
Abstract
Rothmund-Thomson syndrome (RTS; also known as poikiloderma congenitale) is a rare, autosomal recessive genetic disorder characterized by abnormalities in skin and skeleton, juvenile cataracts, premature ageing and a predisposition to neoplasia1,2,3,4. Cytogenetic studies indicate that cells from affected patients show genomic instability often associated with chromosomal rearrangements causing an acquired somatic mosaicism5,6,7,8,9. The gene(s) responsible for RTS remains unknown. The genes responsible for Werner10 and Bloom11 syndromes (WRN and BLM, respectively) have been identified as homologues of Escherichia coli RecQ, which encodes a DNA helicase12 that unwinds double-stranded DNA into single-stranded DNAs. Other eukaryotic homologues thus far identified are human RECQL (Refs 13, 14), Saccharomyces cerevisiae SGS1 (Refs 15,16) and Schizosaccharomyces pombe rqh1+ (ref. 17). We recently cloned two new human helicase genes, RECQL4 at 8q24.3 and RECQL5 at 17q25, which encode members of the RecQ helicase family18. Here, we report that three RTS patients carried two types of compound heterozygous mutations in RECQL4. The fact that the mutated alleles were inherited from the parents in one affected family and were not found in ethnically matched controls suggests that mutation of RECQL4 at human chromosome 8q24.3 is responsible for at least some cases of RTS.
Similar content being viewed by others


Main
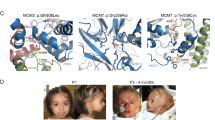
The coding sequence of RECQL4, consisting of 3,627 bases and encoding a protein with 1,208 amino acids, has been published18; exon and intron junctions have also recently been identified (unpublished data). We amplified all exon regions of RECQL4 from patients by PCR and compared their sequences with those of normal individuals (Fig. 1). These studies revealed that both RTS patients (with apparently healthy parents) in a pedigree of Mexican ancestry had the same compound heterozygous mutations. One mutation, referred to as mut-1, is a deletion of 7 bases (GGCCTGC; nt 1,650-1,656; Fig. 1c, left) resulting in an early termination codon (TGA) 14 bases downstream. Mut-1 was transmitted from the mother of the patients, as detected by amplifying the corresponding DNA sequence (52 bp, residues 1,624-1,675) by PCR and subsequent analysis by gel electrophoresis (Fig. 1b). Another mutation, referred to as mut-2, was inherited from the father and was found by sequencing both RECQL4 cDNA and genomic DNA (Fig. 1c, right). Mut-2 is a C→T substitution at residue 2,269 that altered the original CAG (Gln) to a TAG terminator. Both mut-1 and mut-2 occurred in the helicase domain of RECQ4 and predicted truncated proteins of 60 kD and 82 kD, compared with the intact 133-kD RECQ4 helicase. A summary of the mutations ( Table 1) and the putative truncated protein products ( Fig. 2) are shown.

a, Pedigree of RTS patients and family members. The half-filled square and circle represent the carriers of mutated RECQL4 alleles. The filled square (sibling II.3, male) and circle (sibling II.6, female) represent RTS patients carrying mutations in both alleles of RECQL4. Siblings II.2, II.4 and II.5 are not affected with RTS and were not studied. Sibling II.1 (grey square) is also an RTS patient, but was diagnosed at another institution. b, Mutation analysis of RECQL4 in RTS patients and their parents. DNAs from parents (I.1 and I.2) and RTS patients (II.3 and II.6) were analysed for mut-1 in RECQL4. Lane I.1, father; lane I.2, mother; lane II.3, patient II.3; lane II.6, patient II.6. Our results indicate that the mother has a 7-base deletion (mut-1) in one allele, which was transmitted to the patients. c, Direct nucleotide sequence analysis of mutated regions of RECQL4. The nucleotide sequences of the regions containing mut-1 (residues 1,641-1,672; left) and -2 (residues 2,257-2,280; right) are shown for normal and mutated RECQL4.

'Normal' indicates a complete RECQ4 helicase with 1,208 aa, as predicted from the coding region of cloned RECQL4. The darkened area in the molecule represents a consensus helicase domain present in all RecQ helicases. The lines extending from the carboxy-terminal ends of the truncated molecules indicate the altered translational frame.
