
Abstract
Purpose of the review
The ongoing outbreak of novel coronavirus pneumonia (COVID-19) caused by the 2019 novel coronavirus (SARS-CoV-2) in China is lifting widespread concerns. Thus, therapeutic options are urgently needed, and will be discussed in this review.
Recent findings
Iron-containing enzymes are required for viruses most likely including coronaviruses (CoVs) to complete their replication process. Moreover, poor prognosis occurred in the conditions of iron overload for patients upon infections of viruses. Thus, limiting iron represents a promising adjuvant strategy in treating viral infection through oral uptake or venous injection of iron chelators, or through the manipulation of the key iron regulators. For example, treatment with iron chelator deferiprone has been shown to prolong the survival of acquired immunodeficiency syndrome (AIDS) patients. Increasing intracellular iron efflux via increasing iron exporter ferroportin expression also exhibits antiviral effect on human immunodeficiency virus (HIV). The implications of other metals besides iron are also briefly discussed.
Summary
For even though we know little about iron regulation in COVID-19 patients thus far, it could be deduced from other viral infections that iron chelation might be an alternative beneficial adjuvant in treating COVID-19.
Similar content being viewed by others
Introduction
The ongoing outbreak of pneumonia caused by SARS-CoV-2 in China is eliciting widespread concerns, especially as the virus was recently shown to spread from human to human [1]. This epidemic is calling for national and international attention to develop effective therapeutics including selective vaccines. Nonetheless, no specific therapeutic is yet available, leaving the patients to rely on general and supportive therapies, including oxygen supply, broad-spectrum antiviral medicines (e.g., interferon-α), glucocorticoid, and human serum albumin (HSA) [2]. Most recently, lopinavir/ritonavir, an approved anti-HIV drug, has been recommended for treatment of SARS-CoV-2 infection [3]. Remdesivir, a novel nucleotide analogue prodrug in development for treating Ebola virus and Middle East Respiratory Syndrome (MERS) diseases, has also been reported to relieve the pneumonia symptoms for the first case of SARS-CoV-2 infection, as reported in the USA [4]. Although the drugs are showing a promising efficacy, additional therapeutic options should be explored and considered when taking into account the increasing number of SARS-CoV-2 cases.
Since iron is involved in many fundamental biological processes including DNA/RNA synthesis and ATP generation, viruses essentially rely on iron to replicate in host cells [5]. Thereby, there is an active competition between the virus and the host in fighting for iron. Clinical data have indicated that poor prognosis is related to the condition of iron overload observed in patients with infection of hepatitis B/C (HBV/HCV) viruses [6,7,8]. For even though we know little about iron regulations in SARS-CoV-2 patients, some clues could be obtained from other viral infections (Fig. 1). For example, iron supplementation was shown to increase the mortality in HIV-infected patients, irrespective of the severity of anemia, suggesting a detrimental role of excess iron in HIV infection [9]. HIV-1 replication is dependent on host cell enzymes, some of which are involved in transcription, viral mRNA translation, and viral assembly that require iron [5]. HIV-1 viral load dropped dramatically in a hemochromatosis patient who underwent venesection, suggesting an iron deprivation–mediated control of HIV-1 replication [10].

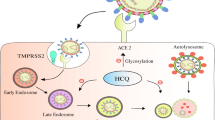
Life cycle of coronaviruses under iron replete and deficiency conditions. a Sufficient intracellular iron levels support coronavirus replication, whereas b iron deficiency undermines its replication process by interfering with viral transcription, translation, assembly, and exocytosis. CoVs enter into host cells via binding to various receptors and disassemble to release viral genome and nucleocapsid. Transcription and translation of viral genes yield viral genomic RNA and structure proteins (e.g., S, E, M, and N). After further processing and assembly in endoplasmic reticulum (ER) and Golgi, new CoVs are constructed. Finally, the new formed virions are exocytosed by fusing with virus-containing vesicles. The whole process of viral replication requires iron-containing enzymes and consumes abundant ATP. Iron is a critical participant for mitochondria to produce ATP. In short, adequate iron enables the virus to complete its replication process, and otherwise iron deficiency impairs this process
To this end, iron depletion can have marked anti-HIV effect. HIV-1 transcription and replication were inhibited by number of iron chelators including 2-hydroxy-1-naphthylaldehyde benzoyl hydrazine (311) and ICL670 (also known as deferasirox or exjade) [11], 2-benzoylpyridine 4-allyl-3-thiosemicarbazone (Bp4aT) and 2-benzoylpyridine 4-ethyl-3-thiosemicarbazone (Bp4eT) [12], and PPYeT and PPYaT [13]. In a recent study, ex vivo inhibition of HIV-1 in peripheral blood mononuclear cells (PBMCs) obtained from patients with sickle cell disease (SCD) was linked to the increased expression of ferroportin and reduced intracellular iron levels [14]. Thus, we can consider the potential of iron chelation as an alternative beneficial adjuvant in treating SARS-CoV-2 infection that we discuss below.
