
Abstract
Eicosapentaenoic acid:docosahexaenoic acid (EPA:DHA) 6:1, an omega-3 polyunsaturated fatty acid formulation, has been shown to induce a sustained formation of endothelial nitric oxide (NO) synthase-derived NO, a major vasoprotective factor. This study examined whether chronic intake of EPA:DHA 6:1 prevents hypertension and endothelial dysfunction induced by angiotensin II (Ang II) in rats. Male Wister rats received orally corn oil or EPA:DHA 6:1 (500 mg kg−1 per day) before chronic infusion of Ang II (0.4 mg kg−1 per day). Systolic blood pressure was determined by tail cuff sphingomanometry, vascular reactivity using a myograph, oxidative stress using dihydroethidium and protein expression by immunofluorescence and western blot analysis. Ang II-induced hypertension was associated with reduced acetylcholine-induced relaxations of secondary branch mesenteric artery rings affecting the endothelium-dependent hyperpolarization (EDH)- and the NO-mediated relaxations, both of which were improved by the NADPH oxidase inhibitor VAS-2870. The Ang II treatment induced also endothelium-dependent contractile responses (EDCFs), which were abolished by the cyclooxygenase (COX) inhibitor indomethacin. An increased level of vascular oxidative stress and expression of NADPH oxidase subunits (p47phox and p22phox), COX-1 and COX-2, endothelial NO synthase and Ang II type 1 receptors were observed in the Ang II group, whereas SKCa and connexin 37 were downregulated. Intake of EPA:DHA 6:1 prevented the Ang II-induced hypertension and endothelial dysfunction by improving both the NO- and EDH-mediated relaxations, and by reducing EDCFs and the expression of target proteins. The present findings indicate that chronic intake of EPA:DHA 6:1 prevented the Ang II-induced hypertension and endothelial dysfunction in rats, most likely by preventing NADPH oxidase- and COX-derived oxidative stress.
Similar content being viewed by others
Introduction
Cardiovascular diseases including coronary heart diseases and stroke remain the leading cause of death worldwide both in developed and developing countries, with hypertension being a major risk factor.1 The development of major cardiovascular diseases is associated early in the process with the induction of an endothelial dysfunction characterized by a reduced formation of vasoprotective factors including nitric oxide (NO) and endothelium-dependent hyperpolarization (EDH), and often also by the development of endothelium-dependent contracting responses.2, 3 Despite the fact that the nature of the factors involved in endothelium-dependent contractile responses (EDCFs) depends on the type of pathology, and the vascular bed and species studied, a major role has been attributable to vasoconstrictor prostanoids derived from the arachidonic acid cascade via the cyclooxygenase (COX) pathway.3 Moreover, previous studies have indicated a key role of both the circulating and the local angiotensin systems in the induction of endothelial dysfunction in experimental models of hypertension, atherosclerosis and diabetes, and in aging-related endothelial dysfunction, and also in patients with cardiovascular risk factors.4, 5, 6, 7, 8, 9, 10 Angiotensin II (Ang II) is thought to contribute to endothelial dysfunction by inducing vascular oxidative stress subsequent to the upregulation of the expression of NADPH oxidase,11, 12 which, in turn, promotes uncoupling of endothelial NO synthase (eNOS),13 alteration of calcium-dependent K channels involved in EDH14 and an increased expression of COXs involved in EDCFs.15, 16
Although current first line antihypertensive treatments effectively reduce systolic blood pressure in hypertensive patients, they seem to restore the protective endothelial function only to some extent.17 Indeed, a partially improved endothelial function has been observed in hypertensive patients in response to calcium channel antagonists, and also in conduit arteries in response to angiotensin-converting enzyme inhibitors and angiotensin type 1 receptor antagonists.17, 18 Several epidemiological and primary and secondary prevention studies have indicated that dietary intake of omega-3 polyunsaturated fatty acids (PUFAs), including the two major compounds eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), is able to reduce the risk of cardiovascular diseases.19, 20, 21 Moreover, dietary supplementation with omega-3 PUFA-rich fish or fish oil significantly reduced blood pressure in hypertensive patients22, 23, 24 and improved endothelium-dependent vasodilatation in patients with coronary artery diseases,25 in heart transplanted patients26 and in hypertensive type 2 diabetic patients.27 We as well as several other groups have shown that formulations of purified EPA and DHA induce potent and sustained endothelium-dependent relaxations of arterial rings by stimulating the endothelial formation of NO and, often also, EDH.28, 29, 30 Moreover, the ability of omega-3 PUFAs to induce endothelium-dependent relaxations has been shown to depend on both the purity and ratio of EPA:DHA, and that the most active formulations include EPA:DHA 6:1 and 9:1.28 The characterization of the signal transduction pathway leading to sustained eNOS activation has indicated the involvement of the redox-sensitive Src/PI3-kinase/Akt pathway and the subsequent phosphorylation of the eNOS activator site Ser 1177.28 Moreover, as omega-3 PUFAs have been shown to redirect the COX-dependent formation of prostanoids towards three-series rather than two-series, and three-series prostanoids such as TxA3 are less potent vasoconstrictors than two-series prostanoids such as TxA2, they may be of interest to prevent the induction of EDCFs.31, 32
Therefore, we have tested the hypothesis that chronic intake of the EPA:DHA 6:1 formulation prevents the Ang II-induced hypertension and endothelial dysfunction in rats.
Methods
Ethics statement
This study conforms to the Guide of care and the use of laboratory animals published by the US National Institutes of Health (Bethesda, MD, USA; NIH publication number 85–23, revised 1996). The present protocol was approved by the local Ethics Committee (Comité Régional d’Ethique en Matière d’Expérimentation Animale de Strasbourg, approval AL/01/09/09/05).
Preparation of omega-3 PUFAs products
Highly purified EPA and DHA were provided by Pivotal Therapeutics, Inc (Woodbridge, ON, Canada). The EPA:DHA 6:1 (w/w) ratio was prepared and adjusted to the relative purity of each compound under nitrogen flux to avoid oxidation of the omega-3 PUFAs. Thereafter, the formulation is aliquoted in amber glass vials under nitrogen, and stored at 4 °C until use.
