
Abstract
Leishmaniasis is a group of diseases with a spectrum of clinical manifestations ranging from cutaneous ulcers to visceral leishmaniasis, which results from the bite of an infected sandfly to human. Attempts to develop an effective vaccine have been shown to be feasible but no vaccine is in active clinical use. This study adopts a Reverse Vaccinology approach to identify common vaccine candidates from both highly pathogenic species Leishmania major and Leishmania infantum. Total proteome of both species were compared to identify common proteins, which are further taken for sub-cellular localization and transmembrane helices prediction. Plasma membrane proteins having only one transmembrane helix were first identified and analyzed which are non-homologous in human and mouse in order to avoid molecular mimicry with other proteins. Selected proteins were analyzed for their binding efficiency to both major histocompatibility complex (MHC) class I and class II alleles. As a result, 19 potential epitopes are screened in this study using different approaches, which can be further verified through in vivo experiments in MHC compatible animal models. This study demonstrates that Reverse Vaccinology approach has potential in discovering various immunogenic antigens from in silico analysis of pathogen’s genome or proteome instead of culturing the whole organism by conventional methods.
Similar content being viewed by others

Introduction
Leishmaniasis, a vector-borne disease caused by obligate intracellular protozoa, is characterized by vast diversity and by specificity within that diversity. The disease is endemic in focal areas of ~90 countries in the tropics, subtropics, and southern Europe, in settings that range from deserts to rain forests and from rural to urban areas. Infection in humans is caused by ~20 Leishmania species (Leishmania and Viannia subgenera), which are transmitted by ~30 species of phlebotomine sandflies Phlebotomus (Old World) and Lutzomyia (New World). Leishmania are digenetic protozoa which inhabit two highly specific hosts, the sandfly, where they grow as motile flagellated promastigotes in the gut, and the mammalian macrophage, where they survive and grow intracellularly as non-flagellated amastigotes in the phagolysosome. Amid this diversity, particular parasite, vector, and host species maintain the transmission cycle in a given setting [1, 2].
Traditionally, leishmaniasis has been classified into three groups according to the clinical manifestations of disease. Cutaneous leishmaniasis is, by and large, a self-limiting, but chronic skin ulcer developing at the site of the sandfly bite, which may take months to heal. Mucocutaneous leishmaniasis initially causes similar skin ulcers that heal, but subsequently lesions reappear, primarily in the mucous tissue of the nose and mouth. These are often accompanied by secondary infections and massive tissue destruction. Visceral leishmaniasis is a very severe systemic disease, with the organisms homing to the liver, spleen, and bone marrow. Visceral leishmaniasis is usually fatal if not treated [2]. The immune response to Leishmania infection is dependent on both the species of the parasite infecting the host and the genetics of the host. The severity of disease caused by a particular species may vary markedly between individuals; hence, one species of Leishmania can cause more than one clinical syndrome [3]. There are several key components involved in the immune response to Leishmania. The outcome of infection is largely dependent on the ability of the host to mount a protective T-helper-1 (Th1) response versus the ability of the parasite to evade and manipulate the host's immune system [4]. Macrophages and effector molecules, dendritic cells (DC), T-helper cells (CD4þ T cells), cytotoxic T cells (CD8þ T cells), natural killer (NK) cells, and cytokines are all, in one way or another, considered to play important roles in the immune response to Leishmania infection [5].
Although chemotherapeutic treatments for the leishmaniasis exist, the drugs are costly, limited, and toxic [6]. Furthermore, available treatments are threatened by drug resistance, which is reviewed by Croft et al. [7]. Even with treatment, various disease forms can cause lifelong disfigurement and scarring. Thus, as with all infectious diseases, prevention of leishmaniasis is superior to a cure. A prophylactic vaccination would prove to be the most effective strategy to control infection and spreading of this group of diseases [8]. However, despite substantial effort spent in developing a vaccine, there is currently no licensed vaccine against human leishmaniasis [9]. One of the requirements of an “ideal” anti-leishmanial vaccine is for it to be effective against more than one Leishmania species in order to protect individuals in areas where cutaneous and visceral leishmaniasis, for example, coexist. Several studies [10–23] highlight the complexity of the problem and the difficulties facing the design of a pan-Leishmania vaccine. Factors, such as virulence, genetic differences between Leishmania species as well as host genetic factors controlling the response to different Leishmania species [24] suggest that such a vaccine may not be feasible. However, the availability of the genome sequence of Leishmania major, Leishmania infantum, and other species may allow us to identify the genes responsible for the different disease phenotypes. This may lead to the identification of shared antigens, which could be incorporated in a pan-Leishmania vaccine [25].
