, HSP47 expression is greater in DAD than in UIP tissue [17]. Consistent with these results, IPF patients with anti-HSP47 autoantibodies have more near-term lung function impairment and 6-month mortality compared to negative IPF patients.
In another study, α-defensins—a family of mammalian neutrophil peptides with various biological functions—are among the most up-regulated genes in AE-IPF in gene expression microarray analysis [18]. Corresponding peripheral blood protein levels are elevated in patients with AE-IPF in the absence of infection. Furthermore, serum levels of the ST2 protein [a member of the interleukin-1-receptor family expressed mainly in T-helper type-2 cells and induced by proinflammatory proteins such as tumor necrosis factor (TNF)-alpha and/or interleukin-1 beta] are also significantly higher in patients with AE-IPF than in controls [19].
However, as shown by Collard et al. [20] in a study detecting the plasma biomarker profile of AE-IPF and comparing it to profiles of stable IPF and ARDS, AE-IPF is characterized by increased type II alveolar epithelial cell injury proliferation markers, such as Krebs von den Lunden (KL-6) and surfactant protein (SP)-D, endothelial cell injury markers (i.e., von Willebrand factor) and coagulation abnormalities, such as protein C, thrombomodulin or plasminogen activator inhibitor (PAI)-1. Interestingly, there is little evidence about the role of type 1 alveolar epithelial cell injury or substantial acute inflammation.
Incidence
The reported incidence of AE-IPF varies widely among studies, but it is now recognized as a relatively common event [2]. Data from the placebo arms of large clinical trials find an incidence of AE-IPF of 6–16 % over a period of 1 year [21–23]. The incidence is lower (2.3 %) in another trial [24].
In another randomized, controlled trial of anticoagulant therapy in IPF involving 56 patients followed for approximately 3 years, 32 (57 %) were re-hospitalized for AE-IPF, out of whom 53 % died [25]. Of note, subjects were initially enrolled in this study during an admission to the hospital, suggesting that the study population was enriched for prior episodes of AE-IPF or rapidly progressive disease. The incidence of AE-IPF may vary greatly with the severity of disease, the delay since the diagnosis of IPF, diagnostic criteria used and efforts made to rule out infection. This is illustrated by data from a recent high-quality IPF trial, in which only about half the cases of acute deterioration suspected by investigators to be AE-IPF were eventually confirmed to be AE-IPF by an adjudication committee [23].
Although data from trials are informative, participants are generally selected as having early rather than advanced disease, with possibly low progression of disease as compared to the general IPF population, therefore possibly underestimating the frequency of AE-IPF. Nevertheless, somewhat comparable results were obtained in a retrospective review of an observational cohort of 147 patients with biopsy-proven IPF, with 11 meeting the criteria for AE-IPF [26]. The 2-year incidence of AEs is 9.6 %, and AE-IPF-related mortality is 78 %; the incidence of AE-IPF in this study with a biopsy-confirmed diagnosis of IPF may differ from those in which IPF was diagnosed without biopsy due to potential selection bias. The time from the first visit to the onset of AE-IPF ranges between 3 and 60 months. The retrospective design of this study may have underestimated the incidence rate, as 14 additional cases of acute worsening could not be included due to missing data. This south-Korean study suggests potential higher mortality in the Asian population compared to Caucasian patients, which might be linked to genetic susceptibility. Mortality may vary, however, with the diagnostic criteria used, the population studied, comorbidities, potential genetic factors and, most of all, the time at which mortality is assessed. Whether earlier diagnosis and management might improve outcome is unknown.
In a recent larger retrospective study of 461 patients with IPF (269 of which were biopsy proven) from the same group, the 1- and 3-year incidences of AE-IPF are 14.2 and 20.7 %, respectively [27]. Using a different approach, Martinez et al. [28] evaluated the clinical course of patients with mild to moderate IPF and found that an apparent acute clinical deterioration preceded death in 47 % of patients who died from IPF. Autopsy findings also show that AE are the most common immediate cause of death in patients with IPF [29]. Collectively, these observations suggest that the incidence of AE-IPF may be underestimated in IPF clinical trials and may represent a significant proportion of lethal events.
