
Abstract
T cell activation requires extracellular stimulatory signals that are mainly mediated by T cell receptor (TCR) complexes. The TCR recognizes antigens on major histocompatibility complex molecules with the cooperation of CD4 or CD8 coreceptors. After recognition, TCR-induced signaling cascades that propagate signals via various molecules and second messengers are induced. Consequently, many features of T cell-mediated immune responses are determined by these intracellular signaling cascades. Furthermore, differences in the magnitude of TCR signaling direct T cells toward distinct effector linages. Therefore, stringent regulation of T cell activation is crucial for T cell homeostasis and proper immune responses. Dysregulation of TCR signaling can result in anergy or autoimmunity. In this review, we summarize current knowledge on the pathways that govern how the TCR complex transmits signals into cells and the roles of effector molecules that are involved in these pathways.
Similar content being viewed by others
Introduction
As antigens enter the body, they are processed and presented by major histocompatibility complex (MHC) molecules expressed on the surface of antigen-presenting cells and recognized by T cell receptors (TCRs) on the surface of T cells. TCR signaling, in cooperation with signaling pathways induced by cytokines, costimulatory molecules, chemokines, integrins, and metabolites, drives the differentiation of activated T cells into specific T cell subtypes1. This results in the generation of various types of T cells with different specialized functions. Effector T cells fight against pathogens at initial exposure, and memory T cells provide defense against future infection. CD4+ T cells can differentiate into specialized effector subtypes, including T helper type 1 (Th1), Th2, Th17, follicular helper T, and regulatory T (Treg) cells. These subtypes regulate the immune response to address diverse types of pathogens. By generating specific T cell subtypes, the immune system can fine-tune itself and protect against inappropriate activation. It must achieve a delicate balance of sufficient activation to control infectious agents while preventing autoimmunity. Thus precise regulation of the T cell activation process is crucial for overall immune homeostasis2. Recent data suggest that TCR signaling is crucial for T cell differentiation and memory. How the fate of T cell differentiation is regulated has been widely investigated3. T cells are part of the adaptive immune system and fight against various infections and cancers. However, abnormal T cell function can cause autoimmune and inflammatory diseases. Naive T cells are initially activated through their TCRs by antigen/MHC complexes expressed by antigen-presenting cells. Subsequent signals, including environmental cues and signaling through CD28 or other costimulatory receptors, are required for T cell activation. Various signaling pathways, including the Ras-extracellular signal-related kinase (ERK)-activator protein (AP)-1 pathway, the inositol triphosphate (IP3)-Ca2+-nuclear factor of activated T cells (NFAT) pathway, the protein kinase C (PKC)θ-IĸB kinase (IKK)-nuclear factor (NF)-κB pathway, and the tuberous sclerosis complex (TSC)1/2-mammalian target of rapamycin (mTOR) pathway, are involved in TCR signaling (Fig. 1). Furthermore, several membrane proteins, such as lymphocyte function-associated antigen 1 and linker for activation of T cells, regulate T cell activation and function. For example, lymphocyte function-associated antigen 1 mediates TCR-induced T cell migration and activation by recruiting actinin and talin for the polymerization of filamentous actin4,5. Wiskott–Aldrich syndrome protein and cell division control protein 42 are also involved in actin filament polymerization, the activity of which can be regulated by a protein complex composed of VAV1, noncatalytic region of tyrosine kinase adaptor protein, and adhesion and degranulation-promoting adapter protein, which associates with linker for activation of T cells-Src homology (SH)2 domain-containing leukocyte protein 766. However, hematopoietic progenitor kinase 1 inhibits this complex by phosphorylating SH2 domain-containing leukocyte protein 767. Genetic/epigenetic controls also regulate T cell functions and activity8,9,10,11.

ADAP adhesion and degranulation-promoting adapter protein, BCL B cell lymphoma, Cbl casitas B-lineage lymphoma, CaM calmodulin, CaMKIV calcium/calmodulin-dependent protein kinase type IV, CARMA1 caspase recruitment domain-containing membrane-associated guanylate kinase protein 1, Cdc42 cell division control protein 42, CRAC calcium release-activated calcium, CRBN cereblon, CREB cAMP response element-binding protein, CTLA-4 cytotoxic T lymphocyte-associated protein-4, DAG diacylglycerol, DEPTOR DEP domain-containing mTOR-interacting protein, Dlgh1 discs large homolog 1, ERK extracellular signal-related kinase, F-actin filamentous actin, GADD45α growth arrest and DNA damage inducible alpha, GβL G protein beta subunit-like, GRB2 growth factor receptor-bound protein 2, HPK1 hematopoietic progenitor kinase 1, IKK IκB kinase, IP3 inositol triphosphate, IP3R IP3 receptor, JNK c-Jun N-terminal kinase, LAT linker for activation of T cells, Lck lymphocyte-specific protein tyrosine kinase, LFA-1 lymphocyte function-associated antigen-1, MALT1 mucosa-associated lymphoid tissue lymphoma translocation protein 1, MAPK mitogen-activated protein kinase, MEK MAPK/ERK kinase, MEKK MAP kinase kinase kinase, MKK MAP kinase kinase, mTOR mammalian target of rapamycin, NCK noncatalytic region of tyrosine kinase adaptor protein 1, NF-κB nuclear factor-κB, NFAT nuclear factor of activated T cells, PDK1 phosphoinositide-dependent kinase 1, PI3K phosphoinositide 3-kinase, PIP2 phosphatidylinositol 4,5-bisphosphate, PKCθ protein kinase C theta, PLCγ1 phospholipase C gamma 1, Raptor regulatory-associated protein of mTOR, RasGRP Ras guanyl-releasing protein 1, SLP76 Src homology 2 domain containing leukocyte protein 76, SOS son of sevenless, TCR T cell receptor, WASP Wiskott–Aldrich syndrome protein, ZAP-70 zeta chain of TCR-associated protein 70, ZIP Zrt- and Irt-like protein.
