
Abstract
The protozoan parasite Leishmania donovani, a causative agent of visceral leishmaniasis, has evolved several strategies to interfere with the immune system and establish persistent infections that are potentially lethal. In this article, we discuss two mechanisms of immune evasion adopted by the parasite: the induction of immune suppressive IL-10 responses and the generation of poor and functionally impaired CD8+ T-cell responses.
Similar content being viewed by others


Introduction
Leishmaniasis is a vector-borne parasitic disease caused by protozoan of the genus Leishmania. Leishmania parasites exist as extracellular flagellated promastigotes within sandfly vectors and as intracellular amastigotes in infected mammals. Disease manifestation depends on the infecting species and ranges from subclinical infections, to self-healing cutaneous lesions, to life-threatening infections of visceral organs. Visceral leishmaniasis (VL), which is caused by L. donovani and/or L. chagasi/infantum, is characterized by chronic parasitization of the spleen, liver and bone marrow.
Most of the studies on the immunology of VL have been carried out in murine models.
Like in humans, L. donovani infections in mice also result in the parasitization of the spleen, liver and bone marrow. However, the parasite only establishes chronic infection in the spleen and bone marrow [1]. Indeed, infection in the liver is self-resolving within 6–8 weeks and relies on the development of a Th1-dominated granulomatous response [2], characterized by high IFN-γ production. The Th1 response is induced by IL-12 secreted by dendritic cells (DCs) [3] and is crucial for parasite control and resolution of the disease [3, 4].
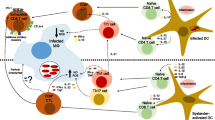
Although disease resolution occurs in the liver, the spleen and the bone marrow remain chronically infected with Leishmania [5]. As yet we know very little about the immune response in the spleen and the requirements for controlling parasite growth in this organ. IL-12, Th1 responses and CD8+ T cells are definitely involved in partially limiting parasite growth during chronic infection [4, 6]. In contrast, TNF-α, which is a key cytokine essential for parasite clearance in the liver [7], is responsible for the disruption of the microarchitecture of the splenic marginal zone that occurs during chronic infection [5, 8]. This disruption severely hampers the migration of DCs [8] and naïve T-cells to the T-cell area [9], contributing to the immune suppression observed at later stages of infection. Furthermore, the spleen mounts a regulatory response dominated by CD4+ T-cells co-producing IL-10 and IFN-γ [10], cells that have also been observed in human VL patients [11]. To date, the strategies adopted by Leishmania to evade protective immune responses in the spleen and the reasons why IFN-γ-producing Th1 responses are unable to control parasite multiplication in the spleen remain unknown. Of numerous mechanisms of immune evasion that have been proposed in the past [12–15], the induction of immune suppressive IL-10 responses is the most prominent one.
IL-10 in experimental VL
IL-10 has been implicated as a key regulator during infection with a variety of parasitic, bacterial, viral and fungal pathogens (reviewed in [16–18]). Although IL-10 responses are typically generated to balance excessive Th1 and CD8+ T-cell responses and prevent immunopathology, overproduction of IL-10 has been shown to inhibit proinflammatory responses leading to susceptibility to infectious pathogens such as Malaria [19, 20], Leishmania [21, 22], lymphocytic choriomeningitis virus (LCMV) [23, 24] and Mycobacteria spp. [25].
Various studies have now shown that one of the major contributing factors to disease progression in leishmaniasis is indeed IL-10. IL-10 receptor blockade in mice infected with L. donovani nearly abrogates infection [26]; and Il10 −/− mice are highly resistant to VL [27]. Several cell populations have been shown to express IL-10 during Leishmania infections including, natural regulatory T cells (Tregs) [12], Th1 cells [10, 13], NK cells [28], macrophages [29], B cells [28] and DCs [30]. Nevertheless, the cellular sources of IL-10 responsible for mediating disease progression and parasite persistence remain unknown.
Chronic infections in humans and in mice have been recently associated with the generation of CD4+ T-cells co-expressing IL-10 and IFN-γ [10, 11]. These cells also appear to be involved in parasite persistence in a model of cutaneous leishmaniasis [13] and to display strong host-protective functions in Toxoplasma infections [31]. Leishmania-specific IFN-γ+ IL-10+ Th1 responses are first detected 3 weeks after infection in L. donovani-infected mice [10]. Because of the lack of a specific marker characterizing these cells, formal evidence that they are in fact the major players in the induction of disease progression is still lacking.
Regulatory T cells (Tregs) have also been shown to be involved in parasite persistence during chronic Leishmania infections [12]. However, IL-10 production by these cells or an increase in Tregs frequency during chronic disease was never detected in L. donovani infections [10]. We do not exclude, though, that these cells could play an IL-10-independent role in the establishment of persistent infection in VL.