Risk factors
Several risk factors for AE-IPF have been identified. AE may occur at any time during the course of IPF, although they are more common in patients with more progressive disease such as those with decline in FVC at 6 months after baseline [30]. There is no clear association with age. A smoking history may protect from AE-IPF [27], although AE-IPF appear to be more common in males [26], but data are conflicting regarding smoking. In one study, a comparison of clinical features between IPF patients with and without AE-IPF shows no difference in age, smoking status, oxygenation and lung function at the time of diagnosis [26]. Ambient air pollution, particularly the increase in exposure to ozone and nitrogen dioxide emanating largely from motor vehicles, increases the risk of AE-IPF [15]. Pulmonary hypertension at the time of evaluation for lung transplantation increases the risk of subsequent AE in IPF, likely through endothelial dysfunction [31]. Data from several studies suggest seasonal variations in AE-IPF, with an increased frequency during winter months [32–34]. Patients with a history of AE-IPF have a higher risk of subsequent AE-IPF events [34]. Gastroesophageal reflux is also a risk factor for AE-IPF, as administration of proton pump inhibitors significantly reduces the incidence of AE-IPF [13]. Pepsin levels, a marker of gastric aspiration, are elevated in BAL fluid from patients during AE-IPF compared with those obtained in stable disease [12]. Symptomatic reflux may explain a higher risk of AE in patients with asymmetrical IPF [35].
Several observations suggest that patients undergoing surgical lung biopsy, or even surgical procedures in organs other than the lungs, may be at higher risk for AE-IPF, although distinguishing these cases from postsurgical acute respiratory distress syndrome can be difficult [36, 37]. The exact mechanism by which surgical procedures may trigger an AE is presently unknown. Patients with more severe disease (particularly with low DLco) or requiring mechanical ventilation seem to be at higher risk of AE after surgery [38]. There have been case reports of AE-IPF developed after BAL procedure [39]. However, in a number of those cases, BAL or lung biopsy may have been performed in subjects with an already accelerated course of disease who would be more prone to develop an AE-IPF.
AE-IPF were also observed in patients receiving chemotherapy for lung cancer, during the immediate post-operative period after lung resection for lung cancer, or following radiotherapy [40–42, 43, 44]. AE has been reported in patients with the syndrome of combined pulmonary fibrosis and emphysema [8], especially during treatment for lung cancer, and following radiation therapy or surgery [45], with a particularly high risk of death in this population with severely impaired gas exchange. In fact, chemotherapeutic agents can cause acute interstitial pneumonia in subjects without underlying respiratory diseases, with a high incidence in some series [46].
Clinical, imaging and pathology findings
AE-IPF can occur at any time during the course of the disease and may also be the mode of presentation of IPF [47]. Several retrospective studies evaluated the clinical characteristics in patients with AE-IPF [26, 27, 34, 48, 49]. The mean age is around 70 years as expected for IPF patients. The great majority of the subjects present with dyspnea and severe hypoxemia, and some with fever. Cough and flu-like symptoms can be present. Medical history should focus on smoking habits, toxic exposure and prescribed medications. Physical findings include tachypnea, cyanosis, digital clubbing, bilateral inspiratory crackles and edema of the lower extremities.
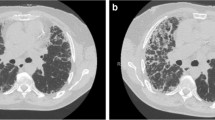
The short-term prognosis is dismal, with a median survival of about 2 months from the onset of the AE [27]. There is great heterogeneity in the degree of pulmonary functional impairment, time from the IPF diagnosis, need for supplemental oxygen, use of corticosteroids or immunosuppressive drugs prior to the acute event, and degree of hypoxemia at presentation. Many patients show severe respiratory failure and would theoretically be eligible for mechanical ventilation [50]. On high-resolution computed tomography (HRCT), characteristic findings of AE-IPF are new-onset ground glass opacities or airspace consolidation superimposed on an underlying UIP pattern. [51, 52] Different patterns have been described, with higher mortality and less response to therapy in subjects with diffuse ground glass attenuation than in those with peripheral or disseminated (patchy) opacities, likely reflecting disease extent rather than distinct pathogenic processes [51] (Fig. 1).

High-resolution computed tomography of the chest in a patient with AE-IPF. Upper panel UIP pattern with basal subpleural reticulation, traction bronchiectasis and mild honeycombing; lower panel 2 months later, superimposed ground glass opacities are visible, with architectural distortion
Lung pathology in AE-IPF is characterized by a combination of underlying UIP pattern associated with superimposed acute lung injury with diffuse alveolar damage (Fig. 2) or organizing pneumonia features [52–55]. In addition to AE-IPF, the pattern of diffuse alveolar damage is observed in patients with adult respiratory distress syndrome (ARDS), a syndrome associated with different diagnoses, most of them from known causes (such as sepsis, pneumonia or trauma), and in idiopathic acute interstitial pneumonia, a distinct entity also characterized by rapidly progressive hypoxemia, mortality of 50 % or more and no proven treatment [56–59]. Survivors of AE-IPF usually have a good long-term prognosis (similar to ARDS survivors), but some experience recurrences or chronic, progressive functional deterioration. [59].