With the advent of genome era, drastic revolution has emerged in the vaccine development strategies and has catalyzed a shift from conventional culture-based approaches to genome-based vaccinology. The application of genome analysis to vaccine development has given rise to a concept termed as Reverse Vaccinology which initiated with a positive feedback loop in terms of development and application of novel approaches to the field of vaccinology. It relies on the combined use of immunological and genomic information to identify relevant protein antigens for diagnostic or vaccine purposes [26]. In our study, we have identified few peptide sequences which can be used as common vaccine targets from both L. major and L. infantum with the help of Reverse Vaccinology approach.
Materials and Methods
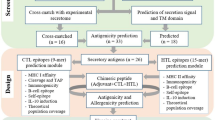
The workflow of our approach has been shown in Fig. 1. Steps are explained below:

Workflow of Reverse Vaccinology approach to identify potential vaccine candidates from L. mojor and L. infantum. Ptn proteins; PM plasma membrane proteins; EC extra cellular proteins; TMH transmembrane helix
-
1.
Data acquisition
Total protein sequences from L. major and L. infantum were downloaded from NCBI Genome Project in the form of flat file. Genome information is presented in Table 1. Homologous proteins from both organisms were screen by stand-alone BLAST program [27]. In order to consider each homologous sequence, it is important to identify bidirectional best hits which were taken by keeping first genome as reference genome and second genome as comparative and the same procedure applied vice versa.
Table 1 Summary of the L. major, L. infantum, and L. braziliensis genomes -
2.
Sub-cellular localization and transmembrane helix prediction
For better confirmation, all selected proteins were analyzed first by PSORT [28] for sub-cellular localization followed by transmembrane helix prediction using TMHMM [29]. Plasma membrane and extra cellular proteins screened from PSORT were given to TMHMM and those proteins having only one transmembrane helix were selected further. It has been reported that 250 out of 600 vaccine candidates from N. meningitides B failed to be cloned and expressed due to the presence of more than one transmembrane spanning region [30]. Therefore, proteins having multiple transmembrane helices were removed before doing further analysis.
-
3.
Identification of non-homologous proteins to human and mouse
Cell surface and secreted proteins resulted from previous steps were checked for their homology with human and mouse using stand-alone BLAST tool. Proteins which are non-homologous to human and mouse are considered further in order to prevent possible auto-immune response during experimental validation.
-
4.
Prediction of major histocompatibility complex (MHC) class I binding epitopes
BIMAS [31] helped to identify peptides of all proteins those bind to MHC class I molecules with a good binding affinity, which is based on the half-time of dissociation of the β2 microglobulin from HLA (human leukocyte antigen). A cutoff value of ≥100 was chosen for peptide selection. SYFPEITHI [32] and ProPred1 [33] are the other algorithms which predict binding of nonameric peptides to multiple MHC class I HLA alleles. The MHC alleles which were selected for this analysis and found to bind with majority of proteins are HLA_0201, HLA-A2, HLA-A 0205, HLA-Cw 0602, HLA-A2.1, HLA-A3, HLA-B14, HLA-B 5401, and HLA-B 5102.
-
5.
Prediction of MHC class II binding epitopes
The analysis for class II HLA binding was carried out to predict the binding affinity for MHC class II alleles DRB1_0101, DRB1_0102, DRB1_1101, DRB1_1104, DRB1_1501, DRB1_1502, DRB1_0402, DRB1_0404, DRB1_0405, DRB1_1301, and DRB1_1302 using ProPred [34]. The objective of using more than one tool was to select only those peptides with positive binding score with multiple methods. This would reduce chances of failure in experiments.
-
6.
Exclusion of “self” peptides
A vaccine candidate with similar sequence to the host (e.g., human or mouse) is likely to be a poor immunogen due to epitope mimicry, or if an immune response is triggered, cause autoimmunity in the host [35–37]. To consider this aspect, all predicted class I and class II HLA peptides were searched again in human and mouse genome database using BLAST and peptides which show no similarity are selected as the final set of vaccine candidates.
Results and Discussion
By adopting in silico analysis, we have identified about 19 proteins which show high potential for being common vaccine candidates of L. major and L. infantum. Figure 2 represents overall statistics of the process.