In vivo treatment of rats
Thirty-two male Wistar rats (10 weeks old) were randomly divided into four groups of eight rats each. Rats received daily by gavage 500 mg kg−1 per day of either EPA:DHA 6:1 or corn oil (control) for 5 weeks. After 1 week, rats underwent sham surgery (sham rats) or surgery with implantation of an osmotic mini-pump infusing Ang II (0.4 mg kg−1 per day) for 4 weeks as described previously.5 Briefly, rats were anesthetized with sodium pentobarbital (50 mg kg−1, i.p.; Centravet, Velaine-en-Haye, France). A 1 cm incision was made in the midscapular region, and an osmotic mini-pump (Alzet 2004, Charles River Laboratories, Saint-Germain-sur-l’Arbresle, France) was implanted. Pumps contained Ang II (Enzo Life Sciences, Villeurbanne, France), which was dissolved in saline solution to obtain an infusion rate of 0.4 mg kg−1 per day. Sham-operated rats underwent an identical surgical procedure without pump implantation. After surgery, rats were housed in a thermo-neutral environment, on a 12:12 h photoperiod and were provided food and drinking water ad libitum.
Determination of the plasma level of lipids
Analyses of total blood fatty acid levels, and calculation of the Omega Score (EPA+DHA+Docosapentaenoic acid), arachidonic acid/EPA ratio and n-6/n-3 ratio were performed at the central laboratory of the University Health Network (Specialty Laboratory, Toronto, ON, Canada), accredited by the College of American Pathologists’ Laboratory Accreditation Program. The fatty acid composition of whole blood was determined on 200 μl of sample after lipid extraction by a modification of the method of Bligh and Dyer.33 The total lipid fraction was then methylated with 12% (w/w) boron trifluoride in methanol by incubation at 90 °C for 25 min to produce fatty acid methyl-esters. After cooling, the fatty acid methyl-esters were extracted with hexane, washed with water, dried under nitrogen and dissolved in hexane. The fatty acid composition was then determined by gas-liquid chromatography performed on a 100 m Varian Select FAME CP7420 capillary column (0.25 mm i.d.), using an Agilent Technologies 6890N series gas chromatograph equipped with a split/splitless mode injector, and a flame ionization detector (Agilent Technologies Canada Inc., Mississauga, ON, Canada). The injector and detector were maintained at 280 °C and 300 °C, respectively, and samples were analyzed by multilevel temperature programing in the range of 90–265 °C with ultra-high purity grade helium as the carrier gas. The percent composition of fatty acids was calculated from the individual peak areas using appropriate standards. The procedure was routinely validated by proficiency testing using gas chromatography–mass spectrometry.
Blood pressure measurements
Systolic blood pressure was measured in conscious rats twice a week for a total of 4 weeks, by using a tail-cuff sphygmomanometer connected to a computerized system (BP-2000 Blood pressure analysis system, Visitech Systems, Inc., Apex, NC, USA).
Vascular reactivity studies
To determine the effect of the EPA:DHA 6:1 treatment on the endothelial function, vascular reactivity studies were performed on secondary branch mesenteric artery rings using a DMT wire myograph (Danish Myo Technology A/S, Aarhus, Danemark). Briefly, secondary branch mesenteric arteries of rats were cleaned of connective tissue, cut into rings (2–3 mm in length), mounted onto two stainless steel wires and suspended in organ baths containing oxygenated Krebs bicarbonate solution (composition in mM: NaCl 119, KCl 4.7, KH2PO4 1.18, MgSO4 1.18, CaCl2 1.25, NaHCO3 25 and D-glucose 11, pH 7.4, 37 °C) for the determination of changes in isometric tension. After the equilibration period, rings were exposure to Krebs bicarbonate solution containing a high concentration of potassium (80 mM) until reproducible contractile responses were obtained. Thereafter, rings were contracted with phenylephrine (PE, 1 μM) to ~80% of the maximal contraction induced by the high potassium solution before addition of acetylcholine (ACh, 1 μM) to test the endothelial function. After washout and a 30 min equilibration period, rings were again contracted with PE before the construction of a concentration-relaxation curve to either ACh, levcromakalim (Lev, an ATP-sensitive K+ channel opener) or sodium nitroprusside (a NO donor). In some experiments, rings were exposed to an inhibitor for 30 min before being contracted with PE. To study NO-mediated relaxation, rings were incubated in the presence of indomethacin (an inhibitor of COXs) and charybdotoxin plus apamin (inhibitors of IKCa and SKCa, respectively) to prevent the formation of vasoactive prostanoids and EDH-mediated relaxation, respectively. The EDH-mediated relaxation was studied in rings incubated with indomethacin and Nω-nitro-L-arginine (L-NA, an eNOS inhibitor) to prevent the formation of vasoactive prostanoids and NO, respectively. Relaxations were expressed as the percentage of the reversal of the contraction to PE. To study EDCFs, rings were exposed to L-NA and apamin plus charybdotoxin for 30 min to prevent the formation of NO and EDH. This was done prior to determination of a concentration–contraction curve to ACh.
Immunofluorescence studies
To characterize the mechanism underlying the Ang II-induced endothelial dysfunction, the expression level of several proteins involved in NO, EDH and EDCFs responses were determined by immunofluorescence in sections of the secondary branch mesenteric artery. For this purpose, frozen arteries embedded in Tissue-Tek OCT (Sakura 4583, Leiden, The Netherlands) were cryosectioned at 14 μm. Sections were air dried for 15 min and stored at −80 °C until use. Sections were first fixed with paraformaldehyde at 4% (Electron Microscopy Sciences, Hatfield, PA, USA), washed and treated with either 10% milk or 5% goat serum in phosphate-buffered saline containing 0.1% Triton X-100 for 1 h at room temperature to block nonspecific binding. Mesenteric artery sections were then incubated overnight at 4 °C with an antibody directed against either eNOS (1/50, cat: 610297, BD Transduction Laboratories, Le Pont de Claix, France), arginase-1 (1/100, cat: 610708, BD Transduction Laboratories), small and intermediate conductance calcium-activated potassium channels (SKCa, 1/200, cat: APC-025; IKCa, 1/200, cat: APC064, Alomone Labs, Jerusalem, Israel), connexin 37 (1/100 to 1/200, cat: CX37B12-A, Alpha Diagnostic International, San Antonio, TX, USA), Ang II type 1 receptor (AT1R, 1/ 400, sc-1177, Santa Cruz Biotechnology, Clinisciences, Nanterre, France), AT2R (1/50, sc-9040, Santa Cruz Biotechnology), NADPH oxidase sub-units p47phox (1/200, sc-14015, Santa Cruz Biotechnology) and p22phox (1/200, sc-20781, Santa Cruz Biotechnology), COX-1 (1/250, ab109025, Abcam, Paris, France) and COX-2 (1/200, sc-1745, Santa Cruz Biotechnology). For negative controls, the primary antibody was omitted. Sections were then washed with phosphate-buffered saline, incubated with the fluorescent secondary antibody (1/400, Alexa 633-conjugated goat anti-rabbit or anti-mouse IgG, A-21070 and A-21050, Thermo Fisher, Illkirch, France) for 2 h at room temperature in the dark before being washed with phosphate-buffered saline and mounted in Dako fluorescence mounting medium (Dako S3023, Les Ulis, France) and cover-slipped before being evaluated by confocal microscopy using a confocal laser-scanning microscope (Leica TSC SPE, Mannheim, Germany). Quantification of fluorescence levels was performed using Image J software (version 1.49p for Windows, US National Institutes of Health).