Postmortem pathologic examination of the right lung in a patient who died from acute exacerbation of IPF. a Macroscopic examination, showing dense, liver-like aspect of the lung, with reticulation on the surface. b Microscopic view of the lower lobe showing chronic fibrosis with UIP pattern with honeycombing and cystic changes and diffuse alveolar damage with hyperplastic pneumocytes (H&E stain magnification ×40) (courtesy of Dr Giulio Rossi, Modena, Italy)
Acute exacerbations are an independent prognostic factor in IPF [30, 31]. More than half of the AE events are lethal, with mortality greater than 90 % in patients requiring ventilatory assistance [5].
Diagnosis
The current criteria for AE-IPF can be found in Table 1. The diagnosis requires ruling out various potential causes of clinical worsening, including especially lung infection, pulmonary embolism and left heart failure. Further causes of acute respiratory distress syndrome may be easily identified due to the clinical context [60].
Chest HRCT is key to assess parenchymal abnormalities as new occurrence of ground glass attenuation superimposed on the underlying UIP pattern. Ground glass attenuation may indeed be a hallmark of the underlying process of diffuse alveolar damage that develops in the setting of the ongoing fibroproliferative process. However, it is not specific for AE-IPF and can be also related to infections. CT pulmonary angiography is the gold standard for diagnosing pulmonary embolism and should be performed in the workup of any IPF patient with unexplained rapidly developing respiratory failure to rule out pulmonary embolism [61].
Ruling out infection in patients with suspected AE-IPF is challenging [62]. AE-IPF may follow a genuine episode of infection and some temporal overlap between the two conditions may occur. Attempts should be made to identify bacteria, opportunistic pathogens and respiratory viruses in respiratory samples, keeping in mind that negative microbiology does not exclude a possible infection. Noninvasive molecular methods, including urinary antigens for S. pneumoniae or Legionella as well as blood cultures and sputum cultures, are generally indicated. Performing bronchoscopy and bronchoalveolar lavage can be useful to exclude other causes of acute respiratory failure, especially infection. It is often difficult or impossible to perform due to severe dyspnea or acute respiratory failure and does not yield the diagnosis of AE-IPF itself. Therefore, the benefit:risk balance of bronchoscopy must be carefully evaluated at an individual level. The diagnostic approach of AE-IPF, and particularly the most appropriate algorithm to rule out infection and diagnose AE-IPF early, is a topic of review at the international level to provide better guidance to clinicians.
Transthoracic echocardiography is definitely useful to exclude congestive heart failure as a possible cause of acute clinical deterioration and may reveal indirect signs of pulmonary hypertension.
Although open-lung biopsy could secure the diagnosis of AE-IP as described above, it is contraindicated in most cases when AE-IPF is the suspected diagnosis [53]. Open-lung biopsy might be considered following careful benefit:risk assessment in subjects requiring mechanical ventilation and once intubated, when other diffuse parenchymal lung diseases have not been yet ruled out and when preexisting IPF had not been diagnosed prior to AE. In these cases, a UIP pattern is discovered at lung biopsy along with diffuse alveolar damage or an organizing pneumonia pattern [47].
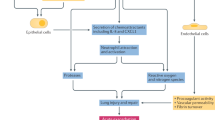
There is only very limited evidence regarding the best diagnostic approach and severity assessment of AE-IPF; nevertheless, it is conceivable that diagnosing AE-IPF at a very early stage might allow a better workup of all the diagnostic procedures and result in an improved outcome. Therefore, a timely and accurate approach with HRCT is warranted in all patients with IPF with rapid worsening of dyspnea. This may also allow a better understanding of the natural course of AE-IPF (see Fig. 3).