Characteristics and Life Cycle of Coronaviruses
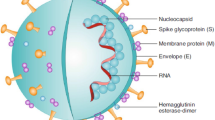
CoVs represent the largest group of Coronaviridae, belonging to the Nidovirales order, whereby positive-sense and single-stranded RNA is enveloped inside. Apart from CoVs, Torovirinae is the other subfamily of Coronaviridae. CoVs are further subdivided into 4 genera: α-CoV, β-CoV, γ-CoV, and δ-CoV on the basis of their phylogenetic clustering. SARS-CoV-2 that causes COVID-19 belongs to the β group [15, 16]. CoVs are named after their “crown-like” appearance observed under electron microscopy with club-shaped peplomers projecting from their surface. CoVs are non-segmented positive-sense RNA viruses containing approximately 30 kilobase (kb) genomes, characterized by the organization of 5′-leader-UTR-replicase-S (Spike)-E (Envelope)-M (Membrane)-N (Nucleocapsid)-3′UTR-poly (A) tail with accessory genes interspersed within the structural genes at the 3′ end of the genome [17]. To penetrate host cells, CoVs can use various cell surface molecules as their receptors, preferentially ectoenzymes, e.g., aminopeptidase N (APN), angiotensin-converting enzyme 2 (ACE2), and dipeptidyl peptidase 4 (DPP4) [18].
The SARS-CoV-2 is the newly discovered member of the coronavirus family. Based on the most recent results of genome sequencing, most contigs of SARS-CoV-2 match to the genome of the lineage B from β-CoV and show more than 85% identity with the genome of a bat Severe Acute Respiratory Syndrome (SARS)–like CoV (bat-SL-CoVZC45, MG772933.1) [19]. Furthermore, it was found that SARS-CoV-2 share 96% identical at the whole-genome level to a bat CoV [20]. Despite the sequence diversity with SARS-CoV S-protein, SARS-CoV-2 S-protein shares the similar receptor binding domain (RBD) domain as the SARS/SARS-like CoV, in support of the strong interaction of SARS-CoV-2 with human ACE2 molecules [21]. More than 80% of ACE2 receptors are expressed in a small population of type II alveolar cells (AT2) [22], suggesting that AT2 cells could be the target cells of SARS-CoV-2.
After binding and fusion with the host cells, CoVs disassemble to liberate the inside contents of the virion into the cytoplasm, including the nucleocapsid and viral RNA. Components of the replication-transcription complex are firstly translated [23]. In addition, abundant sub-genomic negative-sense RNAs are produced. Then, the viral structural proteins (S, E, and M) are translated, inserted into the endoplasmic reticulum (ER), and transported to the ER-Golgi intermediate compartment. After accumulation of adequate viral genomic RNA and structural proteins, the N protein and genomic RNA assemble in cytoplasm to form the helical nucleocapsid. Subsequently, the S, E, and M proteins delivered to the budding compartment interact with nucleocapsid to constitute the assembled virus. Finally, the virus is released from the Golgi and exocytosed to the extracellular compartment from the host cells by fusing with virion-containing vesicles [24] (Fig. 1).
Iron Dependence of Viral Replication
For the host, iron is an essential trace element necessary for many fundamental enzymatic and non-enzymatic reactions and diverse physiological processes, such as mitochondrial function including ATP generation, DNA/RNA synthesis and repair, and cell survival/ferroptosis [25]. Iron is also essential for viral replication. In the context of HIV-1 infection, iron is involved in several key steps of virus replication. In the reverse transcription of viral RNA into DNA, the required dNTPs are generated by RNRs which are an iron-dependent proteins [26]. NF-κB can be activated by iron via generating reactive oxygen species (ROS) [27]. IкB kinase activation depends on iron efflux [28,29,30], which increases NF-кB levels and contributes to the activation of HIV-1 promoter [31]. Nuclear export of new transcribed viral RNA is also iron-dependent [32]. Finally, an iron-binding ATPase, ATP binding cassette subfamily E member 1 (ABCE1), is involved in the assembly of the Gag capsid proteins into mature HIV-1 virions [33].