Leishmania-specific antibodies have also been shown to induce IL-10 expression by macrophages through ligation of FcγRs with IgG-opsonized parasites [29]. Since humoral responses are first detected at day 21 pi [32], which coincides with the loss of control on parasite growth in the spleen, it is possible that antibody-induced IL-10 expression by macrophages also contributes to the establishment of a persistent infection in the spleen of L. donovani-infected mice. Indeed, antibody-induced macrophage-derived IL-10 has been shown to play a pivotal role in disease progression in a low dose model of L. major [33]. Moreover, FcγrIII −/− mice are also resistant to infection with L. mexicana [34]. This literature is in agreement with studies in L. donovani-infected B-cell deficient mice (μMT mice). Interestingly, μMT mice are significantly less susceptible to L. donovani and are able to cure infection within 35 days [35]. However, this increased resistance may not only rely on the absence of antibody production and therefore lack of induction of IL-10 by macrophages. B cells have also been reported to express IL-10 during VL [28] and could theoretically contribute to parasite persistence. Although IL-10+ Th1 responses are thought to be the major source of IL-10 during chronic VL, the contribution of B-cell and macrophage-derived IL-10 to parasite persistence requires further clarification.
Since Il10 −/− mice show a certain degree of resistance to L. donovani during acute infection [27], it is possible that the downregulation of leishmanicidal responses at different stages of infection may be caused by different sources of IL-10. Several cells were recently shown to express IL-10 mRNA during acute infection; these include NK cells, CD4 T cells, CD8 T cells and B cells. Nevertheless, the relative amount of IL-10 produced by each cell population and its contribution toward establishing Leishmania susceptibility are yet to be determined.
IL-10 in human VL
In humans, plasma levels of IL-10 are strongly linked to susceptibility to a variety of infectious diseases, such as malaria [36], leprosy [37] and HIV [38]. Persistent L. donovani infections are also associated with increased levels of IL-10 in the serum of VL patients [39], however, the cellular sources of IL-10 have been elusive. Recently, a study demonstrated that individuals with VL display an accumulation of IL-10 mRNA in CD25−Foxp3− T cells within the spleen [11]. Besides the technical limitations involved in human studies, inter-individual variations in IL-10 production due to single nucleotide polymorphisms (SNPs) in the IL-10 promoter [40–42] also restrict our understanding of the mechanism involved. This inter-individual difference in IL-10 production may be one of the possible factors responsible for the variation in susceptibility to infection with visceralizing Leishmania species observed in humans [43]. Indeed, the IL10-819C/C genotype was lately found to be associated with higher levels of IL-10 [44] and with increased susceptibility to L. braziliensis infection. To overcome these obstacles, we recently employed a novel experimental model to study human IL-10 expression in VL using transgenic Il10 −/−/hIL10BAC mice [45]. In these mice, human IL-10 is under genomic regulatory control, although we cannot rule out the possibility that there are control elements missing in the human IL10 BAC. This topic is discussed in more detail by Hedrich et al. in this issue. Sequence analysis of the human IL-10 BAC revealed a SNP haplotype block (IL-10-819T), which has previously been associated with low IL-10 production [46]. Il10 −/−/hIL10BAC mice rapidly cleared L. donovani infection and displayed the same resistance phenotype as Il10 −/− mice. Interestingly, Il10 −/−/hIL10BAC mice were able to recapitulate IL-10 production in myeloid cells and were therefore resistant to endotoxin-induced shock; yet they failed to generate antigen-specific, IL-10-secreting Th1 responses following L. donovani infection. This observation suggests that IL-10 expression may be regulated in a cell-specific manner and that T-cell-derived IL-10 seems to be the major factor involved in the establishment of persistent infections. Nevertheless, we still need to identify all cellular sources of human IL-10 in our model in order to determine the cell-type-specific contribution to disease pathogenesis during L. donovani infection.
L. donovani and CD8+ T-cell responses
Another mechanism of evasion of host-protective immunity is the capacity of L. donovani to escape CD8+ T-cell responses [14]. Unlike the experimental model of cutaneous leishmaniasis, in which the role of CD8+ T cells has been controversial in the past [47–50], CD8+ T cells have clearly been shown to be essential for the control of primary infections [2, 6, 51–53] and to be the main mediators of resistance to rechallenge in the mouse model of VL [2]. These cells have also been associated with cure in VL patients [54]. Several vaccination studies involving various Leishmania species have further highlighted the critical role played by CD8+ T cells in the host’s-protective immunity [32, 55–59]; however, the mechanisms involved remain elusive.