In addition to these mutations, we found another set of compound heterozygous mutations in the cells of an RTS patient of European descent deposited in the cell bank of the National Institute of Aging (AG05013). These mutations were a 2-base deletion (mut-3) and a G→T substitution at the junction of intron 12 and exon 13 that destroys the splicing acceptor sequence (mut-4). Both mutations were associated with a translational frameshift ( Table 1 and Fig. 2). The sequence analysis of mRNA produced from the mut-4 allele revealed that the region corresponding to exon 13 was absent in the shortened transcripts (data not shown). To investigate if these mutations occur in normal individuals, we extended our DNA analysis to ethnically matched controls (132 chromosomes from individuals of Mexican descent for mut-1 and -2; and 114 chromosomes from individuals of European descent for mut-3 and -4). We carried out direct sequencing of the corresponding regions amplified from the chromosomal DNAs using PCR. No mutations were found in the chromosomes examined, consistent with our conclusion that these mutations are rare among normal individuals.
To determine if these RTS patients carried additional mutations in WRN and BLM, we analysed the entire sequence of the cDNA from poly(A)+ RNA of sibling II.3 and AG05013 cells. No mutations were found in WRN and BLM (data not shown), suggesting thatthese two genes are not involved in RTS and implying that intact WRN or BLM in RTS patients do not complement the defect(s) caused by the RECQL4 mutation.
Mutational analysis of DNAs from 7 clinically diagnosed RTS patients revealed mutations in RECQL4 in 3 patients, including siblings II.3 and II.6. We are currently observing the broad clinical phenotypes of the four remaining patients to determine their diagnoses. The fact that we detected mutations in RECQL4 in 3 of 7 RTS patients indicates that RECQL4 is involved in the pathogenesis of some RTS cases; however, this also implies that the remaining patients may have problems in other unknown gene(s) that cause similar clinical phenotypes. Future studies with genetic diagnosis using RECQL4 should define a more homogeneous subgroup of RTS. Although the biological functions of the RecQ helicase family have not been elucidated, these multiple helicases, as 'guardian angels' of the genomes of higher eukaryotes, are certain to be involved in maintaining the integrity of chromosomal DNA. Further studies are also needed to clarify if RECQ4 helicase is involved in suppressing hyper-recombination of the genome, as was shown for WRN and BLM helicases19.
Methods
Patient cell lines.
Cell lines were established by skin biopsy from patients with RTS diagnosed from clinical symptoms. The following patients were investigated: L9552914-J, L6474163-L (II.3 and II.6 on the pedigree, respectively) and D8903644-K were provided by N.M.L.; TC4398 by R.W.M.; and AG05013, AG05139 and AG03587A by the Aging Cell Repository of the National Institute of Aging.
RNA preparation, sequencing and mutation analysis.
Total RNA was extracted from cultured cells by the acid guanidinium-phenol-chloroform method, and poly(A)+ RNAs purified using Oligotex-dT30 beads (Takara). Poly(A)+ RNAs from fibroblasts of RTS patients were converted to cDNAs by reverse transcriptase. We amplified the entire coding region of RECQL4 from each subject from the cDNAs by PCR using primers Q4A6 (5´-AGATTCGCTGGACGATCGCAAGCG-3´) and Q4A8 (5´-GTCACTGCCCTAGCCTCTGACAAC-3´). They were cloned in pCR2.1 vector plasmid (Invitrogen) and sequenced by the dideoxy-chain termination method using an ABI 373A sequencer (Applied Biosystems). Genomic sequences for each mutation were analysed by direct sequencing of each PCR fragment encompassing the mutation. We prepared the primers used for identification of mutations 1-4 on the basis of the sequence of RECQL4 genomic structure (S.K., unpublished data): mut-1 (in exon 10), forward primer 5´-GGAAATGTGCTGGGAAAGGAG-3´, reverse primer 5´-ACCAGTGCCTCAGGTGTCAGC-3´; mut-2 (in exon 14), forward primer 5´-CTCGATTCCATTATCATTTACTGC-3´, reverse primer 5´-CTCTTCACAGCCAGGAAGTCC-3´; mut-3 (in exon 15), forward primer 5´-AGAGCTGGTGTCCCCGTGGAC-3´, reverse primer 5´-TGGGAACACGCGCTGTACCAG-3´; mut-4 (in the splicing-junction of exon 13), forward primer 5´-GCCTCACACCAC-TGCCGCCTCTGG-3´, reverse primer 5´-GACAGGCAGATGGTCAGTGGGATG-3´. To detect the mut-1 7-bp deletion, DNA fragments containing a 52-bp region (residues 1,624-1,675) were amplified from genomic DNAs from RTS patients by PCR using primers Q4C1 (5´-TCTGGCCTGCCACCGTGTCTC-3´) and Q4C3 (5´-TGGTCATGCCCGAGTGTATGC-3´). Amplified DNAs were separated electrophoretically on 15% polyacrylamide gels.