ATP hydrolysis is necessary for the unwinding activity of helicases of SARS-CoV and MERS-CoV during the viral replication [34, 35]. Iron is an important component of the complexes I, II, III, and IV as well as cytochromes, which participate in the oxidative phosphorylation in mitochondria to conduct electron transportation in maintaining mitochondrial functions and ATP synthesis [36]. Treatment with deferiprone (DFP) induces apoptosis in HIV-1-infected cells through mitochondrial membrane depolarization, leading to permanent elimination of infected cells in culture [37].
Interplay of Host Iron Metabolism and Viral Intrusion
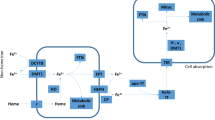
Iron metabolism in host is fine-tuned through regulation of iron absorption in the intestine, iron storage in the liver and spleen, iron transport in blood, iron utilization (mainly in bone marrow for erythropoiesis), and iron recycling by macrophages. Iron is absorbed by duodenal enterocytes and released into plasma by an iron exporter protein, ferroportin, which is expressed on the basolateral side of the duodenal enterocytes. Ferroportin is also expressed in macrophages and essentially governs iron release and recycling [38]. The systemic iron homeostasis is fundamentally orchestrated by the hepcidin-ferroportin axis. Hepcidin is mainly expressed and secreted by hepatocytes, and can bind to its sole receptor, ferroportin. After binding to hepcidin, ferroportin is internalized and degraded, leading to the inhibition of iron absorption from the duodenum and reduction of iron release from macrophages [39]. The cellular iron uptake is primarily mediated by the interaction between iron-bound transferrin and transferrin receptor1 (TfR1) [40].
Hepcidin expression could be regulated by systemic iron availability (iron deficiency and iron overload), inflammatory cytokines (IL-6 and IL-1β), bone morphogenetic proteins (BMP2 and BMP6), and erythropoietic signals (ERFE, GDF15, and TWSG1) [40]. SARS patients exhibited increased amounts of proinflammatory cytokines in serum including IL-1β, IL-6, and IFN-γ coupled to the pulmonary inflammation and extensive lung damage [41]. Infection with SARS-CoV-2 also leads to the increased levels of IL1-β, IFN-γ, IP10 (interferon-inducible protein 10), and MCP1 (monocyte chemotactic protein 1), likely inducing T-helper-1 cell response [3]. Thus, induction of these cytokines could supposedly promote hepcidin production and lead to iron sequestration in macrophages, which warrants future investigations. Of note, macrophages are presumed to be infected by SARS-CoV-2 [42]. Thus, increased iron storage will most likely favor viral replication inside macrophages. Furthermore, viruses can manipulate other iron-related proteins to facilitate their replication and propagation. In the context of human cytomegalovirus (HCMV) infection, homeostatic iron regulator protein (HFE), a competitor of TfR1 to bind to transferrin, is degraded after binding by US2 protein, leading to cellular iron overload [43]. In macrophages infected by HIV-1, the interaction of Nef protein and HFE also induces cellular iron overload [44]. Plus, TfR1 is also used as the receptor during the entry of several types of viruses [45, 46].