Determination of the in situ vascular level of oxidative stress
The redox-sensitive fluorescent dye dihydroethidium (2.5 μM) was applied to 25 μm unfixed cryosections of secondary branch mesenteric artery for 30 min at 37 °C in a light-protected humidified chamber. The sections were then mounted in Dako fluorescence mounting medium and cover-slipped before being evaluated by confocal microscopy using a confocal laser-scanning microscope (Leica TSC SPE). Quantification of fluorescence levels was performed using Image J software (version 1.49p for Windows, US National Institutes of Health).
Western blot analysis
To characterize the mechanism underlying the Ang II-induced endothelial dysfunction, the expression level of several proteins involved in NO, EDH and EDCFs responses were determined by Western blot analysis in the secondary branch of the mesenteric artery. Proteins were extracted from mesenteric artery segments with radioimmunoprecipitation assay lysis and extraction buffer containing phosphatase and protease inhibitors. Proteins (20 μg) were separated on 10% SDS-polyacrylamide gels, using prestained markers for molecular mass determinations, before being transferred onto nitrocellulose membranes. Membranes were blocked with 5% bovine serum albumin on Tris-buffered saline solution and 0.1% Tween 20 for 1 h. Membranes were then incubated with the primary antibody (COX-2, NADPH oxidase sub-unit p22phox and eNOS; dilution of 1:1000) overnight at 4 °C. After washing, membranes were incubated with their corresponding peroxidase-labeled secondary antibody at room temperature for 60 min. Immunoreactive bands were detected by enhanced chemiluminescence substrate solution. Chemiluminescence signal was recorded using an ImageQuant LAS4000 system (GE Healthcare Europe GmbH, Velizy-Villacoublay, France) and analyzed using ImageQuant TL software (version 8.1, GE Healthcare). Membranes were stripped subsequently and reprobed with a mouse polyclonal anti-glyceraldehyde-3-phosphate deshydrogenase antibody for normalization purposes.
Statistical analysis
Values are expressed as means±s.e.m. Statistical analysis was performed using an analysis of variance followed by the Bonferroni post-hoc test as appropriate using GraphPad Prism (version 5 for Microsoft windows, GraphPad Software, Inc., San Diego, CA, USA). Values of P<0.05 were considered to be statistically significant.
Results
Effects of chronic oral intake of EPA:DHA 6:1 treatment on plasma levels of omega-3 PUFAs
After 5 weeks of chronic feeding of rats with EPA:DHA 6:1, the plasma contained significantly higher levels of PUFAs including EPA, DHA and docosapentaenoic acid, resulting in an increased total omega-3 PUFAs content and omega score (Table 1). Moreover, the total omega-6 PUFAs proportion was significantly reduced in groups receiving EPA:DHA 6:1 (Table 1). Thus, the omega-6/omega-3 ratio was significantly reduced in rats receiving the EPA:DHA 6:1 formulation compared with those receiving corn oil (Table 1).
Effects of EPA:DHA 6:1 treatment on Ang II-induced hypertension
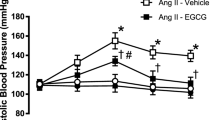
Chronic administration of 0.4 mg kg−1 per day of Ang II to rats caused an increase in systolic blood pressure, which was significant after 3 days and remained elevated throughout the experiment (190.6±8.5 and 120.7±4.2 mmHg for the Ang II and control groups at week 4, respectively; Figure 1). Intake of EPA:DHA 6:1 (500 mg kg−1 per day) partially but significantly prevented the Ang II-induced hypertension, whereas it was without effect in normotensive rats (149.0±7.8 and 115.6±2.8 mmHg at week 4 for the Ang II+EPA:DHA 6:1 group and the EPA:DHA 6:1 group, respectively; Figure 1).

Chronic intake of EPA:DHA 6:1 prevents the Ang II-induced hypertension in rats. Rats received daily by gavage 500 mg kg−1 per day of either EPA:DHA 6:1 or corn oil (control) for 1 week before administration of Ang II (0.4 mg kg−1 per day) using mini osmotic pumps for 3 weeks. Blood pressure was monitored by tail-cuff sphygmomanometry. Results are expressed as means±s.e.m. of 8 rats per group. *P<0.05 vs. Control, #P<0.05 vs. Ang II.