Overview of TCR signaling and T cell development
The TCR complex
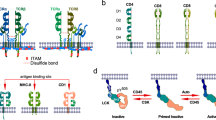
The structural components of the TCR complex were revealed in the 1980s through intense investigation and advances in molecular biology and biochemistry techniques12. The TCR complex consists of TCRα/β chains and CD3γ/δ/ε/ζ subunits, which associate through hydrophobic interactions13,14,15. Somatic VDJ recombination enables the generation of distinct TCRα and TCRβ beta chains, and TCRα/β heterodimers are responsible for antigen recognition by binding to peptide–MHC complexes16,17. CD3 transmits the TCR-triggered signal through immunoreceptor tyrosine-based activation motifs (ITAMs) in its cytoplasmic tail, but it is not directly involved in antigen recognition18,19. ITAMs are tandem duplications of a tyrosine-containing sequence (YXXL/I), and the CD3γ/δ/ε chains each contain one ITAM, while the CD3ζ chain contains three20,21. As a consequence of TCR engagement, ITAM phosphorylation is induced by protein tyrosine kinases (PTKs), which allow other effector molecules to interact with the TCR complex21.
Protein tyrosine kinases
The importance of tyrosine phosphorylation in TCR signaling was revealed by studies using PTK inhibitors22,23. TCR engagement triggers the activation and recruitment of PTKs, including Src family PTKs such as Lck and Fyn and the Syk family PTK zeta chain of TCR-associated protein 70 (ZAP-70)24. Evidence from Lck- or Fyn-null mice shows that Lck is crucial for T cell development, while Fyn is not essential for T cell development, as other Src family kinases can compensate for Fyn25,26,27. Lck is regulated by C-terminal Src kinase-mediated phosphorylation at its Y505 residue, which switches Lck to an inactive state28. The CD45 tyrosine phosphatase dephosphorylates inhibitory phosphorylation at Y505 and dephosphorylates positive regulatory autophosphorylation at Y394, although less efficiently29,30,31. This tight regulation of Lck activity protects against hyperactivation of T cells and autoimmunity, thus maintaining T cell homeostasis29,31. Activated Lck or Fyn phosphorylates the tyrosine residues of the ITAMs in the CD3 subunits. Tyrosine phosphorylation of CD3ζ provides the binding site for ZAP-70 via its SH2 domain, and then Lck or Fyn activates ZAP-70 by phosphorylation24,32. Therefore, recruitment of ZAP-70 to the activated TCR complex results in the formation of a signaling complex at the plasma membrane by recruiting other proteins through phosphorylation or activation24.
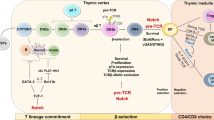
T cell development
T cells develop from thymus-migrant hematopoietic lineage cells, particularly common lymphoid progenitors or lymphoid-primed multipotent progenitors derived from the bone marrow or the fetal liver33,34. Developing T cells in the thymus progress through four double-negative (DN1–4) stages, then a double-positive (DP) stage, and finally mature into single-positive (SP) naive T cells35. DN1–4 cells are distinguished by their expression of CD44 and CD25: DN1, CD44+CD25−; DN2, CD44+CD25+; DN3, CD44−CD25+; and DN4, CD44−CD25−36. At the DN3 stage, a pre-TCR complex that consists of a pre-TCRα chain and a mature β chain first appears. The mature β chain in this complex is a product of somatic DNA rearrangement by recombination activating gene 1/237,38,39,40. T cells with a functional pre-TCR are positively selected by β selection, and they undergo massive proliferation and begin to rearrange the Tcra gene35,41. At the DP stage, both the CD4 and CD8 coreceptors are expressed (CD4+CD8+), and the αβ TCR is formed by replacing the pre-TCRα chain with the TCRα chain42. DP T cells encounter other checkpoints: DP T cells expressing αβ TCRs that recognize their MHC molecules through Tcra rearrangement are positively selected, and self-reactive T cells are deleted through negative selection43,44. In addition, DP T cells with dysfunctional TCRs that cannot receive or transduce TCR-mediated signals undergo apoptosis, while the selected cells further develop into CD4 or CD8 SP cells45.
The strength of TCR signaling and T cell differentiation
TCR stimulation is a fundamental step in most T cell responses. When TCRs are stimulated, the quality or quantity of the resulting signaling is affected by various factors, such as the strength and length of stimulation. Interestingly, differences in the affinities of stimulatory agonists for the TCR are sufficient to cause differences in T cell physiology. When naive CD4+ T cells are subjected to strong TCR stimulation, Th1 cell differentiation is favored over Th2 cell differentiation, both in vitro and in vivo46,47. Conversely, weak TCR signals favor Th2 cell differentiation46,47. Whether differences in TCR signaling strength affect Th17 cell differentiation remains controversial48,49. Importantly, the strength of TCR signaling also regulates Treg cell differentiation. Although thymus-derived Treg cells are induced by a broad range of antigen affinities, high TCR signaling strength preferentially induces thymus-derived Treg cell differentiation50,51. In addition, for peripherally derived Treg cells, a low level of a strong agonism is important for their stable induction52. A longer TCR–pMHC dwell time, as well as a high-affinity TCR, is positively related to follicular helper T cell differentiation53,54. Furthermore, weak TCR stimulation suffices for the generation or enhancement of memory CD8+ T cell function, while a longer TCR–pMHC interaction, high levels of an antigen, or a high affinity antigen are associated with robust proliferation1,55,56.