Antiviral Strategy of Iron Chelation
Iron dependence of viral replication and modulation of host iron metabolism by viruses, as discussed above, signifies the importance of cellular iron homeostasis in viral life cycle and incites the development of iron chelation strategy in treating viral infections. Currently, there are two promising strategies to deplete iron. The first strategy is to deplete iron directly by iron chelators which have strong and selective affinity with iron ions [47, 48]. Some of these iron chelators have been approved by U.S. Food and Drug Administration for clinical use, such as deferoxamine (DFO, Desferal®), deferiprone (DFP, Ferriprox®), and deferasirox (ICL670, Exjade®) [49]. Iron chelators can bind free iron or remove iron from iron-containing proteins [48]. Treatment with higher doses of DFP has been shown to prolong the survival of AIDS patients after HIV-1 infection [50]. Increasing evidence suggests that iron chelators can target HIV-1 replication. The enzymatic activity of ribonucleotide reductase 2 involved in reverse transcription, which contains non-heme iron, is inhibited by DFO and 311 [51]. Bp4aT, Bp4eT, Phenyl-1-pyridin-2yl-ethanone (PPY)-based iron chelators (PPYeT and PPYaT) inhibit HIV-1 transcription by decreasing CDK2 and CDK9 activities, and by upregulating IκBα expression and downregulating nuclear NF-κB [12, 13]. Topical fungicide ciclopirox and the iron chelator DFP inhibit HIV-1 gene expression at the level of transcription initiation by interfering with the hypusine modification of eIF5α [32]. Patients treated with DFP unveiled strong HIV-1 RNA decline while on-drug and also for up to 8 weeks off-drug without viral rebound [52]. DFO and DFP inhibited HIV-1 replication in human PBMCs and macrophages but the inhibition is attributed to a decrease in cell proliferation [53]. Similar to DFO and DFP, oral uptake of bidentate iron chelators, CP502 and CP511 inhibit HIV-1 replication by reducing cellular proliferation [54]. Host cell enlargement induced by viruses, e.g., HCMV, could be inhibited by iron chelators through inhibiting mitochondrial activity and macromolecular synthesis [55]. Nonetheless, these iron chelators may be scrutinized for their antiviral activity against SARS-CoV-2.
The second strategy is to deplete cellular iron through regulating the gene expression involved in iron metabolism. HIV-1 reverse transcription and transcription was suppressed in PBMCs obtained from SCD patients due to increased expression of ferroportin and therefore lowered intracellular iron [14]. On the other hand, hepcidin agonists, such as mini-hepcidin and thiazolidinone derivatives, can reduce systemic iron levels by compromising the function of ferroportin [56]. Nonetheless, the possible applications of agonists to target the hepcidin-ferroportin axis or other iron-related genes in order to achieve antiviral effects still need further exploration, and more efforts are thus urgently needed.
Other Metal-Oriented Therapeutics Against Viral Infection
Apart from iron, cumulative evidence has manifested that other metals (e.g., calcium, zinc, and magnesium) are also involved in the replication process of CoVs. The entry of CoVs into host cells is mediated by the viral S protein. Under this context, previous studies have demonstrated that calcium is indispensable for SARS-CoV S-mediated fusion [57]. The replication of SARS-CoV genome requires RNA-dependent RNA polymerase (RdRp) to synthesize descendant RNAs from a RNA template, which sternly relies on magnesium (Mn2+) for its activity [58]. In the meantime, SARS-CoV RNA dimers, a prerequisite for ribosomal frameshifting, are assembled through “kissing” loop-loop interactions. Nonetheless, to reach more stable formation of loop-loop kissing complex, the presence of Mn2+ appears to be necessary [59]. Regarding zinc (Zn2+), the binding of Zn2+ ions to the metal-binding domain (MBD) of SARS-CoV helicase is essential for its enzymatic activity [60]. Additionally, the maturation of CoVs requires papain-like protease (PLpro), which could cleave the nonstructural polyproteins (pp1a and pp1ab). However, devoid of Zn2+ ions, the stability of the tertiary structure of SARS-CoV PLpro is compromised with diminished activity [61]. Moreover, MERS-CoV PLpro bears a folded structure and potent proteolytic and deubiquitinating activities upon binding with endogenous metal ions [62]. To this end, the above findings collectively suggest that disturbing the viral metal dependence would presumably exhibit antiviral effects.
