
Abstract
Building a mathematical model of population dynamics of pathogens within their host involves considerations of factors similar to those in ecology, as pathogens can prey on cells in the host. But within the multicellular host, attacked cell types are integrated with other cellular systems, which in turn intervene in the infection. For example, immune responses attempt to sense and then eliminate or contain pathogens, and homeostatic mechanisms try to compensate for cell loss. This review focuses on modeling applied to malarias, diseases caused by single-cell eukaryote parasites that infect red blood cells, with special concern given to vivax malaria, a disease often thought to be benign (if sometimes incapacitating) because the parasite only attacks a small proportion of red blood cells, the very youngest ones. However, I will use mathematical modeling to argue that depletion of this pool of red blood cells can be disastrous to the host if growth of the parasite is not vigorously check by host immune responses. Also, modeling can elucidate aspects of new field observations that indicate that vivax malaria is more dangerous than previously thought.
Similar content being viewed by others

Introduction
Population biology is concerned with the dynamics of a population of organisms and influences on those dynamics. For example, consider a herd of zebras on a plain. Lions and hyenas prey on the zebras. If there is a drought, fewer zebras may survive, thus fewer lions and hyenas survive. Lions and hyenas compete against each other, and parasites infect both zebras and predators. A less nutritious grass may invade the plain, etc. From a biophysical standpoint, interactions among the species facilitate the exchange of energy along with nutrients necessary to use that energy. The plain is a substrate on which natural selection acts on the component species and thus shapes their genomes. Mathematical modeling of an ecosystem lets us investigate “what if” situations: given a proposed interaction or process in the ecosystem, what is the logical outcome of the presence of that interaction or process upon the dynamics of the populations in that system? Will these outcomes be robust against perturbations on the system? To quote John Maynard Smith (Maynard Smith 1968), modeling (1) “provides a rapid way of discovering the kind of effect various features—for examples, predation, parasitism, territorial behavior, a long or short period of immaturity—may have on the behavior of a population” and (2) “suggests what needs to be measured before the behavior of any particular species can be understood.”
A group of pathogenic agents in a multicellular host also forms a population. The pathogen is a predator if it attacks a cell type. But multicellular hosts are usually proactive against an invader, mounting immune responses to kill or limit the pathogen and starting processes to repair the tissue damage. From a modeler’s perspective, perhaps the ecosystem is a morass: we may not know all the components of the host’s response, and of those we do know, we may not know which are most important. Host responses may be self-amplifying, self-regulating, and possibly self-harming. They may differ greatly among individuals of the same species due to genetic background or contingency of environmental exposure. The pathogen might have mechanisms to evade or interfere with host responses. But again, we can use mathematical modeling to answer “what-if” questions, and this in turn can guide researchers in the laboratory or clinic. There are two levels of population dynamics in a host-pathogen system: (1) those of individual cells or virions of the invader as well as host cells and responses, and (2) those of both host population and disease incidence, the subject of epidemiology. In this review, I discuss the application of mathematical modeling to a type of human malaria, with the focus on the within-host dynamics of the pathogen. I will also discuss the significance of the results from both public health and evolutionary standpoints.
A conundrum of malaria biology
Malarias are diseases of red blood cells (RBCs) in vertebrates caused by 100+ species of single-cell eukaryotes of the genus Plasmodium, order Haemosporida of the class Apicomplexa. Birds, reptiles, and mammals are targeted (Garnham 1966; Perkins and Austin 2009). Apicomplexans have a set of specialized organelles, together called the apical complex, that enables them to penetrate the cells of their hosts. All known Apicomplexans are intracellular parasites, and some are human disease agents (Cryptosporidium parvum, Toxoplasma gondii) or serious livestock pathogens (Babesia spp., Theileria spp.). At least five Plasmodium spp. infect humans regularly, causing disease characterized by repeating bouts of severe fever and anemia [see Cox-Singh and Singh (2008) for commentary]. Part of the life cycle of these species is spent in mosquitoes of genus Anopheles. The mosquitoes serve as vectors that spread the disease from human to human by picking up the gametocytes, the sexual forms of the parasite that mate within the mosquito gut. Mosquitoes transmit to humans the haploid descendants of the mated forms, the sporozoites, which then enter liver cells. The form of parasite that infects RBCs is the merozoites, which are released initially from the liver as the descendants of sporozoites but then later by the bursting of infected RBCs. [Baer et al. (2007) is an illustrative study of the initial merozoite release.] After a merozoite enters an RBC, its nucleus undergoes rounds of division by schizogony. The infection is sustained among the host’s RBCs in this manner, but some asexual forms become gametocytes in a process that is not well understood. The disease pathology is due to the asexual forms that cannot be transmitted to mosquitoes, although blood transfusion can spread them.