Regulatory mechanisms in TCR signaling
Positive TCR signaling pathways
The Ras-ERK1/2-AP-1 pathway
Ras proteins make up a family of small GTPases expressed in animal cells that includes H-Ras, N-Ras, K-Ras4A, and K-Ras4B57. These isoforms have conserved effector binding domains but different carboxy-terminal regions, which enables them to selectively associate with various cell membranes, resulting in their intracellular compartmentalization57. Ras functions as a binary signal switch: as Ras is switched on, it transmits signals to other proteins, turning on genes involved in cell growth, differentiation, and survival58. If Ras is permanently activated by mutation, it can signal constitutively in the absence of activating signals, resulting in cell transformation59. All Ras isoforms are expressed in lymphocytes and are involved in TCR signaling and T cell development and function60.
The ERK1/2 pathway is a downstream signaling pathway of Ras, and it can be activated by persistent Ras signaling61. ERK1/2 is regulated by a feedback mechanism targeting ERK1/2 itself or its upstream activators. ERK1/2 inactivation is controlled by mitogen-activated protein (MAP) kinase phosphatases, which have dual specificity for Ser/Thr and Tyr residues. ERK1/2 signaling has an important role in controlling T cell development, differentiation, and TCR-induced signal strength62,63.
AP-1 is a basic leucine zipper transcription factor composed of homodimers or heterodimers of Jun, Fos, and activating transcription factor (ATF). AP-1 activity is regulated by extracellular signals that repress or activate AP-1 transcription64,65. For example, the basic leucine zipper ATF-like transcription factor, which belongs to the AP-1 family, can regulate osteoarthritic cartilage destruction by controlling anabolic and catabolic gene expression in chondrocytes66. Basic leucine zipper ATF-like transcription factor/Jun heterodimers can bind to AP-1-binding sites and regulate gene expression. The AP-1 family is also involved in Th17 differentiation67,68.
As upstream signals including TCR, Lck/Fyn, ZAP-70, and growth factor receptor-bound protein 2/son of sevenless are transmitted to Ras, GDP on Ras is exchanged for GTP by son of sevenless69,70. Ras is activated by GTP exchange, resulting in the sequential activation of the kinases Raf, MAP kinase/ERK kinase 1/2, and ERK1/2, resulting in the transcription of c-Fos and JunB. This results in the formation of the AP-1 complex, which induces interleukin (IL)-2 transcription71,72. The c-Jun transcription factor can be activated through the Rac/cell division control protein 42-MAP kinase kinase 4/7-c-Jun N-terminal kinase pathway and related proteins73,74,75. In addition, p38 MAP kinase can also regulate the activity of ATF75,76.
The IP3-Ca2+-NFAT pathway
IP3 is formed when phosphatidylinositol 4,5-bisphosphate is hydrolyzed by phospholipase C. IP3 functions as a second messenger. When IP3 binds to its receptor on the membrane of the endoplasmic reticulum, Ca2+ is released from the endoplasmic reticulum into the cytosol, resulting in the activation of various signaling pathways77. The calcium release-activated calcium channel controls the intracellular Ca2+ concentration in lymphocytes78. Ca2+ is a universal second messenger in T cells. T cell proliferation, differentiation, and effector functions are regulated by Ca2+79. There are two types of Ca2+ signaling pathways in T cells, long term and short term. NFAT is a transcription factor that is activated by Ca2+ influx in T cells and involved in long-term Ca2+ signaling80. It induces gene expression alone or in cooperation with other transcription factors80. When antigen/MHC complexes bind to TCRs, PTKs are activated, resulting in the phosphorylation and activation of phospholipase C-γ1. The membrane phospholipid phosphatidylinositol 4,5-bisphosphate is hydrolyzed, generating IP3 and diacylglycerol (DAG)81. Ca2+ efflux is sensed by stromal interaction molecules 1 and 2. Stromal interaction molecule proteins form clusters known as puncta, which trigger Ca2+ influx via calcium release-activated calcium channels82,83. Ca2+ influx activates the NFAT pathway via Ca2+-related proteins84. An increase in Ca2+ can also facilitate the formation of actin filaments85. A recent study demonstrated that deletion of Crbn, the gene encoding cereblon, in CD4+ T cells increases Kv1.3 channel expression and consequent Ca2+ flux, resulting in stronger NFAT activation after TCR stimulation9, implying that CRBN regulates TCR-induced NFAT activation. In addition, a previous study showed that zinc can regulate TCR signaling pathways and cytokine production by activated T cells86.
The PKCθ-IKK-NF-κB pathway
After T cell activation, the serine/threonine-specific PKCθ is recruited to the immunological synapse87. Then a signaling complex composed of caspase recruitment domain-containing membrane-associated guanylate kinase protein 1 (CARMA1), B cell lymphoma/leukemia 10 (BCL10), and mucosa-associated lymphoid tissue translocation protein 1 (MALT1) is formed in the cytoplasm88. PKCθ was the first PKC family member to be found to be recruited to the immunological synapse. It plays an integral role in activating a range of signaling cascades, leading to transcriptional regulation in T cells88,89,90,91,92,93,94,95.