Literally, versatile metal-oriented therapeutics besides iron chelators have been searched to target diseased conditions for centuries. For instance, bismuth compounds have been used clinically as medicines to treat various gastrointestinal diseases. Bismuth (Bi3+) ions strikingly compete with Zn2+ ions for the MBD of helicase, leading to compromised enzyme activities and severe deficiencies in viral replication [63]. As metal chelators, aryl diketoacids (ADK) have been verified to inhibit enzymes, such as HIV-1 integrase and HCV RdRp, in that ADKs function to repress the NTPase/helicase activities of SARS-CoV by mimicking Bi3+ ions [64]. Mercury (Hg2+) ions and Zn2+ ions act to inhibit viral proteases, such as 3C-like protease (3CLpro) of norovirus, PLpro of SARS-CoV, HCMV protease, and HCV NS3 protease [62]. Of note, the 3CLpro plays a vital role in viral protein maturation for SARS-CoV. In fact, their metal-conjugated compounds, including phenylmercuric acetate (PMA), toluene-3,4-dithiolato zinc (TDT), and Nethyl-N-phenyldithiocarbamic acid zinc (EPDTC), elicit great inhibition on SARS-CoV 3CLpro. As a pharmaceutical excipient, PMA is widely used in parenteral and topical pharmaceutical formulations. Further, zinc acetate is added as a supplement to the drug in treating Wilson’s disease [65]. Zn2+ ions could directly impair viral RNA synthesis, due to its strong suppression on both the replication and transcription complexes [66]. As summarized above, an array of other metals besides iron incredibly account for the functions and activities of enzymes involved in CoVs’ replication, which underpins rational metal-oriented therapeutic development against CoVs.
Conclusion and Perspectives
Iron is crucial for both the host and the pathogen. Iron supply is required for the replication of many viruses, most likely including CoVs, and viruses rely on intracellular iron for their propagation. Emerging studies indicate that iron manipulation, such as iron chelation, is a promising adjuvant therapy in treating viral infection. While the emerging viral infection by SARS-CoV-2 is much less understood compared with HIV-1 or SARS-CoV and MERS-CoV, based on the previous studies, it is plausible that deprivation of iron supply to the virus could serve as a beneficial adjuvant in treating the SARS-CoV-2 infection, with the prerequisite of adequate understandings on one’s iron status, such as serum iron and ferritin levels, and globin content. Meanwhile, other metal-oriented therapeutics could also be reasonably conceived for the antiviral purpose.
Data Availability
Not applicable.
Abbreviations
- 311:
-
2-hydroxy-1-naphthylaldehyde benzoyl hydrazine
- 3CLpro :
-
3C-like protease
- ABCE1:
-
ATP binding cassette subfamily E member 1
- ACE:
-
angiotensin-converting enzyme 2
- ADK:
-
aryl diketoacids
- AIDS:
-
acquired immunodeficiency syndrome
- APN:
-
aminopeptidase N
- AT2:
-
small population of type II alveolar cells
- BMP:
-
bone morphogenetic proteins
- Bp4aT:
-
2-benzoylpyridine 4-allyl-3-thiosemicarbazone
- Bp4eT:
-
2-benzoylpyridine 4-ethyl-3-thiosemicarbazone
- COVID-19:
-
novel coronavirus pneumonia
- CoVs:
-
coronaviruses
- DFO:
-
deferoxamine
- DFP:
-
deferiprone
- DPP4:
-
dipeptidyl-peptidase 4.
- E:
-
envelope
- EPDTC:
-
Nethyl-N-phenyldithiocarbamic acid zinc
- ER:
-
endoplasmic reticulum
- HCMV:
-
human cytomegalovirus
- HFE:
-
homeostatic iron regulator protein
- HIV:
-
human immunodeficiency virus
- HSA:
-
human serum albumin
- IP10:
-
interferon-inducible protein 10
- M:
-
membrane
- MBD:
-
metal-binding domain
- MCP1:
-
monocyte chemotactic protein 1
- MERS:
-
Middle East respiratory syndrome
- N:
-
nucleocapsid
- PBMC:
-
peripheral blood mononuclear cells
- PLpro :
-
papain-like protease
- PMA:
-
phenylmercuric acetate
- PPY:
-
phenyl-1-pyridin-2yl-ethanone
- RdRp:
-
RNA-dependent RNA polymerase
- ROS:
-
reactive oxygen species
- S:
-
spike
- SARS:
-
severe acute respiratory syndrome
- SARS-CoV-2:
-
the 2019 novel coronavirus
- SCD:
-
sickle cell disease
- TDT:
-
toluene-3,4-dithiolato zinc
- TfR1:
-
transferrin receptor1
References
Chan JF, Yuan S, Kok KH, To KK, Chu H, Yang J, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020.
Working Group of Novel Coronavirus PUMCH. Diagnosis and clinical management of 2019 novel coronavirus infection: an operational recommendation of Peking Union Medical College Hospital (V2.0). Zhonghua Nei Ke Za Zhi. 2020;59(3):186–8.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H, et al. First Case of 2019 Novel Coronavirus in the United States. N Engl J Med. 2020;382(10):929–36.