We have recently characterized the antigen-specific CD8+ T-cell responses in L. donovani-infected mice using transgenic parasites expressing the model antigen ovalbumin [14]. In this study, we showed that L. donovani evades CD8+ T-cell responses by limiting clonal expansion (200- to 10,000-fold less expansion than in other infection models [60–64]) and by inducing functional exhaustion during chronic disease. Furthermore, effector CD8+ T cells appear to undergo only 5–7 rounds of division, and only 20% of the cells that survive clonal contraction display an effector phenotype (80% of the cells express markers that are typical for central memory CD8+ T cells). Since strong clonal expansion is needed to control infection and determines the magnitude of the memory response [61], it would be of interest to identify the mechanisms by which L. donovani parasites interfere with the expansion of parasite-specific CD8+ T cells. Reasons for limited expansion may be multifactorial, either resulting from a reduced capacity of dendritic cells to process and present antigen and/or due to a suppressive environment.
One of the main questions that arise is whether infected DCs can process and present Leishmania antigens, provide adequate co-stimulation and secrete the cytokines that are necessary for the priming of naïve CD8+ T cells. Since Leishmania preferentially resides in macrophages, most of the studies in the past have mainly investigated the interaction between the parasite and macrophages. In these cells, Leishmania appears to interfere with the cells’ antigen-presenting capacity by disrupting lipid rafts [65], inhibiting phagosome maturation [66, 67] and cleaving epitopes by secreting endopeptidases [68]. Recent studies, however, have indicated that Leishmania may also interfere with the antigen cross-presenting machinery of dendritic cells [69] and actively downregulate various signaling events in DCs (reviewed in [70]). These observations suggest that infected DCs may not be able to optimally deliver the 3 signals necessary for effective priming of CD8+ T cells and that limited expansion of antigen-specific CD8+ T cells could result from antigen paucity and from poor activation of antigen-presenting DCs. A recent study, though, has shown that DCs infected in vitro with L. braziliensis, despite producing high amounts of TNF-α, fail to upregulate activation markers upon infection [71]. Yet, bystander, uninfected DCs upregulate activation markers, produce IL-12 and are more efficient in presenting antigen than control DCs. The authors suggest that this bystander DC activation together with increased TNF-α production by infected DCs could be responsible for the exaggerated T-cell responses observed in some patients infected with L. braziliensis. It is possible that a similar mechanism also operates during VL and bystander DCs overstimulate effector CD8+ T cells rendering them more susceptible to apoptosis. This would explain the low percentage of effector CD8+ T cells that survive clonal contraction and also the high percentage of multifunctional cells observed already during expansion, cells that are typically seen at later stages of infection (reviewed in [72]). Further investigations are needed to dissect the strategies utilized by the parasite to subvert DC activation and antigen processing.
The splenic environment may also contribute in controlling antigen-specific CD8+ T-cell expansion. The cytokine environment in the spleen has been extensively studied in the mouse model of VL. L. donovani induces a strong proinflammatory response at the onset of infection that is characterized by the production of key proinflammatory cytokines such as TNF-α, IFN-γ, and IL-12 [3, 4]. IL-12 can be detected as early as 5 h after infection and is mainly produced by splenic CD8+ DCs [73]. IL-12 production, though, has been shown to be transient, with the levels of IL-12 being similar to those observed in naïve mice by day 3 post-infection, probably because of DCs exhaustion [3]. Cytokines such as IL-12 and type I IFN have been shown to be crucial for stimulating strong clonal expansions of antigen-specific CD8+ T cells and for the development of their effector functions [74, 75]. A reduced exposure to inflammatory cytokines results in either impaired effector functions or limited clonal expansion, depending on the timing of the deprivation of cytokines [76, 77]. Thus, lack of IL-12 production by splenic DCs could also contribute to limited expansion of Leishmania-specific CD8+ T cells, since maximal antigen-presenting capacity by splenic DC is observed at day 6 after infection. The effect of IL-12 deprivation during CD8+ T cell priming could be acting synergistically with the presence of anti-inflammatory cytokines. In fact, IL-10 mRNA is readily detectable at 24 h after infection with L. donovani [28]. In experimental VL, IL-10 appears to be expressed mainly by natural killer cells (NK) during the first 2 weeks of infection [28]. However, other cell types, such as T cells [10, 12, 22, 28], macrophages [29], DCs [78–80], and B cells [28], have also been reported to express this cytokine following Leishmania infection. IL-10 is a potent anti-inflammatory molecule that exerts immunosuppressive effects on antigen-presenting cells and is able to directly restrict the magnitude of CD8+ T-cell expansion [81]. However, the role of IL-10 during the priming of CD8+ T cells is still controversial. This cytokine has also been shown to enhance priming of CD8+ T cells [82, 83]. Recently, another immunosuppressive cytokine, TGF-β, has been shown to control effector CD8+ T-cell number during clonal expansion, by lowering Bcl-2 expression and selectively promoting the apoptosis of short lived effector cells [84]. Although TGF-β appears to be involved in the establishment of chronic infection in several Leishmania models [85, 86], it does not seem to play an important role during L. donovani infections [87], suggesting that it is probably not involved in controlling the expansion of CD8+ T-cell responses.