Two species, Plasmodium falciparum and Plasmodium vivax, cause the bulk of the public health burden of the malarias. [For review, see White and Breman (2008) and Miller et al. (2002).] P. vivax attacks mainly reticulocytes, which are RBCs that have aged 36 h or less after differentiation from erythroblasts and that still retain traces of a nucleus. This parasite infects ≈108 people per year in both temperate and tropical areas. Infection is temporarily incapacitating, but as I discuss below, its true public health burden is only now becoming apparent. P. falciparum can infect RBCs of any age. This species infects ≈5 × 108 people each year and causes >106 deaths per year, mostly in tropical regions and especially in sub-Saharan Africa. Severe anemia and organ complications, especially in the brain, cause death or long term disability. Figure 1 shows a human RBC parasitized with this species.

Lurking among human RBCs (stained red) is one infested with the schizont form of Plasmodium falciparum. Violet stains show DNA in parasitic nuclei (arrow), which were produced by schizogony after a single-nucleus form of the parasite invaded this erythrocyte. Image credit: 3D confocal microscopy and image processing performed by Drs. Fuyuki Tokumasu and James Dvorak (deceased), Laboratory of Malaria and Vector Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. Used by permission of Dr. Tokumasu
In 2003, Ellis McKenzie of the National Institutes of Health, Bethesda, approached me with a conundrum: It was assumed that P. vivax caused less severe disease than P. falciparum because a mere 1.25% of RBCs are reticulocytes (Krogstad 1995). Yet data from neurosyphilis patients who were infected with P. vivax showed that this parasite has a rather large growth rate early in infection, with parasites from a single infected RBC infecting up to 15 other RBCs (McKenzie et al. 2002). (Before penicillin came into use, the induction of antibacterial fever by malaria was the standard of treatment for neurosyphilis.) This was similar to the initial growth rate of parasitemia seen in P. falciparum infections in neurosyphilis patients (Collins and Jeffery 1999) and other patient groups (White 1997). Could P. vivax cause severe anemia by depleting the reticulocyte population, thus choking off replacements for aging RBCs removed by the spleen? If so, what makes P. vivax supposedly less dangerous than P. falciparum? We set about modeling the dynamics of the parasite population in an hypothetical patient to address this question. This review describes the results of our efforts.
Incidentally, there are many fine theoretical studies of the within-host population dynamics of malaria parasites that attempt to address several aspects of malaria infection. For a selection, see Gravenor et al. (1995), Hoshen et al. (2000), Gravenor et al. (2002), Rouzine and McKenzie (2003), Haydon et al. (2003), Maire et al. (2006), Chiyaka et al. (2009), and Thibodeaux (2010).
Starting with Malthus
We first considered the following question: what would happen if parasitemia grew to the maximum level constrained only by availability of RBCs, with no immune response from the host against the parasite? This situation resembles Malthusian growth of the within-host parasite population, although Mathus was primarily concerned with the effects of human population growth on economies. But the basic idea is that a population grows unchecked until limited by availability of resources, and the growth can lead to catastrophe for the population if the host dies due to anemia. (However, if a mosquito has picked up some of the gametocytes, the catastrophe is not for the Plasmodium species itself. The within-host population is sacrificed.) The 1798 essay of Malthus had a great impact on biologists in the 19th century and is considered one of the founding works in population biology (Malthus 2003). In some ways, the models that I describe are akin to the logistics equation introduced by Malthus’s younger contemporary P.F. Verhulst to account for population growth limited by a resource (Hoppensteadt 1982). Before introducing the models, I must introduce two important concepts.
Concept I: basic reproduction rate
The lifetime τ μ of a free merozoite is ≈10 min (Johnson et al. 1980), much shorter than the development time of the parasite within RBCs (≈2 days for both P. vivax and P. falciparum). The difference between these time scales allows us to derive, by statistical arguments, the average number of descendants an infected RBC has after one development cycle, R, early in the infection before an immune response can respond (or if there is no immune response). Assuming that all vulnerable RBCs are equally likely to be invaded, we introduce a merozoite-RBC binding affinity ζ, so that the rate of infection of RBCs per unit volume of blood, Inf r , is
where μ is the density of merozoites in the blood, and V is the density of vulnerable RBCs (reticulocytes for P. vivax, all RBCs for P. falciparum). Making an additional assumption that early in the infection V changes slowly during one cycle of the within-RBC parasite, we obtain
Here p is the average number of merozoites released from an infected RBC. [The details of the derivation are given in McQueen and McKenzie (2004)]. Direct photographic evidence of bursting infected RBCs suggests that p ≈ 16 (Glushakova et al. 2005). As already mention in the section “A conundrum of malaria biology” above, R for both species at the beginning of infection is ≈15. Thus, the asexual forms of both species are very effective at propagating themselves before host immune reactions can intervene, with a characteristic time to invade an RBC ζV ≈ 24 s. I call the initial value of R the basic reproductive rate, R 0. Given V, τ μ , and p, knowing R 0 determines ζ. As infection progresses, R declines from R 0 because (1) the vulnerable RBC population is depleted and (2) the host mounts an immune response against the parasite.