Effects of EPA:DHA 6:1 treatment on Ang II-induced endothelial dysfunction
To determine the effect of the antihypertensive EPA:DHA 6:1 treatment on the endothelial function, vascular reactivity studies were performed on secondary branch mesenteric artery rings. In rings precontracted by PE (1 μM), ACh induced concentration-dependent relaxations, which were significantly reduced at 0.1 to 1 μM in the Ang II-treated group (Figure 2a). To study NO-mediated relaxations, rings were incubated in the presence of indomethacin (an inhibitor of COXs) and charybdotoxin plus apamin (inhibitors of IKCa and SKCa, respectively) to prevent the formation of vasoactive prostanoids and EDH-mediated relaxation, respectively, whereas the EDH-mediated relaxation was studied in the presence of indomethacin and L-NA (an eNOS inhibitor) to prevent the formation of vasoactive prostanoids and NO, respectively. Compared with the control group, the NO-mediated relaxation was slightly but significantly reduced in the Ang II group (Figure 2b), whereas the EDH-mediated relaxation was markedly reduced (Figure 2c). The EPA:DHA 6:1 treatment prevented the Ang II-induced endothelial dysfunction as indicated by similar NO- and EDH-mediated relaxations to those of the control group (Figure 2b and c). Moreover, the incubation of rings with VAS-2870, a NADPH oxidase inhibitor, prevented the Ang II-induced impairment of both the NO- and EDH-mediated relaxations, indicating a key determinant role of NADPH oxidase-derived oxidative stress (Figure 2d and e).

EPA:DHA 6:1 prevents the Ang II-induced endothelial dysfunction in the secondary branch of the mesenteric artery. (a–c) Rings with endothelium were contracted with 1 μM PE before the addition of increasing concentrations of ACh. (b) NO-mediated relaxations were studied in the presence of indomethacin (10 μM) and charybdotoxin plus apamin (100 nM each) to prevent the formation of vasoactive prostanoids and EDH-mediated relaxations, respectively. (c) EDH-mediated relaxations were studied in the presence of indomethacin and Nω-nitro-L-arginine (300 μM) to prevent the formation of vasoactive prostanoids and NO, respectively. (d and e) To assess the role of NADPH oxidase-derived oxidative stress in Ang II-induced endothelial dysfunction, NO- (d) and EDH-mediated (e) relaxations were performed in the presence of VAS-2870, a NADPH oxidase inhibitor (1 μM). (f) The function of the vascular smooth muscle was assessed in rings with endothelium contracted with 1 μM PE before the construction of a concentration-relaxation curve to sodium nitroprusside (SNP), a NO donor, in the presence of charybdotoxin plus apamin (100 nM each), Nω-nitro-L-arginine (300 μM) and indomethacin (10 μM) to prevent the contribution of EDH, NO and vasoactive prostanoids, respectively. Results are expressed in % relaxations as means±s.e.m. of eight rats per group. *P<0.05 vs. Control, #P<0.05 vs. Ang II.
To study the potential role of EDCFs in the Ang II-induced endothelial dysfunction, rings were subjected to increasing concentrations of ACh in the presence of L-NA, apamin and charybdotoxin to prevent the formation of NO and EDH, respectively. ACh induced small concentration-dependent contractile responses in rings with endothelium from the control group, which were significantly increased in the Ang II group (Figure 3a). EPA:DHA 6:1 treatment prevented the Ang II-induced increase in EDCFs but was without effect in the control group (Figure 3a). The Ang II-induced contractile response to ACh was abolished in the presence of indomethacin (Figure 3b), indicating the involvement of COXs-derived vasocontractile prostanoids.

EPA:DHA 6:1 prevents the Ang II-induced EDCFs in the secondary branch of the mesenteric artery. EDCFs were studied in the presence of charybdotoxin plus apamin (100 nM each) and Nω-nitro-L-arginine (300 μM) to prevent the formation of EDH and NO, respectively. Rings with endothelium were subjected to increasing concentrations of ACh in the absence (a) or presence (b) of indomethacin (10 μM) to prevent the formation of vasoactive prostanoids. Results are expressed in % relaxations as means±s.e.m. of 8 rats per group. *P<0.05 vs. Control, #P<0.05 vs. Ang II. (c) The function of the vascular smooth muscle was assessed in rings with endothelium subjected to increasing concentrations of PE in the presence of charybdotoxin plus apamin (100 nM each), Nω-nitro-L-arginine (300 μM) and indomethacin (10 μM) to prevent the contribution of EDH, NO and vasoactive prostanoids, respectively. Results are expressed in mN mm−1 as means±s.e.m. of eight rats per group.
Neither the Ang II treatment nor the EPA:DHA 6:1 treatment significantly affected the function of the vascular smooth muscle as indicated by similar concentration-dependent contractile responses to PE (Figure 3c) and concentration-dependent relaxations to sodium nitroprusside, a NO donor, in arterial rings exposed to indomethacin, L-NA, apamin and charybdotoxin to prevent the contribution of vasoactive prostanoids, NO and EDH, respectively (Figure 2f).
Effects of EPA:DHA 6:1 treatment on Ang II-induced vascular oxidative stress and expression of target proteins involved in NO, EDH and EDCFs responses
To characterize the mechanisms underlying the Ang II-induced endothelial dysfunction and the protective effect of the EPA:DHA 6:1 treatment, the expression level of several proteins involved in NO, EDH and EDCFs responses were determined in sections of secondary branch mesenteric arteries by immunofluorescence. The Ang II treatment was associated with an increased expression level of eNOS, indicating most likely a compensatory mechanism, and of arginase 1, an enzyme that can limit the bioavailability of the eNOS substrate L-arginine thereby promoting eNOS uncoupling (Figure 4). The Ang II treatment was also associated with a reduced expression of connexin 37 and SKCa, and an increased expression of both COX-1 and COX-2 (Figure 4). The EPA:DHA 6:1 treatment prevented the Ang II-induced alterations of target proteins involved in the NO, EDH and EDCFs pathways (Figure 4). Since an increased vascular level of oxidative stress induced by the AT1R-mediated activation and expression of NADPH oxidase, contributes to Ang II-induced hypertension and endothelial dysfunction,11 the vascular level of oxidative stress was assessed using the redox-sensitive probe dihydroethidium, and the expression level of NADPH oxidase subunits (p22phox and p47phox) and Ang II receptors by immunofluorescence in secondary branch of mesenteric arteries. The Ang II treatment was associated with an increased vascular level of oxidative stress and expression of NADPH oxidase subunits p22phox and p47phox and also of both AT1R and AT2R; all these effects were prevented by the EPA:DHA 6:1 treatment (Figure 4).