It is not yet clear whether antigen-specific CD8+ T-cell responses originated in this type of environment have intrinsic defects as a result of defective priming, and thus become dysfunctional over the course of infection. Alternatively, but not mutually exclusive, the induction of exhausted T cells could results from other factors, such as the suppressive splenic environment during chronic infection, decreased levels of IL-21 [88, 89] and/or the constant presence of high levels of antigen [90]. Functionally impaired CD8+ T-cell responses are generated in many chronic infections in humans and in mice, for example hepatitis B [91], hepatitis C [92], HIV [93], LCMV [94]. The exact mechanisms responsible for the generation of exhausted responses are being extensively investigated. Several pathways have been suggested to result in T-cell exhaustion. The inhibitory receptor programed death 1 (PD-1) and its ligand B7-H1 have been shown to control the initiation and reversion of anergy, to inhibit T-cell functions, and to be the key pathway in the induction of exhaustion [95–97]. During experimental VL, conventional CD11chi splenic DC increasingly express the inhibitory molecule B7-H1 and fail to upregulate the co-stimulatory molecule CD80 [14]. Upregulation of B7-H1 on DC has been observed during several chronic infections and in a wide range of tumors [97–100]. B7-H1 is definitely involved in the induction of T-cell exhaustion in VL, as in vivo blockade of B7-H1 during chronic L. donovani infection increased the survival of antigen-specific CD8+ T cells [14]. Since B7-H1 is thought to inhibit T-cell proliferation by ligation with the PD-1 receptor [101] and to induce programed cell death of effector T cells through ligation with a yet unknown receptor [102], increased survival of CD8+ T cells after B7-H1 blockade may result from restoration of the proliferative capacity or inhibition of induced cell death of effector CD8+ T cells. Interestingly, in contrast to what has been recently reported in the literature [96, 97], in vivo blockade of B7-H1 during chronic VL only transiently restored the functional capacity of exhausted antigen-specific CD8+ T-cells. This suggests that additional pathways may be involved in the suppression of cytokine production by CD8+ T-cells during chronic L. donovani infection. Exhausted CD8+ T cells have been shown to co-express multiple inhibitory receptors under the regulation of the transcriptional repressor Blimp-1 [103, 104]. Thus, various inhibitory pathways may control T-cell exhaustion. Indeed, the simultaneous blockade of PD-1 and LAG3 [104] and/or PD-1 and CTLA-4 [105] showed synergistic effects in lymphocytic choriomeningitis virus (LCMV) and hepatitis C virus (HCV) infection, respectively.
The suppressive splenic environment during chronic VL could also act synergistically with the various inhibitory pathways. A study has demonstrated a synergistic effect between TGF-β and the B7-H1/PD-1 axis in suppressing CD8+ T-cell responses [106]. Furthermore, TGF-β has also been shown to mediate virus-specific CD8+ T-cell deletion during chronic LCMV infection [107]. Although TGF-β does not seem to play an important role during chronic L. donovani infections [87], it is expressed during other Leishmania infections [85, 108] and may therefore play a role in these models. However, IL-10 is highly expressed in both mouse and human VL [11, 26–28, 39, 44, 109]. This cytokine has been shown to inhibit expansion and suppress cytokine production in several chronic infections [24, 81, 110]. Thus, IL-10 could potentially synergistically act with the B7-H1/PD-1 axis in the inhibition of CD8+ T-cell responses and be responsible for the suppression of cytokine production observed during chronic VL.
B7-H1 blockade results in a significant decrease in the parasite burden in L. donovani-infected mice. The mechanism of protection is not clear and might not merely rely on IFNγ production, as blockade failed to fully restore cytokine production. In a previous study, we have shown that therapeutic intervention with antigen-specific CD8+ T-cells in mice chronically infected with L. donovani dramatically reduced the parasite burden [111]. Our recent findings consolidated these results by showing that OT-I CD8+ T-cells rescued from cell death by blocking in vivo B7-H1 or by superinfecting mice with recombinant vaccinia virus expressing SIINFEKL resulted in significant host protection in mice infected with ovalbumin-transgenic L. donovani [14]. Many chronic infections generate dysfunctional CD8+ T-cell responses, and the rescue of these responses results in host protection. Thus, it is important to understand the pathways responsible for the induction of exhaustion and deletion of antigen-specific T cells.