Proposed diagnostic algorithm for acute exacerbation of idiopathic pulmonary fibrosis. (Modified from [14])
Management
Potential preventive strategies for AE-IPF
As recent trials have demonstrated the efficacy of two antifibrotic drugs to reduce the decline in lung function in patients with IPF, there is hope that new treatments for the chronic progression of IPF will also translate in reducing the occurrence of AE-IPF. Indeed, nintedanib, that acts as a triple tyrosine kynase inhibitor, is able to reduce the annual rate of decline in FVC in patients with IPF, and FVC greater than 50 % of predicted value and carbon monoxide diffusion capacity comprised between 30 and 79 % of predicted, and increases time to the first adjudicated AE-IPF, in a pre-specified sensitivity analysis of pooled data [23], consistent with earlier findings [22] and providing proof-of-concept that drug therapy can prevent this deadly event. In a double blind, randomized controlled trial evaluating pirfenidone versus placebo, AE-IPF events occurred exclusively in the placebo group, prompting early termination of the study [63]. The number of AE-IPF or time to clinical worsening including AE-IPF was not significantly different between patients receiving pirfenidone or placebo in phase III trials, although pirfenidone significantly reduced the decline in FVC over time and improved survival at 1 year [64–66]. In future, therapeutic strategies that protect the respiratory epithelium should be evaluated [18]. Gastroesophageal reflux should be treated very actively, even if asymptomatic; clinical trials are needed to evaluate whether treatment of reflux may reduce the risk of AE-IPF.
Current therapeutic approaches
Currently, there is no treatment with demonstrated efficacy for AE-IPF, and therefore no evidence-based management strategy. This is in part due to the lack of a consistent approach to AE-IPF based on pathogenesis, as well as to the lack of randomized controlled trials, which are challenging to conduct because of unpredictable onset and rapid worsening [67].
However, most groups use high-dose corticosteroids, following international guidelines and based on expert opinion (weak positive recommendation), with no recommendation on what type, dose and duration of steroids should be used [1]. Immunosuppressive therapy may be combined with corticosteroid pulses in severe cases, yet with no clear evidence of efficacy. When used, immunosuppressive therapy generally consists in intravenous pulses of cyclophosphamide [68]; cyclosporine or tacrolimus is sometimes preferred to cyclophosphamide, based on small studies from Asia suggesting improved survival [40, 69, 70].
Anticoagulant therapy, which was suggested to improve survival in patients with IPF partly due to an effect on AE-IPF, eventually proved to be deleterious and should not be used to treat this condition [25, 71]. Alternative strategies to target activation of the coagulation cascade include the administration of recombinant human thrombomodulin, which in a retrospective study shows encouraging results [72].
Overall, evidence is currently lacking to support any particular drug in AE-IPF.
Several small studies have suggested a potential beneficial effect of polymyxin B-immobilized fiber column (PMX) hemoperfusion treatment in patients with AE-IPF, possibly through the adsorption of proinflammatory, profibrotic and proangiogenic cytokines onto PMX [73–79]. Although most of these studies suffer from retrospective noncontrolled design and heterogeneity, they support the hypothesis that extracorporeal techniques might be beneficial. Further studies are currently ongoing using plasma exchanges in patients with AE-IPF, in combination with corticosteroids and rituximab (www.clinicaltrials.gov, NCT01266317, NCT01524068.)
Due to the difficulties in differentiating (idiopathic) AE-IPF from bronchopulmonary infection occurring in the course of IPF with potential overlap, broad-spectrum antibiotics are often administered in subjects with acute worsening of dyspnea [80].
Many AE-IPF patients develop acute respiratory failure. The outcome after mechanical ventilatory support, if used, is dismal. Because the vast majority of these patients will die in the intensive care unit or during the follow-up period, a decision not to intubate is generally taken, especially when infection and potential confounding factors have been ruled out [27, 81, 82]. However, a small subset of patients might be eligible for lung transplantation, and in this case a more aggressive approach in the intensive care unit can be considered [83]. Interestingly, extracorporeal membrane oxygenation (ECMO) was able to be used successfully as a bridge to lung transplantation in 11 patients with acute respiratory failure refractory to all other interventions [84]. In this retrospective study, ECMO was used for a mean of 17 days (range 2–56 days); nine patients were transplanted, out of whom eight were alive 1 year after transplantation. Such encouraging experience highlights how organization of care and early inclusion on the transplant list of potential candidates are paramount to offer this potentially life-saving intervention in the event of AE-IPF.