Concept II: populations consist of aging individuals
To develop a reasonable mathematical model, one has to treat the various constituents of the pathogen-host system (the erythocytes, the parasite, etc.) as populations of aging individuals: that is, on a population average, individuals spend D amount of time in a given stage of development with some standard deviation σ. If simple rate equations were used to model the population levels of the constituents, then population level changes would be like that of radioactive nuclei decaying at random. Yet the development time of an individual parasite within an RBC is 48 h for the two species, with maybe a few hours of variation. Reticulocytes last only 36 h, plus or minus a few hours. Individuals in such populations are not like radioactive nuclei that randomly “pop” with probability per unit time of D −1. To account realistically for aging, we used the technique of compartment ordinary differential equations in which a stage of development of an organism (with duration D) is described as a set of N substages with duration DN −1 that decay into one another. Letting P n be the population in substage n (and ignoring interactions between individuals for a moment):
It can be shown that the standard deviation about D for individuals in the development stage is
I stress that D, σ, and the total population ΣP n are the tangible quantities connected to measurement, not N. The N substage populations are just a fictional construct. This technique was developed in the 1960s for population modeling, with Lloyd adapting it to epidemiology and within-host pathogen population dynamics (Lloyd 2001a, b). [See also supplementary material in McQueen and McKenzie (2004).] We wrote a set of modular code libraries in Java (now in C++) that allowed for rapid modification of the numerical model as needed. It was found that a fourth-order Runge-Kutta method with adaptive time stepping (Cash and Karp 1990) worked very well to solve the ordinary differential equation system arising from the application of Eqs. 3 and 4, even though such systems might contain hundreds of dependent variables.
The erythocyte-limited model
This model was first described in McQueen and McKenzie (2004); it can thought as two developmental chains, one for RBCs and the other for the parasite. The stages of each chain are indicated in Fig. 2. For each such stage, a system of differential equations similar to Eq. 3 describes the population changes; the corresponding values of D and σ are also shown in Fig. 2. The two chains interact through the binding of merozoites to the vulnerable RBCs, as described in Eq. 1. For convenience of the reader, the full system of differential equations that describe this model is given in the Electronic supplementary material (ESM).

The development chains assumed for normal and infected RBCs. Plasmodium malariae attacks human senescent RBCs; see the section “Plasmodium vivax is an insidious pathogen” concerning this parasite
The simulation is started with a small number of merozoites to mimic the primary release of merozoites from the liver. The normal basal production rate for human RBCs is about 1,736 μl−1 h−1; we considered models in which this rate could be amplified to compensate for the RBC loss due to infection. We also considered models with dyserythropoiesis, a decline in the rate of production due to factors of the parasite. [For a review of observed effects of malaria upon erythropoiesis, see Wickramasinghe and Abdalla (2000).] For some sets of simulations, we varied the σ for the erythrocytic stage of the parasite to test the effects of changes in this model parameter upon the results. This will be discussed more in the section “Sensitivity to the standard deviation of the life cycletime of the intraerythrocyte parasite.”
Plasmodium vivax is an insidious pathogen
The erythrocyte-limited model of parasite dynamics predicts that even for a basic reproduction rate R 0 ≤ 2, P. vivax infection would eventually deplete the reticulocyte pool enough to kill the patient: there would not be enough replacements for aged RBCs removed by the spleen (McQueen and McKenzie 2004). Figure 3 shows some typical simulation results. [The results in the figure are slightly different from those presented in McQueen and McKenzie (2004) because (1) σ of the infected RBC stage is 2.4 h instead of 4.8 h as in that paper, and (2) the level of lethal anemia was set lower than in that paper.] One effect apparent in Fig. 3 is that a compensatory response of RBC production would make matters worse for the host with P. vivax infection by lowering the value of R 0 at which death via severe anemia could occur, as well as increasing the overall parasitemia. Conversely, dyserythropoiesis would tend to lower the number of parasites produced in lethal infections. These latter effects would also occur in the resource-limited model of P. falciparum infection, but then there is less effect on the survival of the host (see Fig. S1 in the ESM).