Conclusions
Pathogens have developed sophisticated mechanisms to evade immune responses and to establish chronic infections. Leishmania parasites have evolved several strategies that allow them to survive and persist inside the host; key among these are the induction of immunosuppressive IL-10 responses and the generation of functionally impaired antigen-specific CD8+ T cells. The exact pathways involved in immune escape and in the establishment of persistent infections are mostly unknown. A better understanding of the immune evasive mechanisms adopted by the various pathogens could help in the development of novel therapeutic interventions, which are much needed especially for neglected diseases like leishmaniasis, where resistances to drug therapy are constantly emerging and no effective vaccine is available.
References
Engwerda CR, Kaye PM. Organ-specific immune responses associated with infectious disease. Immunol Today. 2000;21:73–8.
Stern JJ, Oca MJ, Rubin BY, Anderson SL, Murray HW. Role of L3T4+ and LyT-2+ cells in experimental visceral leishmaniasis. J Immunol. 1988;140:3971–7.
Gorak PM, Engwerda CR, Kaye PM. Dendritic cells, but not macrophages, produce IL-12 immediately following Leishmania donovani infection. Eur J Immunol. 1998;28:687–95.
Engwerda CR, Murphy ML, Cotterell SE, Smelt SC, Kaye PM. Neutralization of IL-12 demonstrates the existence of discrete organ-specific phases in the control of Leishmania donovani. Eur J Immunol. 1998;28:669–80.
Smelt SC, Engwerda CR, McCrossen M, Kaye PM. Destruction of follicular dendritic cells during chronic visceral leishmaniasis. J Immunol. 1997;158:3813–21.
Kaye PM, Cooke A, Lund T, Wattie M, Blackwell JM. Altered course of visceral leishmaniasis in mice expressing transgenic I-E molecules. Eur J Immunol. 1992;22:357–64.
Tumang MC, Keogh C, Moldawer LL, Helfgott DC, Teitelbaum R, Hariprashad J, et al. Role and effect of TNF-alpha in experimental visceral leishmaniasis. J Immunol. 1994;153:768–75.
Ato M, Stager S, Engwerda CR, Kaye PM. Defective CCR7 expression on dendritic cells contributes to the development of visceral leishmaniasis. Nat Immunol. 2002;3:1185–91.
Engwerda CR, Ato M, Cotterell SE, Mynott TL, Tschannerl A, Gorak-Stolinska PM, et al. A role for tumor necrosis factor-alpha in remodeling the splenic marginal zone during Leishmania donovani infection. Am J Pathol. 2002;161:429–37.
Stager S, Maroof A, Zubairi S, Sanos SL, Kopf M, Kaye PM. Distinct roles for IL-6 and IL-12p40 in mediating protection against Leishmania donovani and the expansion of IL-10+ CD4+ T cells. Eur J Immunol. 2006;36:1764–71.
Nylen S, Maurya R, Eidsmo L, Manandhar KD, Sundar S, Sacks D. Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+ CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med. 2007;204:805–17.
Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–7.
Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(−)Foxp3(−) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–97.
Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stager S. B7-H1 blockade increases survival of dysfunctional CD8(+) T cells and confers protection against Leishmania donovani infections. PLoS Pathog 2009;5:e1000431.
Sacks D, Sher A. Evasion of innate immunity by parasitic protozoa. Nat Immunol. 2002;3:1041–7.
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765.
Mege JL, Meghari S, Honstettre A, Capo C, Raoult D. The two faces of interleukin 10 in human infectious diseases. Lancet Infect Dis. 2006;6:557–69.
Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–7.
Omer FM, de Souza JB, Riley EM. Differential induction of TGF-beta regulates proinflammatory cytokine production and determines the outcome of lethal and nonlethal Plasmodium yoelii infections. J Immunol. 2003;171:5430–6.
Wu Y, Wang QH, Zheng L, Feng H, Liu J, Ma SH, et al. Plasmodium yoelii: distinct CD4(+)CD25(+) regulatory T cell responses during the early stages of infection in susceptible and resistant mice. Exp Parasitol. 2007;115:301–4.
Anderson CF, Mendez S, Sacks DL. Nonhealing infection despite Th1 polarization produced by a strain of Leishmania major in C57BL/6 mice. J Immunol. 2005;174:2934–41.
Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med. 2001;194:1497–506.
Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–9.
Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203:2461–72.
Roque S, Nobrega C, Appelberg R, Correia-Neves M. IL-10 underlies distinct susceptibility of BALB/c and C57BL/6 mice to Mycobacterium avium infection and influences efficacy of antibiotic therapy. J Immunol. 2007;178:8028–35.
Murray HW, Lu CM, Mauze S, Freeman S, Moreira AL, Kaplan G, et al. Interleukin-10 (IL-10) in experimental visceral leishmaniasis and IL-10 receptor blockade as immunotherapy. Infect Immun. 2002;70:6284–93.