EPA:DHA 6:1 prevents the Ang II-induced vascular oxidative stress and the up-regulation of eNOS, arginase 1, COX-1 and COX-2, NADPH oxidase, Ang II receptors and the downregulation of SKCa and connexin 37 (Cx37) in the secondary branch of the mesenteric artery. Vascular oxidative stress and protein immunoreactive signals were determined in unfixed cryosections of the secondary branch of the mesenteric artery. The determination of oxidative stress was done by fluorescence histochemistry using the redox-sensitive probe dihydroethidium, and the expression level of eNOS, arginase 1, COX-1, COX-2, SKCa, Cx37, AT1R, AT2R and NADPH oxidase subunits p22phox and p47phox (adventitia) by immunofluorescence and analysed by confocal microscope. Results are expressed as means±s.e.m. of four to five rats per group. *P<0.05 vs. Control, #P<0.05 vs. Ang II.
In order to confirm the immunofluorescence signals and due to the limited amount of tissue, western blot analysis was performed to determine the expression level of eNOS, COX-2 and p22phox in the secondary branch of the mesenteric artery. The Ang II treatment increased the expression level of eNOS, COX-2 and p22phox whereas no such effect was observed in the Ang II+EPA:DHA 6:1 group and in the EPA:DHA 6:1 group (Figure 5).

EPA:DHA 6:1 prevents the Ang II-induced overexpression of eNOS, COX-2 and p22phox in the secondary branch of the mesenteric artery. Protein expression levels of eNOS, COX-2 and the NADPH oxidase subunit p22phox were determined in the secondary branch of the mesenteric artery by Western blot analysis. Results are expressed as means±s.e.m. of four to five rats per group. *P<0.05 vs. Control, #P<0.05 vs. Ang II.
Discussion
The major findings of the present study indicate that the chronic oral intake of the omega-3 PUFAs formulation EPA:DHA 6:1 prevents hypertension and endothelial dysfunction induced by chronic administration of Ang II to rats. The characterization of the mechanisms underlying the Ang II-induced endothelial dysfunction in the secondary resistance branch of the mesenteric artery (<100 μm in diameter) has indicated a pronounced impairment of the EDH-mediated relaxation and also, to some extent, of the NO-mediated relaxation, and the induction of EDCFs, which are all prevented by the EPA:DHA 6:1 treatment. The vasoprotective effect of the EPA:DHA 6:1 treatment involves the prevention of the decreased expression level of connexin 37 and SKCa involved in the EDH-mediated relaxation, and of the increased expression level of eNOS and arginase 1 involved in the NO-mediated relaxation, and of COX-1 and COX-2 involved in EDCFs. It is most likely explained by the prevention of the NADPH oxidase-derived oxidative stress in the arterial wall. Altogether, these findings suggest that EPA:DHA 6:1 may help to improve the endothelial function and, hence, to enhance the protection of the vascular system in hypertension.
The chronic intake of EPA:DHA 6:1 to rats significantly increased plasma levels of omega-3 PUFAs including EPA, DHA and docosapentaenoic acid, an elongated metabolite of EPA, and decreased those of omega-6 PUFAs including arachidonic acid. As a consequence, the omega-6/omega-3 ratio was decreased by about 65% by the EPA:DHA 6:1 treatment. Previous experimental and clinical studies have indicated that reducing the omega-6/omega-3 ratio has a beneficial health effect as indicated by decreased oxidative stress and inflammation markers in rats, and a reduced risk of cardiovascular diseases and cancer in humans.34, 35
Consistent with previous studies, administration of 0.4 mg kg−1 per day of Ang II to rats induced within a few days an increase in systolic blood pressure that remained elevated during the next 2 weeks.5, 7 The chronic intake of EPA:DHA 6:1 markedly reduced the Ang II-induced hypertension by about 60%. Previous studies have also shown that intake of 0.75% EPA and DHA in the diet for 2 weeks in combination with an inhibitor of the soluble epoxide hydrolase significantly prevented the Ang II-induced hypertension in mice36 and 1 or 5% of DHA in the diet for 14 weeks reduced the high systolic blood pressure level in spontaneously hypertensive stroke-prone rats.37 In the present study, EPA:DHA 6:1 was administered at the dose of 0.5 g kg−1 per day to rats, which is equivalent to about 5.67 g per day for a 70 kg human.38 Such a dose is in line with clinical studies indicating a beneficial effect of intake of omega-3 PUFAs on the cardiovascular system including in hypertensive patients with doses ranging from 0.18 up to 10 g per day.19, 22, 23, 24, 39 Indeed, intake of more than 2 g per day of EPA+DHA for at least 3 weeks has been shown to reduce systolic blood pressure by about 1.25 and 4.51 mmHg in normotensive and hypertensive subjects, respectively,22 at least 3 g per day of omega-3 PUFAs for 3 weeks by about 1.5 and 5.5 mm Hg in normotensive subjects and untreated moderately hypertensive patients, respectively,23 and 15 g per day of omega-3 PUFAs for 4 weeks by about 6.5 mmHg in mildly hypertensive patients.24 Thus, the present findings in conjunction with previous ones indicate that omega-3 PUFAs can contribute to reduce high levels of blood pressure in both experimental models of hypertension and also in hypertensive humans.