a Time of death for simulated P. vivax infection, T D , assuming no immune constraints, as a function of basic reproduction rate R0. b Corresponding total number of RBCs parasitized during the infection, IBC INT . The basal results are for an erythocyte production rate at its basal value; comp results are for a production rate up to five times the basal value to compensate for blood loss due to infection; and dys results assume dyserythropoiesis: the production rate reduced by an amount equal to twice the rate of depletion of RBCs by infection. Infections were simulated for 5 years unless the RBC count dropped below 60% of the basal count, at which point the host was assumed to have died from anemia and the simulation was terminated. Each curve is the result of simulations at 1,000 random values for R0 chosen uniformly on the interval [1.01, 15.0]. Note: Log-log scale for both axes; horizontal axis is log (R0 − 1) in order to emphasize the differences between the curves
Since R 0 ≈ 15 in actual infections, even in patients who survive the disease, human immune constraints on the growth of P. vivax must be very tight indeed! Even though the erythropoietic function affects some of the details of the erythrocyte-limited model infection outcome, infection with P. vivax in this model would be lethal even if R 0 were just above 1 regardless of the erythropoietic function.
The importance of the age-structured nature of the attack of P. vivax becomes apparent when examining Fig. 4, which shows results for simulations using parameters for the pathogen Plasmodium malariae, an attacker of senescent human RBCs. [The duration of the infected RBC stage for this pathogen is 72 h instead of 48 h. See McKenzie et al. (2001) for a review of knowledge about this parasite.] No simulated P. malariae infection ended in host death unless there was dyserythropoiesis, even though P. malariae attacks a slightly larger subset of RBCs in this model. Since no other stage of RBC development depends on the senescent cells, depletion of this population would not cause severe anemia unless a secondary effect disrupted the production of RBCs or killed uninfected RBCs.

Panels a and b are the same as in Fig. 3, except that the model pathogen is P. malariae; see the section “Plasmodium vivax is an insidious pathogen” for details. Curves are the same erythropoietic responses as in Fig. 3. Note for a: only simulation with dyserythropoiesis ended with death of the host
If both P. vivax and P. falciparum infect the host in an erythrocyte-limited model, the results depend on which infects first, and their respective values of R 0. Basically, we found that the species with the largest value of R 0 dominates. See McQueen and McKenzie (2006) for more detailed discussion.
Adding host immunity
In real malaria infections, hosts mount aggressive immune responses, both innate and adaptive. For example, the report of Karunaweera et al. (1992) illustrates the quick production (and then decay) of cytokines associated with innate responses in patients infected by P. vivax. [Some good reviews of immune responses to malaria are Engwerda and Good (2005), Karunaweera et al. (2003), and Plebanski and Hill (2000).] To address how the presence of immune responses might affect the virulence of Plasmodium spp., we added additional components to the model to incorporate two types of immune reactions: (1) a fast acting, fast decaying response to mimic the intense innate reactions seen in malarias, and (2) a slowly activating, slowly decaying response to mimic an antibody response. Since some of the innate immune reaction is believed to be triggered by the bursting of the schizonts, the model innate response is triggered when the level of late-stage-infected RBCs is above a threshold (that varies from simulation to simulation). Then a factor is produced that attacks some subset of the parasite; the rate of removal of parasites, C, would be
Here, Att Inn is the attacker level in the blood for the model innate reaction, ξ Inn is the binding affinity of the attacker to the targeted parasites, and P is the subset of parasite population that is attacked. (For example, it could be only infected RBCs that are trophozoites. In another simulation it might be only schizonts.) In a sense, it is the product ξ Inn Att Inn that is the tangible quantity, as it is a rate of removal. The model antibody response is similar, except that a delay is incorporated before the level of the attacker factor grows; a formalism similar to that of Eq. 3 above enforces the delay. The dynamics of both types of immune responses are modeled with a system of differential equations; for details see McQueen and McKenzie (2008). Also, the immune components of the model are expounded in the ESM.