Murphy ML, Wille U, Villegas EN, Hunter CA, Farrell JP. IL-10 mediates susceptibility to Leishmania donovani infection. Eur J Immunol. 2001;31:2848–56.
Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity. 2008;29:295–305.
Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J Exp Med. 2005;201:747–54.
Svensson M, Maroof A, Ato M, Kaye PM. Stromal cells direct local differentiation of regulatory dendritic cells. Immunity. 2004;21:805–16.
Jankovic D, Kullberg MC, Feng CG, Goldszmid RS, Collazo CM, Wilson M, et al. Conventional T-bet(+)Foxp3(−) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–83.
Stager S, Smith DF, Kaye PM. Immunization with a recombinant stage-regulated surface protein from Leishmania donovani induces protection against visceral leishmaniasis. J Immunol. 2000;165:7064–71.
Padigel UM, Farrell JP. Control of infection with Leishmania major in susceptible BALB/c mice lacking the common gamma-chain for FcR is associated with reduced production of IL-10 and TGF-beta by parasitized cells. J Immunol. 2005;174:6340–5.
Thomas BN, Buxbaum LU. FcgammaRIII mediates immunoglobulin G-induced interleukin-10 and is required for chronic Leishmania mexicana lesions. Infect Immun. 2008;76:623–31.
Smelt SC, Cotterell SE, Engwerda CR, Kaye PM. B cell-deficient mice are highly resistant to Leishmania donovani infection, but develop neutrophil-mediated tissue pathology. J Immunol. 2000;164:3681–8.
Peyron F, Burdin N, Ringwald P, Vuillez JP, Rousset F, Banchereau J. High levels of circulating IL-10 in human malaria. Clin Exp Immunol. 1994;95:300–3.
Yamamura M, Uyemura K, Deans RJ, Weinberg K, Rea TH, Bloom BR, et al. Defining protective responses to pathogens: cytokine profiles in leprosy lesions. Science. 1991;254:277–9.
Stylianou E, Aukrust P, Kvale D, Muller F, Froland SS. IL-10 in HIV infection: increasing serum IL-10 levels with disease progression–down-regulatory effect of potent anti-retroviral therapy. Clin Exp Immunol. 1999;116:115–20.
Ghalib HW, Piuvezam MR, Skeiky YA, Siddig M, Hashim FA, el-Hassan AM, et al. Interleukin 10 production correlates with pathology in human Leishmania donovani infections. J Clin Invest. 1993;92:324–9.
Eskdale J, Gallagher G, Verweij CL, Keijsers V, Westendorp RG, Huizinga TW. Interleukin 10 secretion in relation to human IL-10 locus haplotypes. Proc Natl Acad Sci USA. 1998;95:9465–70.
Gibson AW, Edberg JC, Wu J, Westendorp RG, Huizinga TW, Kimberly RP. Novel single nucleotide polymorphisms in the distal IL-10 promoter affect IL-10 production and enhance the risk of systemic lupus erythematosus. J Immunol. 2001;166:3915–22.
Vicari AP, Trinchieri G. Interleukin-10 in viral diseases and cancer: exiting the labyrinth? Immunol Rev. 2004;202:223–36.
Nylen S, Sacks D. Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol. 2007;28:378–84.
Salhi A, Rodrigues V Jr, Santoro F, Dessein H, Romano A, Castellano LR, et al. Immunological and genetic evidence for a crucial role of IL-10 in cutaneous lesions in humans infected with Leishmania braziliensis. J Immunol. 2008;180:6139–48.
Ranatunga D, Hedrich CM, Wang F, McVicar DW, Nowak N, Joshi T, Feigenbaum L, Grant LR, Stager S, Bream JH. A human IL10 BAC transgene reveals tissue-specific control of IL-10 expression and alters disease outcome. Proc Natl Acad Sci USA. 2009.
Reuss E, Fimmers R, Kruger A, Becker C, Rittner C, Hohler T. Differential regulation of interleukin-10 production by genetic and environmental factors—a twin study. Genes Immun. 2002;3:407–13.
Erb K, Blank C, Ritter U, Bluethmann H, Moll H. Leishmania major infection in major histocompatibility complex class II-deficient mice: CD8+ T cells do not mediate a protective immune response. Immunobiology. 1996;195:243–60.
Huber M, Timms E, Mak TW, Rollinghoff M, Lohoff M. Effective and long-lasting immunity against the parasite Leishmania major in CD8-deficient mice. Infect Immun. 1998;66:3968–70.
Belkaid Y, Von Stebut E, Mendez S, Lira R, Caler E, Bertholet S, et al. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J Immunol. 2002;168:3992–4000.
Uzonna JE, Joyce KL, Scott P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med. 2004;199:1559–66.