Phosphoinositide-dependent kinase 1 (PDK1) is an enzyme that is involved in various signaling pathways. It plays a crucial role in T/B cell development and survival, intestinal homeostasis, and immune tolerance96,97,98,99. NF-κB is a well-known target of PDK1. PDK1 is required for TCR-mediated activation of NF-κB and PKCθ; selective deletion of PDK1 in T cells abrogates activation of NF-κB and PKCθ98. Recently, it was shown that PKCθ induces phosphorylation of PDK1100, resulting in T cell activation and TCR-induced NF-κB activation96,97,98,100,101,102. PKCθ is involved in the activation of NF-κB103. When T cells are in the resting state, NF-κB exists in the cytoplasm bound to IκB. When TCR/CD28 ligation occurs, NF-κB signaling is activated. PDK1 efficiently activates PKCθ via the phosphoinositide 3-kinase pathway104. Activated PKC phosphorylates a serine residue located in the membrane-associated guanylate kinase domain of CARMA1105. Then BCL10 and MALT1 are recruited, resulting in the formation of the active CARMA1-BCL10-MALT1 signaling complex106. This promotes IKK complex activation and IκB degradation, which allows NF-κB to translocate to the nucleus, initiating the transcription of genes that are required for T cell activation11,103,107,108,109,110.
CARMA1, also called CARD11, is a scaffold protein that is considered a hallmark of IKK/NF-κB activation. CARMA1 contains several domains, including a caspase recruitment domain and coiled-coil, SH3, guanylate kinase, and PDZ domains106. Except for the PDZ domain, each of these domains is required for CARMA1 to activate NF-κB. CARMA1 is constitutively associated with the plasma membrane and recruited into lipid rafts after TCR stimulation111. CARMA1 activation is mediated by several mechanisms106, including phosphorylation. PKC phosphorylates CARMA1 between its coiled coil and PDZ domains after it is activated by TCR/CD28 ligation. Phosphorylated CARMA1 undergoes a conformational change, enabling it to associate with BCL10 and MALT13.
TSC1/2-mTOR signaling
TSC1 and TSC2 are tumor suppressors. They heterodimerize and regulate downstream signaling112. mTOR is involved in T cell activation, differentiation, and function113. Rapamycin is an immunosuppressant that promotes G1 arrest and inhibits downregulation of the cyclin-dependent kinase inhibitor p27113. Treatment of T cells with rapamycin inhibits their proliferation and leads to anergy. The ability of rapamycin to promote Treg cell generation underlies its ability to induce T cell anergy114. mTOR is activated by various signals, including growth factors, nutrients, and cellular stress signals, and regulates the growth, proliferation, and survival of cells113,115. Two different mTOR complexes, mTORC1 and mTORC2, are involved in mTOR signaling113. mTORC1 and mTORC2 both include the scaffolding proteins Raptor and Rictor. Activation of mTORC1 results in phosphorylation of S6 kinase 1 and translation of 4E-BP1, while activation of mTORC2 results in phosphorylation of the kinase AKT116. Recently, a relationship between TSC1/2 and mTOR was reported. When TSC2 is phosphorylated by AKT, the GAP activity of the TSC1/2 complex is inhibited, resulting in the activation of the small GTPase Rheb and mTORC1 activation113.
Inhibitory TCR signaling pathways
Phosphatases
Phosphorylation and dephosphorylation of TCR signaling molecules affect signaling complex formation and propagation of TCR signals. Similar to CD45 phosphatase, SH2 domain-containing protein tyrosine phosphatase (SHP-1) dephosphorylates Lck at Y394, resulting in its inactivation117. TCR signaling-mediated Lck activation results in SHP-1 activation via activated Lck phosphorylation of Y564 in SHP-1, which in turn dephosphorylates and inactivates Lck118, resulting in the attenuation of both early and late TCR signaling events. TCR signaling induces the expression of an adhesion molecule, carcinoembryonic antigen-related cell adhesion molecule 1, at a later time point119. The phosphorylation of an immunoreceptor tyrosine-based inhibitory motif in carcinoembryonic antigen-related cell adhesion molecule 1 recruits SHP-1, which dephosphorylates Lck, resulting in the termination of TCR signaling120. By contrast, under strong TCR–ligand binding conditions, ERK1/2 phosphorylate Lck at S59, preventing the recruitment of SHP-1 and sustaining TCR signaling for gene expression121. Suppressors of TCR signaling-1 and -2, which are TCR signaling-related phosphatases, dephosphorylate Syk and ZAP-70, respectively122,123. Mice lacking both suppressors of TCR signaling-1 and -2 show T cell hyperproliferation, enhanced activation of TCR signaling, and increased susceptibility to autoimmunity123. Furthermore, PTEN dephosphorylates PIP3, and dual specificity phosphatases dephosphorylate ERK2124,125. These observations suggest that dephosphorylation events in TCR signaling are important for the termination or negative regulation of TCR signaling cascades.
Ubiquitination and degradation
There is increasing interest in understanding the role of proteolytic mechanisms in the regulation of TCR signal transduction. Proteolysis is primarily caused by proteasomal or lysosomal processes. Many short-lived proteins selectively undergo ubiquitination before their proteasomal degradation. Ubiquitination results from the conjugation of ubiquitin to proteins through a series of enzymatic reactions126. Ubiquitination is initiated when the E1 ubiquitin-activating enzyme releases ubiquitin from the inactive state. Active ubiquitin is then transferred to an E2 ubiquitin-conjugating enzyme. Finally, the E3 ubiquitin ligase transfers activated ubiquitin from the E2 enzyme to the target protein. Thus the E3 ligase facilitates the actual attachment of ubiquitin to the substrate and therefore controls the specificity of substrate targeting127. Although many types of E3 ligases have been reported, the mechanisms determining their substrate specificity are not clearly understood128.