Drakesmith H, Prentice A. Viral infection and iron metabolism. Nat Rev Microbiol. 2008;6(7):541–52.
Kaufmann SH, McMichael AJ. Annulling a dangerous liaison: vaccination strategies against AIDS and tuberculosis. Nat Med. 2005;11(4 Suppl):S33–44.
Thursz M. Iron, haemochromatosis and thalassaemia as risk factors for fibrosis in hepatitis C virus infection. Gut. 2007;56(5):613–4.
Galli A, Svegliati-Baroni G, Ceni E, Milani S, Ridolfi F, Salzano R, et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology. 2005;41(5):1074–84.
Haider BA, Spiegelman D, Hertzmark E, Sando D, Duggan C, Makubi A, et al. Anemia, iron deficiency, and Iron supplementation in relation to mortality among HIV-infected patients receiving highly active antiretroviral therapy in Tanzania. Am J Trop Med Hyg. 2019;100(6):1512–20.
Greaves DE, Griffiths WJ, Lever AM. Does venesection reduce HIV viral load in patients with hereditary haemochromatosis? Antivir Ther. 2013;18(1):135–8.
Debebe Z, Ammosova T, Jerebtsova M, Kurantsin-Mills J, Niu X, Charles S, et al. Iron chelators ICL670 and 311 inhibit HIV-1 transcription. Virology. 2007;367(2):324–33.
Debebe Z, Ammosova T, Breuer D, Lovejoy DB, Kalinowski DS, Kumar K, et al. Iron chelators of the di-2-pyridylketone thiosemicarbazone and 2-benzoylpyridine thiosemicarbazone series inhibit HIV-1 transcription: identification of novel cellular targets--iron, cyclin-dependent kinase (CDK) 2, and CDK9. Mol Pharmacol. 2011;79(1):185–96.
Kumari N, Iordanskiy S, Kovalskyy D, Breuer D, Niu X, Lin X, et al. Phenyl-1-Pyridin-2yl-ethanone-based iron chelators increase IkappaB-alpha expression, modulate CDK2 and CDK9 activities, and inhibit HIV-1 transcription. Antimicrob Agents Chemother. 2014;58(11):6558–71.
Kumari N, Ammosova T, Diaz S, Lin X, Niu X, Ivanov A, et al. Increased iron export by ferroportin induces restriction of HIV-1 infection in sickle cell disease. Blood Adv. 2016;1(3):170–83.
Peng X, Xu X, Li Y, Cheng L, Zhou X, Ren B. Transmission routes of 2019-nCoV and controls in dental practice. Int J Oral Sci. 2020;12(1):9.
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–74.
Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020.
Fehr A, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol (Clifton, NJ). 2015;1282:1–23.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China. N Engl J Med. 2019:2020.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–27.
Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63(3):457–60.
Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020. (in press).
Chan JFW, Lau SKP, To KKW, Cheng VCC, Woo PCY, Yuen K-Y. Middle East respiratory syndrome coronavirus: another zoonotic Betacoronavirus causing SARS-like disease. Clin Microbiol Rev. 2015;28(2):465–522.
Stadler K, Masignani V, Eickmann M, Becker S, Abrignani S, Klenk H-D, et al. SARS—beginning to understand a new virus. Nat Rev Microbiol. 2004;1:209–18.
Khodour Y, Kaguni LS, Stiban J. Iron-sulfur clusters in nucleic acid metabolism: varying roles of ancient cofactors. Enzymes. 2019;45:225–56.
Romeo AM, Christen L, Niles EG, Kosman DJ. Intracellular chelation of Iron by bipyridyl inhibits DNA virus replication: ribonucleotide reductase maturation as a probe of intracellular iron pools. J Biol Chem. 2001;276(26):24301–8.
Xiong S, She H, Takeuchi H, Han B, Engelhardt JF, Barton CH, et al. Signaling role of intracellular Iron in NF-κB activation. J Biol Chem. 2003;278(20):17646–54.
Chen L, Xiong S, She H, Lin SW, Wang J, Tsukamoto H. Iron causes interactions of TAK1, p21ras, and phosphatidylinositol 3-kinase in caveolae to activate IkappaB kinase in hepatic macrophages. J Biol Chem. 2007;282(8):5582–8.