The targets, trigger levels, and binding affinities of both types of responses were varied so that simulations were performed for thousands of points in the parameter space of the models. (In addition, we consider several values of σ for the infected erythrocyte stage; see the section “Sensitivity to the standard deviation of the life cycletime of the intraerythrocyte parasite” below.) The main conclusion is that given an immune response with the same capacity to detect and clear parasites, P. vivax infection would be just as severe in terms of risk of lethal outcome as P. falciparum. This can be seen in Fig. 5, which shows the results of simulations with basic reproduction rate R 0 = 15 but with immune responses present. In most of the phase space of the model immune parameters, the immune responses protect the host from death by severe anemia. A compensatory increase in the rate of erythropoiesis to blood cell loss due to infection tends to boost overall parasitemia, especially for P. vivax, although the fraction of phase space in which the host dies is about same as for the case when the erythropoiesis is fixed at the basal rate. On the other hand, dyserythropoiesis tends to reduce on average the burden of parasitemia to which the host can mount an immune response, but increases the fraction of phase space that is lethal to the host, especially for P. vivax. One wonders if the dyserythropoiesis observed in some patients has been shaped by an evolutionary struggle between humans and Plasmodium spp.

a Fraction of points in the parameter space of the model with immune responses in which simulated infection ended with death of host as a function of the type of erythropoietic response. The fraction is of 9,520 simulations: 2,380 combinations of immune response parameters, each with four values of σ for the duration of the infected RBC stage. b The number of parasites produced during infection, IBC INT , averaged over all points in parameter space for those four types of erythropoietic responses. c Symbols used for the two Plasmodium spp. Abbreviations: Comp production rate can go up to five times the basal rate to compensate for RBC loss, Basal erythrocyte production rate held at basal rate, MDys moderate dyserythropoiesis: production rate declines by 4.5× rate of loss of cells due to infection, SDys severe dyserythropoiesis: production rate declines up to 8.5× rate of cell loss due to infection. Host was assumed to die if the RBC count dropped to under 60% of the basal count
Since even models with plausible immune responses predict that P. vivax can cause infections as severe as those of P. falciparum, why does the latter species cause more severe disease on average than the former, especially in people who have no previous exposure to either pathogen? One possible answer is that actual human immune responses to P. vivax are in a part of the immune system phase space in which infections are controlled more readily, either by being detected at a lower threshold than P. falciparum or by more efficient processes of removal of P. vivax-infected cells. Furthermore, immune responses are immensely complex, and perhaps some important component has been left out. For example, we did not consider the effects of antigenic variation in P. falciparum infection, in which the parasite modifies over time some of its proteins exposed to the immune system (Hommel et al. 1983). And RBCs infected with P. falciparum can sequester from the blood by adhering to vascular endothelium (Udomsangpetch et al. 2002), which perhaps gives that species an extra ability to evade immune attack. Although both species produce factors to suppress host responses (Kubata et al. 1998; del Portillo et al. 2004), perhaps P. falciparum can achieve suppression more readily. Yet there is another possibility: maybe severe P. vivax infection is more common than thought.
News from the clinic: severe vivax malaria
As we were doing our theoretical work on the population dynamics of P. vivax, empirical evidence mounted that this pathogen can cause severe disease after all. In Korea, vivax malaria was associated with retinal hemorrhaging (Choi et al. 2004) and splenic infarction (Kim et al. 2007). Kochar et al. (2005) reported 11 patients with severe malaria (renal and cerebral complications, severe anemia, fatal outcomes) from northwestern India in which polymerase chain reaction (PCR) analysis of parasite DNA as well as pathology examination showed P. vivax present, but not P. falciparum [see also Kochar et al. (2007)]. A follow-up report from that region (Kochar et al. 2009) showed that out of ≈1,100 patients admitted to a hospital in Bikaner for malaria, 40 of them had severe malaria in which PCR analysis could only find P. vivax and not P. falciparum. If further investigation can confirm this work, then P. vivax is a more severe public health problem than realized. The question is not academic, as the drug regime for P. vivax disease is somewhat different from that for P. falciparum disease. There are other questions, too, that need investigating: (1) Has severe vivax malaria always been present but cryptic, or are the cases seen in India and Korea due to the emergence of new strains? (2) Do patients with severe vivax malaria have some other health problem that exacerbates the disease? Only further research will show.
Empirical observation is what ultimately defines the truth of the matter, but the construction of a plausible mathematical model of malaria infection can help us understand why P. vivax can cause severe or even lethal disease. Of course, not all the pathology is due to anemia, as it is in the models discussed in this review. Some disease symptoms will be due to self-damage by the host immune responses, and perhaps by toxins released by the parasite (Elsheikha and Sheashaa 2007). Nonetheless, the modeling shows that there is no a priori reason for P. vivax to be harmless just because it attacks only a small subset of RBCs, the reticulocytes. The fact that all RBCs must pass through the reticulocyte stage means that P. vivax has the capability to do immense harm, especially if it can interfere with erythropoiesis in the bone marrow. Many of the vivax malaria patients described by Kochar et al. (2009) had severe anemia, a result understandable from our models.