Ubiquitination is an important process in the regulation of T cell development, activation, and immune tolerance. Therefore, failure to regulate ubiquitination appropriately can lead to autoimmune and inflammatory diseases129. Several E3 ligases, such as casitas B-lineage lymphoma (Cbl), Itch, Deltex, and gene related to anergy in lymphocytes, are known to be involved in the regulation of TCR signaling via ubiquitination-mediated degradation of TCR signaling molecules, including CD3ζ, PKCθ, ZAP-70, phospholipase C-γ1, and phosphoinositide 3-kinase130,131,132,133. Itch ubiquitinates Jun, thereby reducing AP-1 activity134, while Cbl ubiquitinates CD3ζ via an adapter molecule, ZAP-70135. In contrast, Cbl-b and Itch inhibit the association between ZAP-70 and CD3ζ by conjugating K33-linked ubiquitin chains to CD3ζ, which does not lead to its degradation136. T cell anergy is also regulated by E3 ligases, including Cbl-b, c-Cbl, gene related to anergy in lymphocytes, and Itch137. The expression of gene related to anergy in lymphocytes is induced under anergic T cell conditions with decreased IL-2 production71. Roquin1/2 E3 ligases maintain immune tolerance by regulating T cell activation and differentiation appropriately138,139. Furthermore, ubiquitination plays a role in TCR-induced T cell activation. IκB ubiquitination and subsequent degradation of NF-κB are well-known processes140,141. However, before these can occur, NF-κB essential modulator (also known as IKKγ) must be ubiquitinated by the TRAF6 E3 ubiquitin ligase (K63-linked polyubiquitination), which contributes to the activation of the IKK complex142,143,144.
DAG kinases
DAG is an important signaling molecule involved in several signaling cascades. DAG kinases (DGKs) are lipid kinases that convert DAG to phosphatidic acid by phosphorylation, thereby regulating the subcellular DAG level145. Ten isoforms of mammalian DGKs have been identified, among which DGKα and DGKζ act as crucial regulators downstream of the TCR146,147. When DGKζ expression increases in T cells, TCR-induced Ras-ERK signaling is reduced148. In addition, T cells exhibiting loss of DGKα and/or DGKζ show increased TCR-induced signaling, including Ras-MAP kinase/ERK kinase-ERK-AP-1, PKCθ-NF-κB, and mTOR pathway signaling, leading to hyperactivation, impaired induction of anergy, and reduced antiviral responses in CD8+ T cells149,150,151,152,153,154. DGKα/DGKζ double-knockout mice show impaired T cell development, and phosphatidic acid treatment partially rescues T cell development, suggesting that DGKs not only terminate DAG signaling but also initiate phosphatidic acid-mediated signaling155.
T cell development pathways
Transcriptional control of T cell development
Ikaros
Members of the Ikaros transcription factor family, including Helios and Aiolos, possess zinc-finger motifs156,157,158. They are most abundant in hematopoietic lineages and are mainly lymphocyte restricted156,159. Mice with a homozygous mutation in the DNA-binding domain of Ikaros lack T, B, and natural killer cells, as well as their earliest progenitors160. In another study, functionally Ikaros-null mutant mice were generated by deleting the C-terminal region to avoid a dominant-negative effect due to the loss of the DNA-binding domain161. These mice show an absence or large reduction in fetal T and B cells and in adult γδ T, B, natural killer, and thymic antigen-presenting cells and show aberrant proliferation and differentiation into CD4 lineage T cells postnatally161. These studies suggest that Ikaros promotes the differentiation of hematopoietic stem cells into lymphocytes and establishes early branch points in postnatal T cell development160,161 (Fig. 2). In addition, Ikaros regulates checkpoints in T cell development, such as β selection and the transition from the DP to the SP stage162.

CLP common lymphoid progenitor, DN double negative, DP double positive.
GATA-3
GATA transcription factor family proteins contain zinc-finger motifs that recognize the consensus DNA sequence WGATAR (W represents A or T, and R represents A or G)163,164. GATA-3 was first identified as a regulator of the Tcra gene that binds to a Tα3 element in the Tcra enhancer165, but it also has a well-known role in Th2 cell differentiation166,167. In lymphoid lineage cells, GATA-3 expression is restricted to early hematopoietic precursors, immature/mature T cells, and natural killer cells165,168,169. One group assessed the role of GATA-3 in immune cell development using a Rag2−/− blastocyst complementation system because homozygous deletion of Gata3 is embryonic lethal and showed that GATA-3 is crucial for T cell development170. Another study using mice with a conditional deletion in Gata3 in early- or late-stage thymocytes showed arrest of the DN3 population with decreased DN4, DP, and SP populations or impaired differentiation into CD4+ T cells, respectively.171. These results imply that GATA-3 affects β selection and commitment to the CD4 SP lineage171 (Fig. 2). In the choice between the CD4 or CD8 lineage, GATA-3 can directly induce expression of the Zbtb7b gene, which encodes the transcription factor ThPOK172. ThPOK independently inhibits the differentiation of DP thymocytes into CD8 SP cells and promotes differentiation into CD4 SP cells in a GATA-3-dependent manner, suggesting that GATA-3 is an upstream regulator of ThPOK172. On the other hand, the exact role of GATA-3 in β selection is not completely known.