Xiong S, She H, Sung CK, Tsukamoto H. Iron-dependent activation of NF-kappaB in Kupffer cells: a priming mechanism for alcoholic liver disease. Alcohol. 2003;30(2):107–13.
Xiong S, She H, Tsukamoto H. Signaling role of iron in NF-kappa B activation in hepatic macrophages. Comp Hepatol. 2004;3(Suppl 1):S36.
Pereira LA, Bentley K, Peeters A, Churchill MJ, Deacon NJ. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000;28(3):663–8.
Hoque M, Hanauske-Abel HM, Palumbo P, Saxena D, D'Alliessi Gandolfi D, Park MH, et al. Inhibition of HIV-1 gene expression by Ciclopirox and Deferiprone, drugs that prevent hypusination of eukaryotic initiation factor 5A. Retrovirology. 2009;6:90.
Zimmerman C, Klein K, Kiser P, Singh A, Firestein B, Riba S, et al. Identification of a host protein essential for assembly of immature HIV-1 capsids. Nature. 2002;415:88–92.
Jia Z, Liming Y, Ren Z, Wu L, Wang J, Guo J, et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47.
Adedeji A, Lazarus H. Biochemical characterization of Middle East respiratory syndrome coronavirus helicase. mSphere. 2016;1:e00235–16.
Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, et al. Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers. Chem Rev. 2014;114(8):4366–469.
Hanauske-Abel HM, Saxena D, Palumbo PE, Hanauske AR, Luchessi AD, Cambiaghi TD, et al. Drug-induced reactivation of apoptosis abrogates HIV-1 infection. PLoS One. 2013;8(9):e74414.
Ganz T. Molecular control of iron transport. J Am Soc Nephrol. 2007;18(2):394–400.
Liu J, Sun B, Yin H, Liu S. Hepcidin: a promising therapeutic target for Iron disorders: a systematic review. Medicine. 2016;95:e3150.
Hentze M, Muckenthaler M, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. 2010;142:24–38.
Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136(1):95–103.
Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS. J Virol. 2020;94(7).
Arieh S, Laham-Karam N, Schechter C, Yewdell J, Coligan J, Ehrlich R. A single viral protein HCMV US2 affects antigen presentation and intracellular iron homeostasis by degradation of classical HLA class I and HFE molecules. Blood. 2003;101:2858–64.
Drakesmith H, Chen N, Ledermann H, Screaton G, Townsend A, Xu X. HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. Proc Natl Acad Sci U S A. 2005;102:11017–22.
Parker J, Murphy W, Wang D, O'Brien S, Parrish C. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J Virol. 2001;75.
Radoshitzky S, Abraham J, Spiropoulou C, Kuhn J, Nguyen D, Li W, et al. Transferrin receptor 1 is a cellular receptor for New World hemorrhagic fever arenaviruses. Nature. 2007;446:92–6.
Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–67.
Li B, Esposito B, Wang S, Zhang J, Xu M, Zhang S, et al. Desferrioxamine-caffeine shows improved efficacy in chelating iron and depleting cancer stem cells. J Trace Elem Med Biol. 2019;52.
Cappellini MD. Exjade(R) (deferasirox, ICL670) in the treatment of chronic iron overload associated with blood transfusion. Ther Clin Risk Manag. 2007;3(2):291–9.
Costagliola DG, de Montalembert M, Lefrere JJ, Briand C, Rebulla P, Baruchel S, et al. Dose of desferrioxamine and evolution of HIV-1 infection in thalassaemic patients. Br J Haematol. 1994;87(4):849–52.
Green DA, Antholine WE, Wong SJ, Richardson DR, Chitambar CR. Inhibition of malignant cell growth by 311, a novel iron chelator of the pyridoxal isonicotinoyl hydrazone class: effect on the R2 subunit of ribonucleotide reductase. Clin Cancer Res. 2001;7(11):3574–9.
Saxena D, Spino M, Tricta F, Connelly J, Cracchiolo BM, Hanauske AR, et al. Drug-based Lead discovery: the novel ablative antiretroviral profile of deferiprone in HIV-1-infected cells and in HIV-infected treatment-naive subjects of a double-blind, placebo-controlled, Randomized Exploratory Trial. PLoS One. 2016;11(5):e0154842.