Overall statistics of the process. From 8,122 common proteins of L major and L. infantum, only 19 peptides are screened by the process
Around 8,122 proteins were found to be homologous in both L. major and L. infantum using stand-alone BLAST program. These proteins are considered for sub-cellular localization prediction using WolfPsort. Of the 8,122 proteins, 1,350 proteins were predicted as plasma membrane and 910 as extracellular proteins, which were taken for the prediction of number of transmembrane helices present using TMHMM server. Of the 2,260 proteins, 903 sequences were found to be having the transmembrane helix (TMH) ≤ 1 and 1,257 sequences have TMH > 1. Screened transmembrane proteins are taken for human and mouse BLAST analysis to eliminate all possible homologous proteins, which possibly cause auto-immune response while clinical trials in human and mouse.
Binding analysis of the allele HLA A 0201 which is the most studied HLA MHC class 1 allele [38] was selected for predicting the binding efficiency of the non-homologous proteins with BIMAS and SYFPEITHI tools. Out of the 526 proteins, 50 proteins were found not to bind with the above-mentioned allele with the BIMAS at the predicted threshold of T (1/2) ≥ 100. The rest 476 sequences were analyzed for the efficiency of binding. ProPred 1 tool was also used to analyze binding efficiency with all the alleles given in this tool. The MHC alleles which were found to bind with majority of proteins are HLA_0201, HLA-A2, HLA-A 0205, HLA-Cw 0602, HLA-A2.1, HLA-A3, HLA-B14, HLA-B 5401, and HLA-B 5102.
Binding of peptides to class 2 MHC molecules was carried out for the proteins by using ProPred tool. MHC alleles which were found to bind majority of proteins are DRB1_0101, DRB1_0102, DRB1_1101, DRB1_1104, DRB1_1501, DRB1_1502, DRB1_0402, DRB1_0404, DRB1_0405, DRB1_1301, and DRB1_1302. Figure 3 shows number of alleles binding to highest and lowest number of proteins.

Number of HLA class I and class II alleles binding to highest and lowest number of selected peptides
The T cell epitopes were selected based on the fact that those peptides which have affinity for MHC/HLA molecules are more likely to be recognized by the T cell receptors of specific T cells. Furthermore, only those peptides were selected which has the efficiency to bind with both MHC class 1 and class 2 molecules as these would reduce the number of peptides needed for population coverage for vaccine construct. The selected peptides were checked for the molecular mimicry or cross reactivity between self epitopes and pathogen epitopes to avoid the auto-immune responses [37]. Table 2 represents list of peptides are predicted to bind to depicted HLA class I and class II alleles.
Tagatose-6-phosphate kinase-like protein (XP_822202.1) is predicted to be an extracellular protein with one TMH. The epitope sequence selected lies in the outside region of TMH, which proves that amino acid residues can interact with the host and serves to generate the immune response. Extracellular factors produced by Leishmania spp., Trypanosoma cruzi, and Trypanosoma brucei are found to be important in the host–parasite relationship. Furthermore, these extracellular (secreted) proteins increase parasite replication inside macrophages. Materials secreted by the parasite are found to be involved in the process helping the parasite to survive in an environment more favorable for its own development [38]. In addition, previous studies indicate that trypanosomatid secreted factors elicit strong immunity and protection against infection in mice and dogs. Thus secreted factors could be a source of antigens for vaccine development, as demonstrated in the pathogen Mycobacterium tuberculosis [39].
Phosphatidylinositol 3-kinase-like protein (XP_822211.1) is predicted to be a plasma membrane protein having 1 TMH. At a molecular level, L. infantum rapidly induced activation of phosphatidylinositol 3-kinase/Akt and extracellular signal-regulated kinase, whereas no effect was observed in the c-Jun N-terminal kinase and p38 mitogen-activated protein kinase proinflammatory pathways. Moreover, parasites actively promoted cleavage of the nuclear factor-κB p65(RelA) subunit, causing its impairment. The blockade of phosphatidylinositol 3-kinase/Akt by either treatment of bone marrow-derived dendritic cells (DCs) with wortmannin or transfection with an Akt dominant-negative mutant resulted in a strong decrease in infection rates, revealing for the first time a crucial role of this pathway on Leishmania engulfment by DCs. Overall, our data indicate that activation of Akt and impairment of nuclear factor-κB are responsible for immunogenicity subversion of L. infantum-infected DCs [40].