Palliation of end of life secondary to AE-IPF
No studies are available for end of life care in IPF and AE-IPF. Current approaches are extrapolated from other end-stage conditions, such as lung cancer and end-stage respiratory failure. This is even more important considering that most of the AE-IPF patients are not sedated and intubated, and therefore they and their relatives are exposed to dramatic and painful symptoms and experiences.
Palliation of dyspnea and anxiety is generally managed with opioids and benzodiazepines; respiratory depression is obviously a problem and therefore an open discussion with patients and relatives is mandatory to explain the aims of palliation, as recently demonstrated by an ongoing discussion in the UK on end of life treatments [85]. Also, management of cough is important, using various types of drugs to palliate this symptom.
Limited data are available regarding noninvasive mechanical ventilation using spontaneous/timed mode or continuous positive airway pressure (CPAP) mode, but it might be useful to palliate shortness of breath [50].
A summary of the management strategies potentially useful in various stages of AE-IPF is available in Table 2.
Conclusion
Although significant progress has been made recently to better evaluate disease progression in IPF, AE remain unpredictable, causing a rapid deterioration of dyspnea and lung function, with high risk of death and associated significant psychological distress for patients and relatives. Despite progress made in awareness of physicians regarding this dramatic event, diagnosing AE-IPF remains challenging; especially, boundaries between (idiopathic) AE-IPF and infection are still unclear and require further studies to highlight the best flow charts for diagnostic and clinical management. Furthermore, management is complicated by the lack of an effective pharmacological treatment. The current approaches are based on empiric attempts, and some similarities to ARDS.
Conducting clinical trials in AE-IPF is a challenge that now needs to be addressed. Relatively few AE-IPF events were observed in recent IPF clinical trials, indicating that a very large sample size is required to capture a potential reduction in the incidence of AE-IPF in subjects receiving antifibrotic agents aiming at reducing chronic disease progression. However, studies can be conducted to specifically evaluate therapeutic approaches in patients who present a confirmed episode of AE-IPF. Large networks of patients and clinicians will be needed to assess the efficacy of therapeutic interventions, taking into account the influence of confounding factors.
Better guidance to physicians is needed to adapt investigations and criteria to the often severe manifestations that limit diagnostic procedures. International guidelines are currently under revision, with the aim of providing more realistic diagnostic criteria based on noninvasive procedures and account for current limitations in diagnosis and management [86].
The development and validation of biomarkers has been a long-standing objective in the field, which remains an area of research. Biomarkers may contribute to various aspects of the disease course, including early diagnosis, assessment of rate of progression of disease, differentiation between infection and idiopathic disease or prediction of response to therapy.
Improvement in our understanding of the etiology and epidemiology, preventative measures of potential risk factors and clinical predictors are needed. As some novel drugs especially tyrosine kinase inhibitors targeting the epidermal growth factor receptor (EGFR) are associated with high risk of ILD exacerbation, clinicians should exert particular scrutiny when using them. A wide spectrum of new biological drugs is entering clinical practice in oncology; IPF correlates with an higher risk of developing cancer: whether the next antitumoral agents increase the risk of exacerbation is unknown. Continuous post-marketing surveillance is needed to detect potential effects of new drugs on the natural history of IPF. Acute exacerbations of IPF (and of other ILD) is one of the challenges in the years to come for doctors working with this difficult group of diseases. It is the goal of clinical researchers in the field to provide tools and possibly evidence-based guidance to identify therapies for preventing or treating AE-IPF.