Previous studies have indicated that NADPH oxidase-derived formation of superoxide anions plays a pivotal role in the Ang II-induced hypertension and endothelial dysfunction subsequent to the activation of angiotensin type 1 receptors.4, 11, 40, 41, 42 In the present study, NADPH oxidase-derived oxidative stress appears also to contribute to the Ang II-induced endothelial dysfunction since both the NO- and EDH-mediated relaxations were improved in the presence of VAS-2870, an inhibitor of NADPH oxidase. It is also supported by the fact that an increased level of oxidative stress and expression of NADPH oxidase subunits, p22phox and p47phox, as well as AT1R are observed in the arterial wall in the Ang II group. Previous studies have indicated that superoxide anions can chemically react with NO thereby reducing its bioavailability and leading to the subsequent formation of peroxynitrite, which, in turn, promotes uncoupling of eNOS and an enhanced formation of superoxide anions.43 Such a concept is supported by the fact that inhibitors of eNOS decreased the formation of superoxide anions in the arterial wall of Ang II-treated hypertensive rats.44 The increased expression level of arginase 1 in the arterial wall in the Ang II group suggests that this enzyme might also contribute to reduce NO-mediated relaxation by competing with eNOS for the substrate L-arginine.45, 46, 47 In addition, the increased expression level of eNOS protein in the Ang II group indicates most likely a compensatory mechanism to maintain to some extent the NO-mediated relaxation. The fact that in the presence of VAS-2870, EDH-mediated relaxation in the Ang II group was increased suggests the involvement of NADPH oxidase-derived oxidative stress possibly by inactivating the Ca2+-activated K+ channels SKCa and IKCa mediating the hyperpolarization.14
As the vascular level of oxidative stress in the Ang II plus EPA:DHA 6:1 group was similar to that observed in the control group, the vasoprotective effect of EPA:DHA 6:1 most likely involves its ability to prevent the pro-oxidant effect of Ang II. Indeed, EPA:DHA 6:1 prevented the Ang II-induced up-regulation of the expression of the pro-oxidant enzymes NAPDH oxidase and COXs, and also of AT1R in the arterial wall. In addition, previous studies have shown that omega-3 PUFAs can also have an antioxidant effect by directly inhibiting NADPH oxidase activity48 and by scavenging ROS.49
Besides omega-3 PUFAs, polyphenol-rich sources such as red wine and green tea have been shown to improve the endothelial dysfunction in several experimental models of hypertension and in patients with cardiovascular risk factors by targeting vascular oxidative stress (for review, see50). Similarly, the antioxidant vitamin C improved endothelium-dependent vasodilatation in hypertensive patients, whereas no such effect is observed in normotensive subjects.51 Altogether, these findings indicate that several sources of natural products including fish-derived products and fruit and vegetables may help to protect the cardiovascular system and hence delay the development of cardiovascular diseases.
Ang II-induced endothelial dysfunction in the secondary resistance branch of the mesenteric artery involves, besides impaired NO- and EDH-mediated relaxations, also the induction of EDCFs to ACh. As no such contractile responses are observed in the presence of indomethacin, they appear to involve COX-derived vasoconstrictor prostanoids possibly as a consequence of the upregulation of the expression of both COX-1 and COX-2, two redox-sensitive genes.15, 16, 52 COX-derived vasoconstrictor prostanoids have also been shown to contribute to endothelial dysfunction in hypertensive humans, as indomethacin restored the endothelium-dependent vasodilatation to ACh in the forearm of hypertensive patients but not of normotensive subjects.53 Thus, the present findings indicate that EPA:DHA 6:1 effectively prevented the Ang II-induced hypertension and endothelial dysfunction by preventing an impairment of both EDH- and NO-mediated relaxations, and the development of EDCFs.
The fact that the EPA:DHA 6:1 treatment improved the plasma omega-6/omega-3 ratio might also contribute to protect the vascular system. Indeed, a reduction of the omega-6/omega-3 ratio has been shown to promote the production of omega-3-derived anti-inflammatory metabolites such as resolvins.54, 55, 56 Indeed, resolvin D1 prevented the hyperreactivity induced by endothelin-1 and pro-inflammatory cytokines (TNFα and IL-6) in the human pulmonary artery.57 Omega-3 PUFAs also reduced the expression of the pro-inflammatory COX-2 in vascular smooth muscle cells whereas an increased expression was observed in response to arachidonic acid, an omega-6 PUFA.58 As activation of perivascular T cells and the subsequent vascular inflammatory response have been shown to contribute to Ang II-induced hypertension and endothelial dysfunction, omega-3 PUFAs might possibly also contribute to protect the vascular system by limiting the pro-inflammatory response.59, 60
Nevertheless, a limitation of the present study is the fact that corn oil has been chosen as a control oil for isocaloric intake, which has been shown to induce an acute endothelial dysfunction in humans.61
In conclusion, the present findings indicate that chronic intake of EPA:DHA 6:1 is able to prevent the Ang II-induced hypertension and endothelial dysfunction. The beneficial effect of the EPA:DHA 6:1 treatment involves an improved NO- and EDH-mediated relaxations and the prevention of the induction EDCFs most likely by reducing vascular oxidative stress and the expression level of NADPH oxidase and COXs in the arterial wall.
References
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB American Heart Association Statistics C, Stroke Statistics S. Heart Disease and Stroke Statistics-2016 Update: a report from the American Heart Association. Circulation 2016; 133: e38–e360.
Vanhoutte PM, Shimokawa H, Tang EH, Feletou M . Endothelial dysfunction and vascular disease. Acta Physiol (Oxford, Engl) 2009; 196: 193–222.
Vanhoutte PM, Shimokawa H, Feletou M, Tang EH . Endothelial dysfunction and vascular disease-a 30th anniversary update. Acta Physiol (Oxford, Engl) 2017; 219: 22–96.
Dal-Ros S, Bronner C, Auger C, Schini-Kerth VB . Red wine polyphenols improve an established aging-related endothelial dysfunction in the mesenteric artery of middle-aged rats: role of oxidative stress. Biochem Biophys Res Commun 2012; 419: 381–387.
Dal-Ros S, Bronner C, Schott C, Kane MO, Chataigneau M, Schini-Kerth VB, Chataigneau T . Angiotensin II-induced hypertension is associated with a selective inhibition of endothelium-derived hyperpolarizing factor-mediated responses in the rat mesenteric artery. J Pharmacol Exp Ther 2009; 328: 478–486.
Idris Khodja N, Chataigneau T, Auger C, Schini-Kerth VB . Grape-derived polyphenols improve aging-related endothelial dysfunction in rat mesenteric artery: role of oxidative stress and the angiotensin system. PLoS ONE 2012; 7: e32039.
Kane MO, Etienne-Selloum N, Madeira SV, Sarr M, Walter A, Dal-Ros S, Schott C, Chataigneau T, Schini-Kerth VB . Endothelium-derived contracting factors mediate the Ang II-induced endothelial dysfunction in the rat aorta: preventive effect of red wine polyphenols. Pflugers Arch 2010; 459: 671–679.
Lee JO, Oak MH, Jung SH, Park DH, Auger C, Kim KR, Lee SW, Schini-Kerth VB . An ethanolic extract of Lindera obtusiloba stems causes NO-mediated endothelium-dependent relaxations in rat aortic rings and prevents angiotensin II-induced hypertension and endothelial dysfunction in rats. Naunyn-Schmiedeberg Arch Pharmacol 2011; 383: 635–645.