XP_001687567.1 is a surface antigen protein and is predicted to be an extracellular/secreted protein. This protein has one TMH, these are expressed in the infective stages of parasite and is thought that over expression of these proteins results in avirulence due to selective depletion of specific lipid species and decreased expression of the major surface glycoprotein GP63 [40].
Phosphoglycan beta 1,3 galactosyltransferase 4 (XP_822217.1, XP_822221.1, and XP_001686570.1) and glycosomal membrane protein (XP_843475.1) are phosphoglycan family molecules and comprises of glycolipids and glycoproteins containing repeating units of Gal(/31-4)Man(α1-)PO4 with or without additional glycan side chains. They are involved in preventing complement mediated lysis in promastigote, serving as a ligand for receptor mediated endocytosis of the parasite by the macrophage and inhibiting phagosome–endosome fusion [41].
Proteophosphoglycan ppg4 (XP_843162.1), proteophosphoglycan ppg5 (XP_843163.1), and proteophosphoglycan ppg1 (XP_843164.1) are secreted glycoconjugates which form a highly viscous mesh within which the parasite lies embedded and are thought to contribute to the formation of the vacuole in the macrophages [42].
Tuzin protein (XP_001686384.1) is involved in macrophage binding and show structural similarity not sequence identity with T. cruzi. Receptor-type adenylate cyclase a-like protein (XP_001686897.1) is cAMP (cyclic adenosine monophosphate) signaling proceeds along very similar pathways in all kinetoplastid pathogens (T. cruzi, the Leishmania, and T. brucei). Their adenylyl cyclases are structurally very different from the human enzymes and appear to function as enzyme-linked cell surface receptors. They might represent the major sensory apparatus of the kinetoplastids, guiding much of their environmental sensing and host/parasite interaction. The cAMP-specific phosphodiesterases of the kinetoplastids are rather similar to those of human cells and might function in similar ways [43].
Five hypothetical proteins XP_822224.1, XP_001685357.1, XP_001685522.1, XP_001685181.1, and XP_822244.1 have been found in the selected epitopes. Though they are predicted to be epitopes by the MHC class 1 and class 2 tools, their detail functional analysis can be known in the further studies.
Conclusion
Vaccine candidates selected for Leishmaniasis caused by L. major and L. infantum seems to be the potential one through the analysis made by Reverse Vaccinology approach, anyhow, verification of these candidates is to be justified through the confirmation made by wet lab analysis. In silico approach towards the vaccine development is a novel and integrative method of the available Bioinformatics tools which holds to be a promise in the renaissance brought in the vaccine development. From 8,122 common proteins, only 19 epitopes are screened in this study using different approaches. Further detail study is to be done for making out the best epitopes screened on the basis of available multiple analytical tools and wet lab analysis.
Future Prospects
The main objective of our study was to predict potential vaccine candidate for both highly virulent species of Leishmania using various computational algorithms. In future, in vivo and in vitro studies can be carried out to check immune response against predicted epitopes. Moreover, molecular modeling and energy calculations can be carried out to find efficiency of HLA-peptide binding.
References
Fauci, A. (2008). Harrison's principles of internal medicine (17th ed.). NY: McGraw-Hill Medical.
Handman, E. (1999). Advances in Parasitology, 44, 1–39.
Awasthi, A., Mathur, R., & Saha, B. (2004). Indian Journal of Medical Research, 119, 238–258.
Vanloubbeeck, Y., & Jones, D. (2004). Annals of the New York Academy of Sciences, 1026, 267–272.
Liese, J., Schleichera, U., & Bogdan, C. (2008). Immunobiology, 213, 377–387.
Murray, H., et al. (2005). Lancet, 366, 1561–1577.
Croft, S., Sundar, S., & Fairlamb, H. (2006). Clinical Microbiology Reviews, 19, 111–126.
Rodriguez-Cortes, A., et al. (2007). Vaccine, 25, 7962–7971.
Reithinger, R., et al. (2007). The Lancet Infectious Diseases, 7, 581–596.
Adler, S., & Gunders, A. E. (1964). Transactions of the Royal Society of Tropical Medicine and Hygiene, 58, 274–277.
Mauel, J., & Behin, R. (1982). In S. Cohen & K. S. Waren (Eds.), Immunity to Parasitic Infections (pp. 3443–3463). Oxford: Blackwell Sci.