Notch
Notch was first identified as a regulator of cell fate decisions during neuronal and epidermal cell differentiation in Drosophila173. In mammals, it is a transmembrane receptor of the Delta/Serrate/Lag-2 family that interacts with membrane-associated ligands, Jagged 1/Serrate 1, Jagged 2/Serrate 2, Delta 1, Delta 2, and Delta 3174. Interaction between cells expressing Notch and cells expressing Delta/Serrate/Lag-2 ligands causes proteolytic cleavage of Notch, which migrates to the nucleus and releases intracellular domains that interact with recombination signal-binding protein for immunoglobulin kappa J region, leading to gene regulation175,176. Targets of activated recombination signal-binding protein for immunoglobulin kappa J region are incompletely characterized. One known target is hairy and enhancer of split 1, which is upregulated by Notch and acts as a transcriptional repressor.
Further evidence for the role of Notch in T cell lineage determination comes from experiments in which mice were reconstituted with bone marrow stem cells expressing constitutively active Notch1. The differentiation of stem cells into B cells was completely blocked in these mice, which developed a thymus-dependent population of T cells in the bone marrow177. In contrast, deletion of Notch1 or inhibition of Notch1 signaling in hematopoietic stem cells drives B cell development, while T cell development is blocked178,179,180. Thus lymphocyte progenitor cells develop into T cells via Notch signaling, but without these signals, the B cell fate is chosen (Fig. 2).
Conclusions
The signals initiated by the activated TCR complex play essential roles in T cell-mediated immune responses. In recent decades, extensive efforts by researchers and advances in molecular, genetic, and biochemical techniques have made it possible to elucidate the structure and signaling molecules of the TCR complex. Engagement of the TCR complex is a prerequisite for the initiation of the TCR signaling cascades that were summarized in this review. TCR signaling is important for many aspects of T cell regulation, including development, differentiation, activation, proliferation, and survival. Therefore, TCR signaling must be tightly regulated. In this regard, therapeutics have been developed that target the TCR complex, mainly for immune suppression. For example, muromonab-CD3 (orthoclone OKT3) is the first mouse anti-human CD3 monoclonal antibody to be used in the clinic. It binds to CD3ε in circulating T cells and elicits immune suppression by inducing apoptosis. In an attempt to reduce its side effects related to its mouse origin, chimeric or humanized anti-CD3 monoclonal antibodies, such as otelixizumab, teplizumab, and visilizumab, have been developed and are under clinical trials for the treatment of various diseases.
The regulation of TCR signaling is a complicated process and is controlled by a large number of effector molecules, and there are still many aspects of T cell activation and development that are poorly understood. The integration of TCR-induced signaling and CD28-induced signaling is relatively well understood, but the effect of imbalances between these two signaling cascades on T cell differentiation and function is not well understood. For example, strong CD28 signaling blocks Th17 differentiation. Thus there are unknown regulatory mechanisms controlling T cell-mediated immune responses. A more comprehensive understanding of these processes will enable us to therapeutically modulate immune responses for the treatment of autoimmune disease and other immune-related diseases.
References
Daniels, M. A. & Teixeiro, E. TCR signaling in T cell memory. Front. Immunol. 6, 617 (2015).
Courtney, A. H., Lo, W. L. & Weiss, A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem. Sci. 43, 108–123 (2018).
Huang, W. & August, A. The signaling symphony: T cell receptor tunes cytokine-mediated T cell differentiation. J. Leukoc. Biol. 97, 477–485 (2015).
Walling, B. L. & Kim, M. LFA-1 in T cell migration and differentiation. Front. Immunol. 9, 952 (2018).
Evans, R. et al. Integrins in immunity. J. Cell Sci. 122, 215–225 (2009).
Billadeau, D. D., Nolz, J. C. & Gomez, T. S. Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol. 7, 131–143 (2007).
Shui, J. W. et al. Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat. Immunol. 8, 84–91 (2007).
Lee, S. et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J. Allergy Clin. Immunol. 137, 327–330 (2016).
Kang, J. A. et al. Epigenetic regulation of Kcna3-encoding Kv1.3 potassium channel by cereblon contributes to regulation of CD4+ T-cell activation. Proc. Natl Acad. Sci. USA 113, 8771–8776 (2016).
Oh, H. & Ghosh, S. NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 252, 41–51 (2013).
Coudronniere, N., Villalba, M., Englund, N. & Altman, A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc. Natl Acad. Sci. USA 97, 3394–3399 (2000).
Smith-Garvin, J. E., Koretzky, G. A. & Jordan, M. S. T cell activation. Annu. Rev. Immunol. 27, 591–619 (2009).
Hedrick, S. M., Cohen, D. I., Nielsen, E. A. & Davis, M. M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. 1984. J. Immunol. 175, 2771–2775 (2005).
Malissen, M. et al. Mouse T cell antigen receptor: structure and organization of constant and joining gene segments encoding the beta polypeptide. Cell 37, 1101–1110 (1984).
Borst, J. et al. The delta- and epsilon-chains of the human T3/T-cell receptor complex are distinct polypeptides. Nature 312, 455–458 (1984).
Saito, T. & Germain, R. N. Predictable acquisition of a new MHC recognition specificity following expression of a transfected T-cell receptor beta-chain gene. Nature 329, 256–259 (1987).
Dembic, Z. et al. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature 320, 232–238 (1986).
Letourneur, F. & Klausner, R. D. Activation of T cells by a tyrosine kinase activation domain in the cytoplasmic tail of CD3 epsilon. Science 255, 79–82 (1992).
Samelson, L. E., Patel, M. D., Weissman, A. M., Harford, J. B. & Klausner, R. D. Antigen activation of murine T cells induces tyrosine phosphorylation of a polypeptide associated with the T cell antigen receptor. Cell 46, 1083–1090 (1986).