References
Raghu G, Collard HR, Egan JJ et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183:788–824
King TE Jr, Pardo A, Selman M (2011) Idiopathic pulmonary fibrosis. Lancet 378:1949–1961
Cottin V (2014) Current approaches to the diagnosis and treatment of idiopathic pulmonary fibrosis in Europe: the AIR survey. Eur Respir Rev 23:225–230
Selman M, Carrillo G, Estrada A et al (2007) Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One 2:e482
Collard HR, Moore BB, Flaherty KR et al (2007) Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 176:636–643
Kondoh Y, Taniguchi H, Kawabata Y, Yokoi T, Suzuki K, Takagi K (1993) Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest 103:1808–1812
Park IN, Kim DS, Shim TS et al (2007) Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest 132:214–220
Tachikawa R, Tomii K, Ueda H et al (2012) Clinical features and outcome of acute exacerbation of interstitial pneumonia: collagen vascular diseases-related versus idiopathic. Respir Int Rev Thorac Dis 83:20–27
Olson AL, Huie TJ, Groshong SD et al (2008) Acute exacerbations of fibrotic hypersensitivity pneumonitis: a case series. Chest 134:844–850
Baughman RP, Lower EE (2013) Frequency of acute worsening events in fibrotic pulmonary sarcoidosis patients. Respir Med 107:2009–2013
Wootton SC, Kim DS, Kondoh Y et al (2011) Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183:1698–1702
Lee JS, Song JW, Wolters PJ et al (2012) Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J 39:352–358
Lee JS, Collard HR, Anstrom KJ et al (2013) Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med 1:369–376
Johannson K, Collard HR (2013) Acute exacerbation of idiopathic pulmonary fibrosis: a proposal. Curr Respir Care Rep 2:233–240
Johannson KA, Vittinghoff E, Lee K, et al (2013) Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur Respir J 43:1124–1131
Kurosu K, Takiguchi Y, Okada O et al (2008) Identification of annexin 1 as a novel autoantigen in acute exacerbation of idiopathic pulmonary fibrosis. J Immunol 181:756–767
Kakugawa T, Yokota S, Ishimatsu Y et al (2013) Serum heat shock protein 47 levels are elevated in acute exacerbation of idiopathic pulmonary fibrosis. Cell Stress Chaperones 18:581–590
Konishi K, Gibson KF, Lindell KO et al (2009) Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 180:167–175
Tajima S, Oshikawa K, Tominaga S, Sugiyama Y (2003) The increase in serum soluble ST2 protein upon acute exacerbation of idiopathic pulmonary fibrosis. Chest 124:1206–1214
Collard HR, Calfee CS, Wolters PJ et al (2010) Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 299:L3–L7
King TE Jr, Albera C, Bradford WZ et al (2009) Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet 374:222–228
Richeldi L, Costabel U, Selman M et al (2011) Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 365:1079–1087
Richeldi L, du Bois RM, Raghu G et al (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–2082
Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G (2014) Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 370:2093–2101
Kubo H, Nakayama K, Yanai M et al (2005) Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest 128:1475–1482
Kim DS, Park JH, Park BK, Lee JS, Nicholson AG, Colby T (2006) Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J 27:143–150
Song JW, Hong SB, Lim CM, Koh Y, Kim DS (2011) Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 37:356–363
Martinez FJ, Safrin S, Weycker D et al (2005) The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 142:963–967
Daniels CE, Yi ES, Ryu JH (2008) Autopsy findings in 42 consecutive patients with idiopathic pulmonary fibrosis. Eur Respir J 32:170–174
Kondoh Y, Taniguchi H, Katsuta T et al (2010) Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 27:103–110
Judge EP, Fabre A, Adamali HI, Egan JJ (2012) Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur Respir J 40:93–100
Collard HR, Yow E, Richeldi L, Anstrom KJ, Glazer C (2013) Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res 14:73
Olson AL, Swigris JJ, Raghu G, Brown KK (2009) Seasonal variation: mortality from pulmonary fibrosis is greatest in the winter. Chest 136:16–22
Simon-Blancal V, Freynet O, Nunes H et al (2012) Acute exacerbation of idiopathic pulmonary fibrosis: outcome and prognostic factors. Respiration 83:28–35
Tcherakian C, Cottin V, Brillet PY et al (2011) Progression of idiopathic pulmonary fibrosis: lessons from asymmetrical disease. Thorax 66:226–231