Georgiou NA, van der Bruggen T, Oudshoorn M, Nottet HS, Marx JJ, van Asbeck BS. Inhibition of human immunodeficiency virus type 1 replication in human mononuclear blood cells by the iron chelators deferoxamine, deferiprone, and bleomycin. J Infect Dis. 2000;181(2):484–90.
Georgiou NA, van der Bruggen T, Oudshoorn M, Hider RC, Marx JJ, van Asbeck BS. Human immunodeficiency virus type 1 replication inhibition by the bidentate iron chelators CP502 and CP511 is caused by proliferation inhibition and the onset of apoptosis. Eur J Clin Investig. 2002;32(Suppl 1):91–6.
Crowe W, Maglova L, Ponka P, Russell J. Human cytomegalovirus-induced host cell enlargement is iron dependent. Am J Physiol Cell Physiol. 2004;287:C1023–30.
Liu J, Liu W, Liu Y, Miao Y, Guo Y, Song H, et al. New thiazolidinones reduce iron overload in mouse models of hereditary hemochromatosis and beta-thalassemia. Haematologica. 2019;104(9):1768–81.
Lai AL, Millet JK, Daniel S, Freed JH, Whittaker GR. The SARS-CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J Mol Biol. 2017;429(24):3875–92.
Ahn DG, Choi JK, Taylor DR, Oh JW. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch Virol. 2012;157(11):2095–104.
Ishimaru D, Plant EP, Sims AC, Yount BL Jr, Roth BM, Eldho NV, et al. RNA dimerization plays a role in ribosomal frameshifting of the SARS coronavirus. Nucleic Acids Res. 2013;41(4):2594–608.
Seybert A, Posthuma CC, van Dinten LC, Snijder EJ, Gorbalenya AE, Ziebuhr J. A complex zinc finger controls the enzymatic activities of nidovirus helicases. J Virol. 2005;79(2):696–704.
Chou CY, Chien CH, Han YS, Prebanda MT, Hsieh HP, Turk B, et al. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem Pharmacol. 2008;75(8):1601–9.
Lee CC, Kuo CJ, Hsu MF, Liang PH, Fang JM, Shie JJ, et al. Structural basis of mercury- and zinc-conjugated complexes as SARS-CoV 3C-like protease inhibitors. FEBS Lett. 2007;581(28):5454–8.
Yang N, Tanner JA, Zheng BJ, Watt RM, He ML, Lu LY, et al. Bismuth complexes inhibit the SARS coronavirus. Angew Chem Int Ed Eng. 2007;46(34):6464–8.
Lee C, Lee JM, Lee NR, Jin BS, Jang KJ, Kim DE, et al. Aryl diketoacids (ADK) selectively inhibit duplex DNA-unwinding activity of SARS coronavirus NTPase/helicase. Bioorg Med Chem Lett. 2009;19(6):1636–8.
Hsu JT, Kuo CJ, Hsieh HP, Wang YC, Huang KK, Lin CP, et al. Evaluation of metal-conjugated compounds as inhibitors of 3CL protease of SARS-CoV. FEBS Lett. 2004;574(1–3):116–20.
te Velthuis AJ, van den Worm SH, Sims AC, Baric RS, Snijder EJ, van Hemert MJ. Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog. 2010;6(11):e1001176.
Funding
This work was supported by the international collaboration key grant from the Chinese Academy of Sciences (grant number: 121311KYSB20190010). Dr. Nekhai’s research was funded by NIH Research Grants (1P50HL118006, 1R01HL125005, 1UM1AI26617 and U54MD007597), and the District of Columbia Center for AIDS Research grant (P30AI087714). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Wei Liu: participation in drafting and figure preparation.
Shuping Zhang: original draft preparation.
Sergei Nekhai: participation in revision of the submitted article, funding acquisition.
Sijin Liu: conception and design of the study, review and editing, funding acquisition.
Corresponding authors
Ethics declarations
Conflict of Interests
The authors declare that they have no competing interests.
Ethics Approval
Not applicable.
Human and Animal Rights and Informed Consent
Not applicable.
Code Availability
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Virology
Rights and permissions
About this article

Cite this article
Liu, W., Zhang, S., Nekhai, S. et al. Depriving Iron Supply to the Virus Represents a Promising Adjuvant Therapeutic Against Viral Survival. Curr Clin Micro Rpt 7, 13–19 (2020). https://doi.org/10.1007/s40588-020-00140-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40588-020-00140-w