Lee JO, Auger C, Park DH, Kang M, Oak MH, Kim KR, Schini-Kerth VB . An ethanolic extract of Lindera obtusiloba stems, YJP-14, improves endothelial dysfunction, metabolic parameters and physical performance in diabetic db/db mice. PLoS ONE 2013; 8: e65227.
Mehta PK, Griendling KK . Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 2007; 292: C82–C97.
Harrison DG, Cai H, Landmesser U, Griendling KK . Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst 2003; 4: 51–61.
Sunggip C, Kitajima N, Nishida M . Redox control of cardiovascular homeostasis by angiotensin II. Curr Pharm Design 2013; 19: 3022–3032.
Lee DY, Wauquier F, Eid AA, Roman LJ, Ghosh-Choudhury G, Khazim K, Block K, Gorin Y . Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J Biol Chem 2013; 288: 28668–28686.
Behringer EJ, Shaw RL, Westcott EB, Socha MJ, Segal SS . Aging impairs electrical conduction along endothelium of resistance arteries through enhanced Ca2+-activated K+ channel activation. Arterioscler Thromb Vasc Biol 2013; 33: 1892–1901.
Ohnaka K, Numaguchi K, Yamakawa T, Inagami T . Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension 2000; 35 (1 Pt 1): 68–75.
Feletou M, Huang Y, Vanhoutte PM . Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol 2011; 164: 894–912.
Ghiadoni L, Taddei S, Virdis A . Hypertension and endothelial dysfunction: therapeutic approach. Curr Vasc Phrmacol 2012; 10: 42–60.
Li S, Wu Y, Yu G, Xia Q, Xu Y . Angiotensin II receptor blockers improve peripheral endothelial function: a meta-analysis of randomized controlled trials. PLoS ONE 2014; 9: e90217.
Delgado-Lista J, Perez-Martinez P, Lopez-Miranda J, Perez-Jimenez F . Long chain omega-3 fatty acids and cardiovascular disease: a systematic review. Br J Nutr 2012; 107 (Suppl 2): S201–S213.
DiNicolantonio JJ, Niazi AK, McCarty MF, O'Keefe JH, Meier P, Lavie CJ . Omega-3s and cardiovascular health. Ochsner J 2014; 14: 399–412.
Iwamatsu K, Abe S, Nishida H, Kageyama M, Nasuno T, Sakuma M, Toyoda S, Inoue T . Which has the stronger impact on coronary artery disease, eicosapentaenoic acid or docosahexaenoic acid? Hypertens Res 2016; 39: 272–275.
Miller PE, Van Elswyk M, Alexander DD . Long-chain omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid and blood pressure: a meta-analysis of randomized controlled trials. Am J Hypertens 2014; 27: 885–896.
Appel LJ, Miller ER 3rd, Seidler AJ, Whelton PK . Does supplementation of diet with 'fish oil' reduce blood pressure? A meta-analysis of controlled clinical trials. Arch Intern Med 1993; 153: 1429–1438.
Knapp HR, FitzGerald GA . The antihypertensive effects of fish oil. A controlled study of polyunsaturated fatty acid supplements in essential hypertension. N Engl J Med 1989; 320: 1037–1043.
Tagawa H, Shimokawa H, Tagawa T, Kuroiwa-Matsumoto M, Hirooka Y, Takeshita A . Long-term treatment with eicosapentaenoic acid augments both nitric oxide-mediated and non-nitric oxide-mediated endothelium-dependent forearm vasodilatation in patients with coronary artery disease. J Cardiovasc Pharmacol 1999; 33: 633–640.
Fleischhauer FJ, Yan WD, Fischell TA . Fish oil improves endothelium-dependent coronary vasodilation in heart transplant recipients. J Am Coll Cardiol 1993; 21: 982–989.
Woodman RJ, Mori TA, Burke V, Puddey IB, Barden A, Watts GF, Beilin LJ . Effects of purified eicosapentaenoic acid and docosahexaenoic acid on platelet, fibrinolytic and vascular function in hypertensive type 2 diabetic patients. Atherosclerosis 2003; 166: 85–93.
Zgheel F, Alhosin M, Rashid S, Burban M, Auger C, Schini-Kerth VB . Redox-sensitive induction of Src/PI3-kinase/Akt and MAPKs pathways activate eNOS in response to EPA:DHA 6:1. PLoS ONE 2014; 9: e105102.
Omura M, Kobayashi S, Mizukami Y, Mogami K, Todoroki-Ikeda N, Miyake T, Matsuzaki M . Eicosapentaenoic acid (EPA) induces Ca(2+)-independent activation and translocation of endothelial nitric oxide synthase and endothelium-dependent vasorelaxation. FEBS Lett 2001; 487: 361–366.
Stebbins CL, Stice JP, Hart CM, Mbai FN, Knowlton AA . Effects of dietary decosahexaenoic acid (DHA) on eNOS in human coronary artery endothelial cells. J Cardiovasc Pharmacol Ther 2008; 13: 261–268.
Wall R, Ross RP, Fitzgerald GF, Stanton C . Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev 2010; 68: 280–289.
Grimminger F, Mayer K, Kramer HJ, Stevens J, Walmrath D, Seeger W . Differential vasoconstrictor potencies of free fatty acids in the lung vasculature: 2-versus 3-series prostanoid generation. J Pharmacol Exp Ther 1993; 267: 259–265.
Bligh EG, Dyer WJ . A rapid method of total lipid extraction and purification. Can J Biochem Physil 1959; 37: 911–917.
Simopoulos AP . The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed Pharmacother 2002; 56: 365–379.
Dasilva G, Pazos M, Garcia-Egido E, Gallardo JM, Rodriguez I, Cela R, Medina I . Healthy effect of different proportions of marine omega-3 PUFAs EPA and DHA supplementation in Wistar rats: lipidomic biomarkers of oxidative stress and inflammation. J Nutr Biochem 2015; 26: 1385–1392.
Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, Dong H, Iosif AM, Liu JY, Weiss RH, Chiamvimonvat N, Imig JD, Hammock BD . Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J Cardiovasc Pharmacol 2013; 62: 285–297.
Kimura S, Saito H, Minami M, Togashi H, Nakamura N, Ueno K, Shimamura K, Nemoto M, Parvez H . Docosahexaenoic acid attenuated hypertension and vascular dementia in stroke-prone spontaneously hypertensive rats. Neurotoxicol Teratol 2002; 24: 683–693.