Lainson, R., & Bray, R. S. (1966). Transactions of the Royal Society of Tropical Medicine and Hygiene, 60, 526–532.
Lainson, R., & Shaw, J. J. (1966). Transactions of the Royal Society of Tropical Medicine and Hygiene, 60, 533–535.
Lainson, R., & Shaw, J. J. (1977). The Journal of Tropical Medicine and Hygiene, 80, 29–35.
Lujan, R., Chapman, W. L., Jr., Hanson, W. L., & Dennis, V. A. (1990). Journal of Parasitology, 76, 594–597.
Porrozzi, R., et al. (2004). The American Journal of Tropical Medicine and Hygiene, 71, 297–305.
Gicheru, M., Olobo, J. O., & Anjili, C. O. (1997). Experimental Parasitology, 85, 109–116.
Alexander, J., & Phillips, R. S. (1978). Experimental Parasitology, 45, 93–100.
Perez, H., Arredondo, B., & Machado, R. (1979). Experimental Parasitology, 48, 9–14.
Alexander, J. (1982). Transactions of the Royal Society of Tropical Medicine and Hygiene, 76, 646–649.
Alexander, J. (1988). Parasitology, 96, 297–302.
Bebars, M. A., et al. (2000). Journal of the Egyptian Society of Parasitology, 30, 137–156.
Veras, P., et al. (1999). Memorias do Instituto Oswaldo Cruz, 94, 491–496.
McMahon-Pratt, D., & Alexander, J. (2004). Immunological Reviews, 201, 206–224.
Kedzierski, L., Zhu, Y., & Handman, E. (2006). Parasitology, 133, S87–S112.
Rinaudo, C. D., Telford, J. L., Rappuoli, R., & Seib, K. L. J. (2009). Clin Invest, 119, 2515–2525.
Altschul, S. F., et al. (1990). Journal of Molecular Biology, 215, 403–410.
Horton, P., et al. (2007). Nucleic Acid Research, 37, W585–W587.
Krogh, A., Larsson, B., von Heijne, G., & Sonnhammer, E. L. L. (2001). Journal of Molecular Biology, 305(3), 567–580.
Pizza, M., et al. (2000). Science, 287, 1816–1820.
Parker, K. C., Bednarek, M. A., & Coligan, J. E. (1994). Journal of Immunology, 152, 163–175.
Rammensee, H., Bachmann, J., Emmerich, N. P., Bachor, O. A., & Stevanovic, S. (1999). Immunogenetics, 50, 213–219.
Singh, H., & Raghava, G. P. S. (2003). Bioinformatics, 19, 1009–1014.
Singh, H., & Raghava, G. P. S. (2001). Bioinformatics, 17, 1236–1237.
Wilson, C., Tiwana, H., & Ebringer, A. (2000). Microbes and Infection, 2, 1489–1496.
Nachamkin, I., Allos, B. A., & Ho, T. (1998). Clinical Microbiology Reviews, 11, 555–567.
Weber, C. A., Mehta, P. J., Ardito, M., Moise, L., Martin, B., & De Groot, A. S. (2009). Advanced Drug Delivery Reviews, 61, 965–976.
Santarém, N., Silvestre, R., Tavares, J., Silva, M., Cabral, S., Maciel J., Cordeiro-da-Silva, A. (2007). Journal of Biomedicine and Biotechnology 85154
Horwitz, M. A., Harth, G., Dillon, B. J., & Maslesa-Galic, S. (2005). Infection and Immunity, 73(8), 4676–4683.
Neves, B. M., et al. (2010). American Journal of Pathology, 177(6), 2898–2911.
McKean, P. G., Denny, P. W., Knuepfer, E., Keen, J. K., & Smith, D. F. (2001). Cellular Microbiology, 3(8), 511–523.
Peters, C., Stierhof, Y. D., & Ilg, T. (1997). Infection and Immunity, 65(2), 783–786.
Seebeck, T., Schaub, R., & Johner, A. (2004). Current Molecular Medicine, 4(6), 585–599.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
John, L., John, G.J. & Kholia, T. A Reverse Vaccinology Approach for the Identification of Potential Vaccine Candidates from Leishmania spp. Appl Biochem Biotechnol 167, 1340–1350 (2012). https://doi.org/10.1007/s12010-012-9649-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-012-9649-0