Kondoh Y, Taniguchi H, Kitaichi M et al (2006) Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med 100:1753–1759
Ghatol A, Ruhl AP, Danoff SK (2012) Exacerbations in idiopathic pulmonary fibrosis triggered by pulmonary and nonpulmonary surgery: a case series and comprehensive review of the literature. Lung 190:373–380
Park JH, Kim DK, Kim DS et al (2007) Mortality and risk factors for surgical lung biopsy in patients with idiopathic interstitial pneumonia. Eur J Cardiothorac Surg 31:1115–1119
Sakamoto K, Taniguchi H, Kondoh Y et al (2012) Acute exacerbation of IPF following diagnostic bronchoalveolar lavage procedures. Respir Med 106:436–442
Sakamoto S, Homma S, Miyamoto A, Kurosaki A, Fujii T, Yoshimura K, Cyclosporin A (2010) Cyclosporin A in the treatment of acute exacerbation of idiopathic pulmonary fibrosis. Intern Med 49:109–115
Watanabe A, Kawaharada N, Higami T (2011) Postoperative acute exacerbation of IPF after lung resection for primary lung cancer. Pulm Med 2011:960316
Goto T, Maeshima A, Akanabe K, Oyamada Y, Kato R (2011) Acute exacerbation of idiopathic pulmonary fibrosis of microscopic usual interstitial pneumonia pattern after lung cancer surgery. Ann Thorac Cardiovasc Surg 17:573–576
Nagano T, Kotani Y, Fujii O et al (2012) A case of acute exacerbation of idiopathic pulmonary fibrosis after proton beam therapy for non-small cell lung cancer. Jpn J Clin Oncol 42:965–969
Takeda A, Enomoto T, Sanuki N et al (2008) Acute exacerbation of subclinical idiopathic pulmonary fibrosis triggered by hypofractionated stereotactic body radiotherapy in a patient with primary lung cancer and slightly focal honeycombing. Radiat Med 26:504–507
Girard N, Marchand Adam S, Naccache JM et al (2014) Lung cancer in combined pulmonary fibrosis and emphysema: a series of 47 Western patients. J Thorac Oncol 9:1162–1170 (in press)
Minegishi Y, Takenaka K, Mizutani H et al (2009) Exacerbation of idiopathic interstitial pneumonias associated with lung cancer therapy. Intern Med 48:665–672
Sakamoto K, Taniguchi H, Kondoh Y, Ono K, Hasegawa Y, Kitaichi M (2009) Acute exacerbation of idiopathic pulmonary fibrosis as the initial presentation of the disease. Eur Respir Rev Off J Eur Respir Soc 18:129–132
Ambrosini V, Cancellieri A, Chilosi M et al (2003) Acute exacerbation of idiopathic pulmonary fibrosis: report of a series. Eur Respir J 22:821–826
Huie TJ, Olson AL, Cosgrove GP et al (2010) A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: aetiology and outcomes. Respirology 15:909–917
Yokoyama T, Kondoh Y, Taniguchi H et al (2010) Noninvasive ventilation in acute exacerbation of idiopathic pulmonary fibrosis. Intern Med 49:1509–1514
Akira M, Kozuka T, Yamamoto S, Sakatani M (2008) Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 178:372–378
Silva CIS, Muller NL, Fujimoto K et al (2007) Acute exacerbation of chronic interstitial pneumonia: high-resolution computed tomography and pathologic findings. J Thorac Imaging 22:221–229
Churg A, Wright JL, Tazelaar HD (2011) Acute exacerbations of fibrotic interstitial lung disease. Histopathology 58:525–530
Parambil JG, Myers JL, Ryu JH (2005) Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest 128:3310–3315
Dallari R, Foglia M, Paci M, Cavazza A (2004) Acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J 23:792
de Hemptinne Q, Remmelink M, Brimioulle S, Salmon I, Vincent JL (2009) ARDS: a clinicopathological confrontation. Chest 135:944–949
Matthay MA, Ware LB, Zimmerman GA (2012) The acute respiratory distress syndrome. J Clin Invest 122:2731–2740
Mukhopadhyay S, Parambil JG (2012) Acute interstitial pneumonia (AIP): relationship to Hamman-Rich syndrome, diffuse alveolar damage (DAD), and acute respiratory distress syndrome (ARDS). Semin Respir Crit Care Med 33:476–485
Travis WD, Costabel U, Hansell DM et al (2013) An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 188:733–748
Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342:1334–1349
Torbicki A, Perrier A, Konstantinides S et al (2008) Guidelines on the diagnosis and management of acute pulmonary embolism: the Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J 29:2276–2315
Antoniou KM, Cottin V (2012) The challenge of acute exacerbation of pulmonary fibrosis. Respir Int Rev Thorac Dis 83:13–16
Azuma A, Nukiwa T, Tsuboi E et al (2005) Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 171:1040–1047
Taniguchi H, Ebina M, Kondoh Y et al (2010) Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J Off J Eur Soc Clin Respir Physiol 35:821–829