Ahmed S, Colmenares M, Soong L, Goldsmith-Pestana K, Munstermann L, Molina R, et al. Intradermal infection model for pathogenesis and vaccine studies of murine visceral leishmaniasis. Infect Immun. 2003;71:401–10.
Tsagozis P, Karagouni E, Dotsika E. CD8(+) T cells with parasite-specific cytotoxic activity and a Tc1 profile of cytokine and chemokine secretion develop in experimental visceral leishmaniasis. Parasite Immunol. 2003;25:569–79.
Tsagozis P, Karagouni E, Dotsika E. Function of CD8+ T lymphocytes in a self-curing mouse model of visceral leishmaniasis. Parasitol Int. 2005;54:139–46.
Mary C, Auriault V, Faugere B, Dessein AJ. Control of Leishmania infantum infection is associated with CD8(+) and gamma interferon- and interleukin-5-producing CD4(+) antigen-specific T cells. Infect Immun. 1999;67:5559–66.
Rafati S, Kariminia A, Seyde-Eslami S, Narimani M, Taheri T, Lebbatard M. Recombinant cysteine proteinases-based vaccines against Leishmania major in BALB/c mice: the partial protection relies on interferon gamma producing CD8(+) T lymphocyte activation. Vaccine. 2002;20:2439–47.
Rhee EG, Mendez S, Shah JA, Wu CY, Kirman JR, Turon TN, et al. Vaccination with heat-killed leishmania antigen or recombinant leishmanial protein and CpG oligodeoxynucleotides induces long-term memory CD4+ and CD8+ T cell responses and protection against leishmania major infection. J Exp Med. 2002;195:1565–73.
Stager S, Alexander J, Kirby AC, Botto M, Rooijen NV, Smith DF, et al. Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat Med. 2003;9:1287–92.
Gurunathan S, Stobie L, Prussin C, Sacks DL, Glaichenhaus N, Iwasaki A, et al. Requirements for the maintenance of Th1 immunity in vivo following DNA vaccination: a potential immunoregulatory role for CD8+ T cells. J Immunol. 2000;165:915–24.
Gurunathan S, Prussin C, Sacks DL, Seder RA. Vaccine requirements for sustained cellular immunity to an intracellular parasitic infection. Nat Med. 1998;4:1409–15.
Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 1998;8:167–75.
Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–87.
Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28:859–69.
Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–64.
Busch DH, Pilip I, Pamer EG. Evolution of a complex T cell receptor repertoire during primary and recall bacterial infection. J Exp Med. 1998;188:61–70.
Chakraborty D, Banerjee S, Sen A, Banerjee KK, Das P, Roy S. Leishmania donovani affects antigen presentation of macrophage by disrupting lipid rafts. J Immunol. 2005;175:3214–24.
Scianimanico S, Desrosiers M, Dermine JF, Meresse S, Descoteaux A, Desjardins M. Impaired recruitment of the small GTPase rab7 correlates with the inhibition of phagosome maturation by Leishmania donovani promastigotes. Cell Microbiol. 1999;1:19–32.
Desjardins M, Descoteaux A. Inhibition of phagolysosomal biogenesis by the Leishmania lipophosphoglycan. J Exp Med. 1997;185:2061–8.
Garcia MR, Graham S, Harris RA, Beverley SM, Kaye PM. Epitope cleavage by Leishmania endopeptidase(s) limits the efficiency of the exogenous pathway of major histocompatibility complex class I-associated antigen presentation. Eur J Immunol. 1997;27:1005–13.
Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, Collazo-Custodio C, et al. Leishmania antigens are presented to CD8+ T cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. J Immunol. 2006;177:3525–33.
Soong L. Modulation of dendritic cell function by Leishmania parasites. J Immunol. 2008;180:4355–60.
Carvalho LP, Pearce EJ, Scott P. Functional dichotomy of dendritic cells following interaction with Leishmania braziliensis: infected cells produce high levels of TNF-alpha, whereas bystander dendritic cells are activated to promote T cell responses. J Immunol. 2008;181:6473–80.
Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8:107–19.
Maroof A, Kaye PM. Temporal regulation of interleukin-12p70 (IL-12p70) and IL-12-related cytokines in splenic dendritic cell subsets during Leishmania donovani infection. Infect Immun. 2008;76:239–49.
Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–51.
Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–15.
Shaulov A, Murali-Krishna K. CD8 T cell expansion and memory differentiation are facilitated by simultaneous and sustained exposure to antigenic and inflammatory milieu. J Immunol. 2008;180:1131–8.
Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92.
Prina E, Abdi SZ, Lebastard M, Perret E, Winter N, Antoine JC. Dendritic cells as host cells for the promastigote and amastigote stages of Leishmania amazonensis: the role of opsonins in parasite uptake and dendritic cell maturation. J Cell Sci. 2004;117:315–25.