Reagan-Shaw S, Nihal M, Ahmad N . Dose translation from animal to human studies revisited. FASEB J 2008; 22: 659–661.
Enns JE, Yeganeh A, Zarychanski R, Abou-Setta AM, Friesen C, Zahradka P, Taylor CG . The impact of omega-3 polyunsaturated fatty acid supplementation on the incidence of cardiovascular events and complications in peripheral arterial disease: a systematic review and meta-analysis. BMC Cardiovasc Disord 2014; 14: 70.
Yoo SM, Choi SH, Jung MD, Lim SC, Baek SH . Short-term use of telmisartan attenuates oxidation and improves Prdx2 expression more than antioxidant beta-blockers in the cardiovascular systems of spontaneously hypertensive rats. Hypertens Res 2015; 38: 106–115.
Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG . Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 1996; 97: 1916–1923.
Al-Magableh MR, Kemp-Harper BK, Hart JL . Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens Res 2015; 38: 13–20.
Förstermann U . Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 2010; 459: 923–939.
Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Förstermann U, Meinertz T, Griendling K, Munzel T . Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res 2002; 90: E58–E65.
Shatanawi A, Lemtalsi T, Yao L, Patel C, Caldwell RB, Caldwell RW . Angiotensin II limits NO production by upregulating arginase through a p38 MAPK–ATF-2 pathway. Eur J Pharmacol 2015; 746: 106–114.
Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE . Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol 2009; 107: 1249–1257.
Mengal V, Silva PH, Tiradentes RV, Santuzzi CH, de Almeida SA, Sena GC, Bissoli NS, Abreu GR, Gouvea SA . Aliskiren and l-arginine treatments restore depressed baroreflex sensitivity and decrease oxidative stress in renovascular hypertension rats. Hypertens Res 2016; 39: 769–776.
Morre J, Morre DM, Brightmore R . Omega-3 but not omega-6 unsaturated fatty acids inhibit the cancer-specific ENOX2 of the HeLa cell surface with no effect on the constitutive ENOX1. J Diet Suppl 2010; 7: 154–158.
Richard D, Kefi K, Barbe U, Bausero P, Visioli F . Polyunsaturated fatty acids as antioxidants. Pharmacol Res 2008; 57: 451–455.
Schini-Kerth VB, Auger C, Etienne-Selloum N, Chataigneau T . Polyphenol-induced endothelium-dependent relaxations role of NO and EDHF. Adv Pharmacol 2010; 60: 133–175.
Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A . Age-related reduction of NO availability and oxidative stress in humans. Hypertension 2001; 38: 274–279.
Hu ZW, Kerb R, Shi XY, Wei-Lavery T, Hoffman BB . Angiotensin II increases expression of cyclooxygenase-2: implications for the function of vascular smooth muscle cells. J Pharmacol Exp Ther 2002; 303: 563–573.
Taddei S, Virdis A, Mattei P, Salvetti A . Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 1993; 21 (6 Pt 2): 929–933.
Seki H, Tani Y, Arita M . Omega-3 PUFA derived anti-inflammatory lipid mediator resolvin E1. Prostaglandins Other Lipid Mediat 2009; 89: 126–130.
Calder PC . Omega-3 fatty acids and inflammatory processes. Nutrients 2010; 2: 355–374.
Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN . Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem 2003; 278: 14677–14687.
Hiram R, Rizcallah E, Sirois C, Sirois M, Morin C, Fortin S, Rousseau E . Resolvin D1 reverses reactivity and Ca2+ sensitivity induced by ET-1, TNF-alpha, and IL-6 in the human pulmonary artery. Am J Physiol Heart Circ Physiol 2014; 307: H1547–H1558.
Bousserouel S, Brouillet A, Bereziat G, Raymondjean M, Andreani M . Different effects of n-6 and n-3 polyunsaturated fatty acids on the activation of rat smooth muscle cells by interleukin-1 beta. J Lipid Res 2003; 44: 601–611.
Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG . Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II–induced hypertension. Circ Res 2010; 107: 263–270.
Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG . Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 2014; 64: 1108–1115.
Tousoulis D, Papageorgiou N, Antoniades C, Giolis A, Bouras G, Gounari P, Stefanadi E, Miliou A, Psaltopoulou T, Stefanadis C . Acute effects of different types of oil consumption on endothelial function, oxidative stress status and vascular inflammation in healthy volunteers. Br J Nutr 2010; 103: 43–49.
Acknowledgements
We thank Brigitte Pollet and Romain Vauchelles (UMR CNRS 7213, Faculty of Pharmacy, Strasbourg University) for technical assistance in vascular reactivity, and microscopy and image analysis, respectively. ZR is supported by a PhD fellowship from the Higher Education Commission of Pakistan and Gomal University, and FZ by a fellowship from the Higher Education Ministry of Libya. TPR and GCS received grants from the Brazilian National Council for Scientific and Technological Development (CNPq).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Drs Abdur Mirajkar and Azhar Alvi are both employees of Pivotal Therapeutics, Inc. All other authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Niazi, Z., Silva, G., Ribeiro, T. et al. EPA:DHA 6:1 prevents angiotensin II-induced hypertension and endothelial dysfunction in rats: role of NADPH oxidase- and COX-derived oxidative stress. Hypertens Res 40, 966–975 (2017). https://doi.org/10.1038/hr.2017.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2017.72
Keywords
This article is cited by
-
Lycium barbarum glycopeptide alleviates neuroinflammation in spinal cord injury via modulating docosahexaenoic acid to inhibiting MAPKs/NF-kB and pyroptosis pathways
Journal of Translational Medicine (2023)
-
NADPH oxidase family proteins: signaling dynamics to disease management
Cellular & Molecular Immunology (2022)
-
Effects of Docosahexaenoic Acid and Its Peroxidation Product on Amyloid-β Peptide-Stimulated Microglia
Molecular Neurobiology (2020)
-
The association between glucose-6-phosphate dehydrogenase deficiency and abnormal blood pressure among prepregnant reproductive-age Chinese females
Hypertension Research (2019)
-
Dronedarone induces regression of coronary artery remodeling related to better global antioxidant status
Hypertension Research (2019)