Noble PW, Albera C, Bradford WZ et al (2011) Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 377:1760–1769
King TE Jr, Bradford WZ, Castro-Bernardini S et al (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370:2083–2092
Bhatti H, Girdhar A, Usman F, Cury J, Bajwa A (2013) Approach to acute exacerbation of idiopathic pulmonary fibrosis. Ann Thorac Med 8:71–77
Morawiec E, Tillie-Leblond I, Pansini V, Salleron J, Remy-Jardin M, Wallaert B (2011) Exacerbations of idiopathic pulmonary fibrosis treated with corticosteroids and cyclophosphamide pulses. Eur Respir J 38:1487–1489
Inase N, Sawada M, Ohtani Y et al (2003) Cyclosporin A followed by the treatment of acute exacerbation of idiopathic pulmonary fibrosis with corticosteroid. Intern Med 42:565–570
Horita N, Akahane M, Okada Y et al (2011) Tacrolimus and steroid treatment for acute exacerbation of idiopathic pulmonary fibrosis. Intern Med 50:189–195
Noth I, Anstrom KJ, Calvert SB et al (2012) A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 186:88–95
Tsushima K, Yamaguchi K, Kono Y et al (2014) Thrombomodulin for acute exacerbations of idiopathic pulmonary fibrosis: A proof of concept study. Pulm Pharmacol Ther 29:233–240
Enomoto N, Suda T, Uto T et al (2008) Possible therapeutic effect of direct haemoperfusion with a polymyxin B immobilized fibre column (PMX-DHP) on pulmonary oxygenation in acute exacerbations of interstitial pneumonia. Respirology 13:452–460
Seo Y, Abe S, Kurahara M et al (2006) Beneficial effect of polymyxin B-immobilized fiber column (PMX) hemoperfusion treatment on acute exacerbation of idiopathic pulmonary fibrosis. Intern Med 45:1033–1038
Yoshida T, Kodama M, Tamura Y et al (2007) [Direct hemoperfusion with a polymyxin B-immobilized fiber column eliminates neutrophils and improves pulmonary oxygenation—a comparison of two cases with acute exacerbation of idiopathic pulmonary fibrosis]. Nihon Kokyuki Gakkai Zasshi J Jpn Respir Soc 45:890–897
Abe S, Azuma A, Mukae H et al (2012) Polymyxin B-immobilized fiber column (PMX) treatment for idiopathic pulmonary fibrosis with acute exacerbation: a multicenter retrospective analysis. Intern Med 51:1487–1491
Hara S, Ishimoto H, Sakamoto N et al (2011) Direct hemoperfusion using immobilized polymyxin B in patients with rapidly progressive interstitial pneumonias: a retrospective study. Respiration 81:107–117
Oishi K, Mimura-Kimura Y, Miyasho T et al (2013) Association between cytokine removal by polymyxin B hemoperfusion and improved pulmonary oxygenation in patients with acute exacerbation of idiopathic pulmonary fibrosis. Cytokine 61:84–89
Guerin C (2011) Extracorporeal management of acute lung disease. Respiration 81:105–106
Agarwal R, Jindal SK (2008) Acute exacerbation of idiopathic pulmonary fibrosis: a systematic review. Eur J Intern Med 19:227–235
Rangappa P, Moran JL (2009) Outcomes of patients admitted to the intensive care unit with idiopathic pulmonary fibrosis. Crit Care Resusc 11:102–109
Al-Hameed FM, Sharma S (2004) Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J 11:117–122
Gaudry S, Vincent F, Rabbat A et al (2014) Invasive mechanical ventilation in patients with fibrosing interstitial pneumonia. J Thorac Cardiovasc Surg 147:47–53
Dellgren G, Schersten H, Kjellman U (2011) et al [ECMO can be a bridge to lung transplantation. New method saves life in acute pulmonary failure according to a retrospective study]. Lakartidningen 108:1493–1497
One chance to get it right. Improving people’s experience of care in the last few days and hours of life. Leadership Alliance for the Care of Dying People (2014). http://www.gov.uk/government/uploads/system/uploads/attachment_data/file/323188/One_chance_to_get_it_right.pdf
Ryerson CJ, Collard HR (2014) Acute exacerbations complicating interstitial lung disease. Curr Opin Pulm Med 20:436–441
Conflict of interest
Dr Cottin reports personal fees from Acetilon, Bayer, Boehringer Ingelheim, Gilead, GSK, Intermune, Roche and Sanofi, outside the submitted work; Dr Cottin’s wife is an employee of Sanofi Pasteur. Fabrizio Luppi, Stefania Cerri, Sofia Taddei and Giovanni Ferrara have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Luppi, F., Cerri, S., Taddei, S. et al. Acute exacerbation of idiopathic pulmonary fibrosis: a clinical review. Intern Emerg Med 10, 401–411 (2015). https://doi.org/10.1007/s11739-015-1204-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-015-1204-x