Wanasen N, Xin L, Soong L. Pathogenic role of B cells and antibodies in murine Leishmania amazonensis infection. Int J Parasitol. 2008;38:417–29.
Qi H, Popov V, Soong L. Leishmania amazonensis-dendritic cell interactions in vitro and the priming of parasite-specific CD4(+) T cells in vivo. J Immunol. 2001;167:4534–42.
Biswas PS, Pedicord V, Ploss A, Menet E, Leiner I, Pamer EG. Pathogen-specific CD8 T cell responses are directly inhibited by IL-10. J Immunol. 2007;179:4520–8.
Foulds KE, Rotte MJ, Seder RA. IL-10 is required for optimal CD8 T cell memory following Listeria monocytogenes infection. J Immunol. 2006;177:2565–74.
Kang SS, Allen PM. Priming in the presence of IL-10 results in direct enhancement of CD8+ T cell primary responses and inhibition of secondary responses. J Immunol. 2005;174:5382–9.
Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–44.
Anderson CF, Lira R, Kamhawi S, Belkaid Y, Wynn TA, Sacks D. IL-10 and TGF-beta control the establishment of persistent and transmissible infections produced by Leishmania tropica in C57BL/6 mice. J Immunol. 2008;180:4090–7.
Gomes NA, Gattass CR, Barreto-De-Souza V, Wilson ME, DosReis GA. TGF-beta mediates CTLA-4 suppression of cellular immunity in murine kalaazar. J Immunol. 2000;164:2001–8.
Zubairi S, Sanos SL, Hill S, Kaye PM. Immunotherapy with OX40L-Fc or anti-CTLA-4 enhances local tissue responses and killing of Leishmania donovani. Eur J Immunol. 2004;34:1433–40.
Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science. 2009;324:1576–80.
Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science. 2009;324:1572–6.
Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. 2009;106:8623–8.
Reignat S, Webster GJ, Brown D, Ogg GS, King A, Seneviratne SL, et al. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J Exp Med. 2002;195:1089–101.
Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001;75:5550–8.
Goepfert PA, Bansal A, Edwards BH, Ritter GD Jr, Tellez I, McPherson SA, et al. A significant number of human immunodeficiency virus epitope-specific cytotoxic T lymphocytes detected by tetramer binding do not produce gamma interferon. J Virol. 2000;74:10249–55.
Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–93.
Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, et al. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110:180–5.
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7.
Lukens JR, Cruise MW, Lassen MG, Hahn YS. Blockade of PD-1/B7-H1 interaction restores effector CD8+ T cell responses in a hepatitis C virus core murine model. J Immunol. 2008;180:4875–84.
Trabattoni D, Saresella M, Biasin M, Boasso A, Piacentini L, Ferrante P, et al. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood. 2003;101:2514–20.
Chen L, Zhang Z, Chen W, Zhang Z, Li Y, Shi M, et al. B7-H1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J Immunol. 2007;178:6634–41.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–7.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, et al. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity. 2009;31:309–20.
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37.
Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, Gostick E, Price DA, Freeman GJ, Wherry EJ, Chang KM. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog 2009;5:e1000313.
Wei S, Shreiner AB, Takeshita N, Chen L, Zou W, Chang AE. Tumor-induced immune suppression of in vivo effector T-cell priming is mediated by the B7-H1/PD-1 axis and transforming growth factor beta. Cancer Res. 2008;68:5432–8.
Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–57.
Wilson ME, Young BM, Davidson BL, Mente KA, McGowan SE. The importance of TGF-beta in murine visceral leishmaniasis. J Immunol. 1998;161:6148–55.
Karp CL, el-Safi SH, Wynn TA, Satti MM, Kordofani AM, Hashim FA, et al. In vivo cytokine profiles in patients with kala-azar. Marked elevation of both interleukin-10 and interferon-gamma. J Clin Invest. 1993;91:1644–8.
den Haan JM, Kraal G, Bevan MJ. Cutting edge: Lipopolysaccharide induces IL-10-producing regulatory CD4+ T cells that suppress the CD8+ T cell response. J Immunol 2007;178:5429–33.
Polley R, Stager S, Prickett S, Maroof A, Zubairi S, Smith DF, et al. Adoptive immunotherapy against experimental visceral leishmaniasis with CD8+ T cells requires the presence of cognate antigen. Infect Immun. 2006;74:773–6.
Acknowledgments
This work was funded by the start up funds from the Johns Hopkins University School of Medicine to S.S.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stäger, S., Joshi, T. & Bankoti, R. Immune evasive mechanisms contributing to persistent Leishmania donovani infection. Immunol Res 47, 14–24 (2010). https://doi.org/10.1007/s12026-009-8135-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-009-8135-4