Yao, X. R., Flaswinkel, H., Reth, M. & Scott, D. W. Immunoreceptor tyrosine-based activation motif is required to signal pathways of receptor-mediated growth arrest and apoptosis in murine B lymphoma cells. J. Immunol. 155, 652–661 (1995).
Bu, J. Y., Shaw, A. S. & Chan, A. C. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc. Natl Acad. Sci. USA 92, 5106–5110 (1995).
Mustelin, T., Coggeshall, K. M., Isakov, N. & Altman, A. T cell antigen receptor-mediated activation of phospholipase C requires tyrosine phosphorylation. Science 247, 1584–1587 (1990).
June, C. H. et al. Inhibition of tyrosine phosphorylation prevents T-cell receptor-mediated signal transduction. Proc. Natl Acad. Sci. USA 87, 7722–7726 (1990).
Zhang, W., Sloan-Lancaster, J., Kitchen, J., Trible, R. P. & Samelson, L. E. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92, 83–92 (1998).
Stein, P. L., Lee, H. M., Rich, S. & Soriano, P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell 70, 741–750 (1992).
Molina, T. J. et al. Profound block in thymocyte development in mice lacking p56lck. Nature 357, 161–164 (1992).
Appleby, M. W. et al. Defective T cell receptor signaling in mice lacking the thymic isoform of p59fyn. Cell 70, 751–763 (1992).
Bergman, M. et al. The human p50csk tyrosine kinase phosphorylates p56lck at Tyr-505 and down regulates its catalytic activity. EMBO J. 11, 2919–2924 (1992).
McNeill, L. et al. The differential regulation of Lck kinase phosphorylation sites by CD45 is critical for T cell receptor signaling responses. Immunity 27, 425–437 (2007).
Baker, M. et al. Development of T-leukaemias in CD45 tyrosine phosphatase-deficient mutant lck mice. EMBO J. 19, 4644–4654 (2000).
D’Oro, U. & Ashwell, J. D. Cutting edge: the CD45 tyrosine phosphatase is an inhibitor of Lck activity in thymocytes. J. Immunol. 162, 1879–1883 (1999).
Chan, A. C., Iwashima, M., Turck, C. W. & Weiss, A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell 71, 649–662 (1992).
Ghaedi, M. et al. Common-lymphoid-progenitor-independent pathways of innate and T lymphocyte development. Cell Rep. 15, 471–480 (2016).
Kondo, M., Weissman, I. L. & Akashi, K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91, 661–672 (1997).
Germain, R. N. T-cell development and the CD4-CD8 lineage decision. Nat. Rev. Immunol. 2, 309–322 (2002).
Godfrey, D. I., Kennedy, J., Suda, T. & Zlotnik, A. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8- triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J. Immunol. 150, 4244–4252 (1993).
von Boehmer, H. & Fehling, H. J. Structure and function of the pre-T cell receptor. Annu. Rev. Immunol. 15, 433–452 (1997).
Shinkai, Y. et al. Restoration of T cell development in RAG-2-deficient mice by functional TCR transgenes. Science 259, 822–825 (1993).
Groettrup, M. et al. A novel disulfide-linked heterodimer on pre-T cells consists of the T cell receptor beta chain and a 33 kd glycoprotein. Cell 75, 283–294 (1993).
Mombaerts, P. et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992).
Kreslavsky, T. et al. beta-Selection-induced proliferation is required for alphabeta T cell differentiation. Immunity 37, 840–853 (2012).
Carpenter, A. C. & Bosselut, R. Decision checkpoints in the thymus. Nat. Immunol. 11, 666–673 (2010).
Starr, T. K., Jameson, S. C. & Hogquist, K. A. Positive and negative selection of T cells. Annu. Rev. Immunol. 21, 139–176 (2003).
Mancini, S. J. et al. TCRA gene rearrangement in immature thymocytes in absence of CD3, pre-TCR, and TCR signaling. J. Immunol. 167, 4485–4493 (2001).
Zhang, N., Hartig, H., Dzhagalov, I., Draper, D. & He, Y. W. The role of apoptosis in the development and function of T lymphocytes. Cell Res. 15, 749–769 (2005).
Brogdon, J. L., Leitenberg, D. & Bottomly, K. The potency of TCR signaling differentially regulates NFATc/p activity and early IL-4 transcription in naive CD4+ T cells. J. Immunol. 168, 3825–3832 (2002).
Constant, S., Pfeiffer, C., Woodard, A., Pasqualini, T. & Bottomly, K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J. Exp. Med. 182, 1591–1596 (1995).
Gomez-Rodriguez, J. et al. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity 31, 587–597 (2009).
Purvis, H. A. et al. Low-strength T-cell activation promotes Th17 responses. Blood 116, 4829–4837 (2010).
Lee, H. M., Bautista, J. L., Scott-Browne, J., Mohan, J. F. & Hsieh, C. S. A broad range of self-reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity 37, 475–486 (2012).
Moran, A. E. et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 208, 1279–1289 (2011).
Gottschalk, R. A., Corse, E. & Allison, J. P. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J. Exp. Med. 207, 1701–1711 (2010).
Fazilleau, N., McHeyzer-Williams, L. J., Rosen, H. & McHeyzer-Williams, M. G. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 10, 375–384 (2009).
Tubo, N. J. et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153, 785–796 (2013).