GenBank accession number.
RECQL4 cDNA, AB006532.
Accession codes
References
Rothmund, A. Uber cataracten in verbindung mit einer eigenthumlichen hautdegeneration. Arch. Klin. Exp. Ophthal. 4, 159– 182 (1868).
Thomson, M.S. Poikiloderma congenitale. Br. J. Dermatol. 48, 221–234 (1936).
Vennos, E.M., Collins, M. & James, W.D. Rothmund-Thomson syndrome: review of the world literature. J. Am. Acad. Dermatol. 27, 750– 762 (1992).
Vennos, E.M. & James, W.D. Rothmund-Thomson syndrome. Dermatol. Clin. 13, 143–150 (1995).
Ying, K.L., Oizumi, J. & Curry, C.J.R. Rothmund-Thomson syndrome associated with trisomy-8 mosaicism. J. Med. Genet. 27, 258– 260 (1990).
Der Kaloustian, V.M., McGill, J.J., Vekemans, M. & Kopelman, H.R. Clonal lines of aneuploid cells in Rothmund-Thomson syndrome. Am. J. Med. Genet. 37, 336–339 (1990).
Orstavik, K.H., McFadden, N., Hagelsteen, J., Ormerod, E. & Van der Hagen, C.B. Instability of lymphocyte chromosomes in a girl with Rothmund-Thomson syndrome. J. Med. Genet. 31, 570–572 ( 1994).
Miozzo, M. et al. Chromosomal instability in fibroblasts and mesenchymal tumors from 2 sibs with Rothmund-Thomson syndrome. Int. J. Cancer 77, 504–510 (1998).
Lindor, N.M. et al. Rothmund-Thomson syndrome in siblings: evidence for acquired in vivo mosaicism. Clin. Genet. 49, 124–129 (1996).
Yu, C.-E. et al. Positional cloning of the Werner's syndrome gene. Science 272, 258–262 ( 1996).
Ellis, N.A. et al. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83, 655–666 (1995).
Nakayama, K., Irino, N. & Nakayama, H. The recQ gene of Escherichia coli K12: molecular cloning and isolation of insertion mutants. Mol. Gen. Genet. 200, 266–271 ( 1985).
Seki, M. et al. Molecular cloning of cDNA encoding human DNA helicase Q1 which has homology to Escherichia coli RecQ helicase and localization of the gene at chromosome 12p12. Nucleic Acids Res. 22 , 4566–4573 (1994).
Puranam, K.L. & Blackshear, P.J. Cloning and characterization of RECQL, a potential human homologue of the Escherichia coli DNA helicase RecQ. J. Biol. Chem. 269, 29838– 29845 (1994).
Gangloff, S., McDonald, J.P., Bendixen, C., Arthur, L. & Rothstein, R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol. 14, 8391– 8398 (1994).
Watt, P.M., Louis, E.J., Borts, R.H. & Hickson, I.D. Sgs1: a eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and required for faithful chromosome segregation. Cell 81, 253–260 ( 1995).
Stewart, E., Chapman, C.R., Al-Khodairy, F., Carr, A.M. & Enoch, T. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 16, 2682–2692 (1997).
Kitao, S., Ohsugi, I., Goto, M., Furuichi, Y. & Shimamoto, A. Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics 54, 443–452 (1998).
Yamagata, K. et al. Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc. Natl Acad. Sci. USA 95, 8733–8738 (1998).
Acknowledgements
We thank S. Tahara, G.A. Coetzee and B.E. Henderson for advice and for providing us with the chromosomal DNAs from individuals of Mexican and European ancestries. This work was supported by the Drug Organization (The Organization for Drug ADR Relief, R and D Promotion and Product Review) of the Japanese Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kitao, S., Shimamoto, A., Goto, M. et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet 22, 82–84 (1999). https://doi.org/10.1038/8788
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/8788
This article is cited by
-
Continuous millisecond conformational cycle of a DEAH box helicase reveals control of domain motions by atomic-scale transitions
Communications Biology (2023)
-
Clinical challenges in interpreting multiple pathogenic mutations in single patients
Hereditary Cancer in Clinical Practice (2021)
-
Comparison of the fertility of tumor suppressor gene-deficient C57BL/6 mouse strains reveals stable reproductive aging and novel pleiotropic gene
Scientific Reports (2021)
-
RNF8 ubiquitinates RecQL4 and promotes its dissociation from DNA double strand breaks
Oncogenesis (2021)
-
Somatic and germline analysis of a familial Rothmund–Thomson syndrome in two siblings with osteosarcoma
npj Genomic Medicine (2020)