Sensitivity to the standard deviation of the life cycle time of the intraerythrocyte parasite
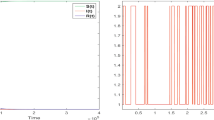
In addition to elucidating how severe anemia can come about, mathematical modeling can help frame other questions concerning basic malaria biology. One interesting phenomenon in human malaria is the synchronization of merozoite release from bursting infected RBCs. Various mechanisms have been proposed for this: (1) different effects of fever during the earlier and late parts of the erythrocytic stage (Kwiatkowski 1995), (2) modulation of the life cycle of the parasite by immune responses (Hoshen et al. 2000; Rouzine and McKenzie 2003), and (3) modulation by the melatonin cycle of the host (Hotta et al. 2000). We found in our simulation that a quickly acting, quickly decaying innate response would induce synchronized release of merozoites as illustrated in Fig. 6; otherwise, bursting of individual parasitized cells becomes out of phase (McQueen and McKenzie 2008). One question we investigated was the sensitivity of simulation results to the standard deviation in the time that parasites spend within RBCs, σ IBC . The value of this quantity by itself does not determine if synchronization occurs, but it does effect the duration of merozoite release from the synchronized bursting of schizonts. In particular, we wanted to know if short duration of release would enhance the parasite levels by allowing an evasion of immune pressures.

An example of a time series of the density of merozoites, μ, from a simulated P. vivax infection. These data are from the same simulation used to illustrate the gray curves in Fig. 1, panels A–D, in McQueen and McKenzie (2008). Both a quickly acting, quickly decaying model innate response and a slowly acting, slowing decaying antibody response are present. Time = 0 marks the beginning of infection
Simulation results indicate that a small value of σ IBC (“tight” synchronization) might not necessarily benefit the parasite; in fact, as is apparent in Fig. 7, it might actually suppress overall parasitemia. Furthermore, the fraction of the model parameter space in which the host dies of anemia is smaller as the standard deviation in the within-host duration decreases, especially for P. vivax infection. From a purely dynamical point of view, a tight synchronization of merozoite release might well benefit the host, not the parasite. However, one effect not included is the possible release of a factor that might suppress immune responses: a quick, overwhelming release of such a factor could boost parasitemia. Another issue is that for the propagation of the parasite genome, it is the survival of the gametocytes and their ability to be taken up by mosquitoes that is important, and we do not know what bearing tight synchronization has on that process. Both of these issues merit further empirical and theoretical investigation.

a Fraction of points in the parameter space of the model with immune responses in which simulated infection ended with death of host as a function of the standard deviation σ IBC in the erythrocytic stage of the parasite. The fraction is of 9,520 simulations: 2,380 combinations of immune response parameters, each with the four types of erythropoietic responses as given in the legend of Fig. 5. b Number of parasites produced during infection, IBC INT , averaged over all points in parameter space for those four values of σ. Squares P. falciparum , circles P. vivax. Host death was assumed if the RBC count dropped to under 60% of the basal count
Some further directions
The production of gametocytes is an area of active research, as much remains to be discovered. For example, are gametocytes produced at a constant rate throughout infection or is their production triggered by some stressor on the within-host parasite population (Carter and Miller 1979)? Indeed, Diebner et al. (2000) used mathematical modeling results to argue that the rate of gametocyte production is highly variable during P. falciparum infection and is modulated by host innate immunity. Furthermore, the actual transition steps from asexual forms to gametocytes are still not known, although careful gene expression analysis is giving us a clearer answer (Eksi et al. 2008). Can modeling help with the understanding of that process? The development of gametocytes differs between P. vivax and P. falciparum (McKenzie et al. 2007); what are the implications of that for their population dynamics (and perhaps on their transmissibility to mosquitoes)? Further modeling could well prove fruitful in understanding the genesis and development of gametocytes.
Finally, in the discussions above, I used severe anemia as a proxy for severity of infection in the hypothetical patient. But one could easily use additional criteria to define an infection in which the host is at major risk of dying or suffering long term disability. For example, the level of cytokines in a model immune response could be used as a proxy for possible cerebral damage. One could also consider a build-up of toxins from the parasite as well. “What-if” questions could be used to elucidate the consequences of plausible processes that may damage the host in cases of malaria.
References
Baer K, Klotz C, Kappe SH, Schnieder T, Frevert U (2007) Release of hepatic Plasmodium yoelii merozoites into the pulmonary microvasculature. PLoS Pathog 3:e171
Carter R, Miller LH (1979) Evidence for environmental modulation of gametocytogenesis in Plasmodium falciparum in continuous culture. Bull World Health Org 57(Suppl 1):37–52
Cash JR, Karp AH (1990) A variable order Runge-Kutta method for initial value problems with rapidly varying right-hand sides. Assoc Comput Machinery Trans Math Softw 16:201222
Chiyaka C, Garira W, Dube S (2009) Effects of treatment and drug resistance on the transmission dynamics of malaria in endemic areas. Theor Popul Biol 75:14–29
Choi HJ, Lee SY, Yang H, Bang JK (2004) Retinal haemorrhage in vivax malaria. Trans R Soc Trop Med Hyg 98:387–389
Collins WE, Jeffery GM (1999) A retrospective examination of the patterns of recrudescence in patients infected with Plasmodium falciparum. Am J Trop Med Hyg 61:4448
Cox-Singh J, Singh B (2008) Knowlesi malaria: newly emergent and of public importance? Trends Parasitol 24:406–410
del Portillo HA, Lanzer M, Rodriguez-Malaga S, Zavala F, Fernandez-Becerra C (2004) Variant genes and the spleen in Plasmodium vivax malaria. Int J Parasitol 34:1547–1554
Diebner HH, Eichner M, Molineaux L, CollinsWE, Jeffery GM, Dietz K (2000) Modelling the transition of asexual blood stages of Plasmodium falciparum to gametocytes. J Theor Biol 202:113–127
Eksi S, Suri A, Williamson KC (2008) Sex- and stage-specific reporter gene expression in Plasmodium falciparum. Mol Biochem Parasitol 160:148–151
Elsheikha HM, Sheashaa HA (2007) Epidemiology, pathophysiology, management and outcome of renal dysfunction associated with plasmodia infection. Parasitol Res 101:1183–1190
Engwerda CR, Good MF (2005) Interactions between malaria parasites and the host immune system. Curr Opin Immunol 17:381–387
Garnham PCC (1966) Malaria parasites and other Haemosporidia. Blackwell Scientific, Oxford
Glushakova S, Yin D, Li T, Zimmerberg J (2005) Membrane transformation during malaria parasite release from human red blood cells. Curr Biol 15:R760R761
Gravenor MB, McLean AR, Kwiatkowski D (1995) The regulation of malaria parasitaemia: parameter estimates for a population model. Parasitology 110:115–122
Gravenor MB, Lloyd AL, Kremsner PG, Missinou MA, English M, Marsh K, Kwiatkowski D (2002) A model for estimating total parasite load in falciparum malaria patients. J Theor Biol 217:137–148
Haydon DT, Matthews L, Timms R, Colegrave N (2003) Top-down or bottom-up regulation of intra-host blood-stage malaria: do malaria parasites most resemble the dynamics of prey or predator? Proc Biol Sci 270:289–298
Hommel M, David PH, Oligino LD (1983) Surface alterations of erythrocytes in Plasmodium falciparum malaria. Antigenic variation, antigenic diversity, and the role of the spleen. J Exp Med 157:1137–1148
Hoppensteadt FC (1982) Mathematical methods of population biology. Cambridge University Press, New York
Hoshen MB, Heinrich R, Stein WD, Ginsburg H (2000) Mathematical modelling of the within-host dynamics of Plasmodium falciparum. Parasitology 121:227–235
Hotta CT, Gazarini ML, Beraldo FH, Varotti FP, Lopes C et al (2000) Calcium-dependent modulation by melatonin of the circadian rhythm in malarial parasites. Nat Cell Biol 2:466–468
Johnson JG, Epstein N, Shiroishi T, Miller LH (1980) Factors affecting the ability of isolated Plasmodium knowlesi merozoites to attach to and invade erythrocytes. Parasitology 80:539–550
Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN (1992) Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci USA 15:3200–3203
Karunaweera ND, Wijesekera SK, Wanasekera D, Mendis KN, Carter R (2003) The paroxysm of Plasmodium vivax malaria. Trends Parasitol 19:188–193
Kim A, Park YK, Lee JS, Chung MH, Kim ES (2007) A case of symptomatic splenic infarction in vivax malaria. Korean J Parasitol 45:55–58
Kochar DK, Saxena V, Singh N, Kochar SK, Kumar SV, Das A (2005) Plasmodium vivax malaria. Emerg Infect Dis 11:132–134
Kochar DK, Pakalapati D, Kochar SK, Sirohi P, Khatri MP, Kochar A, Das A (2007) An unexpected cause of fever and seizures. Lancet 370:908
Kochar DK, Das A, Kochar SK, Saxena V, Sirohi P, Garg S, Kochar A, Khatri MP, Gupta V (2009) Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am J Trop Med Hyg 80:194–198
Krogstad DJ (1995) In: Mandell GL, Bennett JE, Dolin R (eds) Principles and practice of infectious diseases. Churchill Livingstone, New York, p 24152427
Kubata BK, Eguchi N, Urade Y, Yamashita K, Mitamura T, Tai K, Hayaishi O, Horii T (1998) Plasmodium falciparum produces prostaglandins that are pyrogenic, somnogenic, and immunosuppressive substances in humans. J Exp Med 188:1197–1202
Kwiatkowski D (1995) Malarial toxins and the regulation of parasite density. Parasitol Today 11:206–212
Lloyd AL (2001a) The dependence of viral parameter estimates on the assumed viral life cycle: limitations of studies of viral load data. Proc R Soc B Biol Sci 268:847–854
Lloyd AL (2001b) Destabilization of epidemic models with the inclusion of realistic distributions of infectious periods. Proc R Soc B Biol Sci 268:985–993
Maire N, Smith T, Ross A, Owusu-Agyei S, Dietz K, Molineaux L (2006) A model for natural immunity to asexual blood stages of Plasmodium falciparum malaria in endemic areas. Am J Trop Med Hyg 75(2 Suppl):1931
Malthus TR (2003) An essay on the principle of population (reprint of 1798 original). In: Applemann P (ed) An essay on the principle of population. Norton, New York, pp 15–124
Maynard Smith J (1968) Mathematical ideas in biology. Cambridge University Press, London
McKenzie FE, Jeffery GM, Collins WE (2001) Plasmodium malariae blood-stage dynamics. J Parasitol 87:626637
McKenzie FE, Jeffery GM, Collins WE (2002) Plasmodium vivax blood-stage dynamics. J Parasitol 88:521535
McKenzie FE, Jeffery GM, Collins WE (2007) Gametocytemia and fever in human malaria infections. J Parasitol 93:627–633
McQueen PG, McKenzie FE (2004) Age-structured red blood cell susceptibility and the dynamics of malaria infections. Proc Natl Acad Sci USA 101:1961–1966
McQueen PG, McKenzie FE (2006) Competition for red blood cells can enhance Plasmodium vivax parasitemia in mixed-species malaria infections. Am J Trop Med Hyg 75:112–125
McQueen PG, McKenzie FE (2008) Host control of malaria infections: constraints on immune and erythropoeitic response kinetics. PLoS Comp Biol 4:e100149
Miller LH, Baruch D, Marsh K, Doumbo OK (2002) The pathogenic basis of malaria. Nature 415:673679
Perkins SL, Austin CC (2009) Four new species of Plasmodium from New Guinea lizards: integrating morphology and molecules. J Parasitol 95:424–433
Plebanski M, Hill AV (2000) The immunology of malaria infection. Curr Opin Immunol 12:437–441
Rouzine IM, McKenzie FE (2003) Link between immune response and parasite synchronization in malaria. Proc Natl Acad Sci USA 100:3473–3478
Thibodeaux JJ (2010) Modeling erythropoiesis subject to malaria infection. Math Biosci 225:59–67
Udomsangpetch R, Pipitaporn B, Silamut K, Pinches R, Kyes S, Looareesuwan S, Newbold C, White NJ (2002) Febrile temperatures induce cytoadherence of ring-stage Plasmodium falciparum infected erythrocytes. Proc Natl Acad Sci USA 99:11825–11829
White NJ (1997) Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob Agents Chemother 41:14131422
White NJ, Breman JG (2008) Malaria. In: Fauci AS, Braunwald E, Kasper DL et al. (eds) Harrison’s principles of internal medicine, 17e (http://www.accessmedicine.com/content.aspx?aID=2896266). McGraw-Hill, New Yorkk
Wickramasinghe SN, Abdalla SH (2000) Blood and bone marrow changes in malaria. Baillières Best Pract Res Clin Haematol 13:277–299
Acknowledgments
I thank Dr. F. Ellis McKenzie, Forgarty International Center, National Institutes of Health, for insightful discussion. Some of the studies utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). This research was supported by the Intramural Research Program of the NIH, Center of Information Technology.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 830 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
McQueen, P.G. Population dynamics of a pathogen: the conundrum of vivax malaria. Biophys Rev 2, 111–120 (2010). https://doi.org/10.1007/s12551-010-0034-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-010-0034-3