Maru, S., Jin, G., Schell, T. D. & Lukacher, A. E. TCR stimulation strength is inversely associated with establishment of functional brain-resident memory CD8 T cells during persistent viral infection. PLoS Pathog. 13, e1006318 (2017).
Zehn, D., Lee, S. Y. & Bevan, M. J. Complete but curtailed T-cell response to very low-affinity antigen. Nature 458, 211–214 (2009).
Hobbs, G. A., Der, C. J. & Rossman, K. L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129, 1287–1292 (2016).
Rommel, C. & Hafen, E. Ras–a versatile cellular switch. Curr. Opin. Genet. Dev. 8, 412–418 (1998).
Koeffler, H. P., McCormick, F. & Denny, C. Molecular mechanisms of cancer. West. J. Med. 155, 505–514 (1991).
Iborra, S. et al. N-ras couples antigen receptor signaling to Eomesodermin and to functional CD8+ T cell memory but not to effector differentiation. J. Exp. Med. 210, 1463–1479 (2013).
Mebratu, Y. & Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 8, 1168–1175 (2009).
Jia, H., Xu, M., Bo, Y., Li, W. & Zhou, R. Ras-ERK1/2 signaling accelerates the progression of colorectal cancer via mediation of H2BK5ac. Life Sci. 230, 89–96 (2019).
Bertin, S. et al. Dual-specificity phosphatase 6 regulates CD4+ T-cell functions and restrains spontaneous colitis in IL-10-deficient mice. Mucosal Immunol. 8, 505–515 (2015).
Konicek, B. W., Xia, X., Rajavashisth, T. & Harrington, M. A. Regulation of mouse colony-stimulating factor-1 gene promoter activity by AP1 and cellular nucleic acid-binding protein. DNA Cell Biol. 17, 799–809 (1998).
Angel, P. & Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072, 129–157 (1991).
Rhee, J. et al. Inhibition of BATF/JUN transcriptional activity protects against osteoarthritic cartilage destruction. Ann. Rheum. Dis. 76, 427–434 (2017).
Park, S. H. et al. BATF regulates collagen-induced arthritis by regulating T helper cell differentiation. Arthritis Res. Ther. 20, 161 (2018).
Carr, T. M., Wheaton, J. D., Houtz, G. M. & Ciofani, M. JunB promotes Th17 cell identity and restrains alternative CD4(+) T-cell programs during inflammation. Nat. Commun. 8, 301 (2017).
Wiesmuller, L. & Wittinghofer, F. Signal transduction pathways involving Ras. Mini review. Cell. Signal. 6, 247–267 (1994).
Buday, L., Egan, S. E., Rodriguez Viciana, P., Cantrell, D. A. & Downward, J. A complex of Grb2 adaptor protein, Sos exchange factor, and a 36-kDa membrane-bound tyrosine phosphoprotein is implicated in ras activation in T cells. J. Biol. Chem. 269, 9019–9023 (1994).
Anandasabapathy, N. et al. GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity 18, 535–547 (2003).
Samelson, L. E. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu. Rev. Immunol. 20, 371–394 (2002).
Wang, X., Destrument, A. & Tournier, C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim. Biophys. Acta 1773, 1349–1357 (2007).
Kaga, S., Ragg, S., Rogers, K. A. & Ochi, A. Activation of p21-CDC42/Rac-activated kinases by CD28 signaling: p21-activated kinase (PAK) and MEK kinase 1 (MEKK1) may mediate the interplay between CD3 and CD28 signals. J. Immunol. 160, 4182–4189 (1998).
Dodeller, F. & Schulze-Koops, H. The p38 mitogen-activated protein kinase signaling cascade in CD4 T cells. Arthritis Res. Ther. 8, 205 (2006).
Round, J. L. et al. Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF-kappaB transcription factors. Nat. Immunol. 8, 154–161 (2007).
Berridge, M. J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 933-940, 2009 (1793).
Oh-hora, M. & Rao, A. Calcium signaling in lymphocytes. Curr. Opin. Immunol. 20, 250–258 (2008).
Joseph, N., Reicher, B. & Barda-Saad, M. The calcium feedback loop and T cell activation: how cytoskeleton networks control intracellular calcium flux. Biochim. Biophys. Acta 1838, 557–568 (2014).
Oh-hora, M. & Rao, A. The calcium/NFAT pathway: role in development and function of regulatory T cells. Microbes Infect. 11, 612–619 (2009).
Rhee, S. G. & Choi, K. D. Regulation of inositol phospholipid-specific phospholipase C isozymes. J. Biol. Chem. 267, 12393–12396 (1992).
Weidinger, C., Shaw, P. J. & Feske, S. STIM1 and STIM2-mediated Ca(2+) influx regulates antitumour immunity by CD8(+) T cells. EMBO Mol. Med. 5, 1311–1321 (2013).
Zhang, S. L. et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 (2005).
Hesterberg, R. S., Cleveland, J. L. & Epling-Burnette, P. K. Role of polyamines in immune cell functions. Med. Sci. (Basel) 6, 22 (2018).
Liu, C. S. C. et al. Cutting edge: Piezo1 mechanosensors optimize human T cell activation. J. Immunol. 200, 1255–1260 (2018).
Aydemir, T. B., Liuzzi, J. P., McClellan, S. & Cousins, R. J. Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-gamma expression in activated human T cells. J. Leukoc. Biol. 86, 337–348 (2009).
Monks, C. R., Kupfer, H., Tamir, I., Barlow, A. & Kupfer, A. Selective modulation of protein kinase C-theta during T-cell activation. Nature 385, 83–86 (1997).
Thome, M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat. Rev. Immunol. 4, 348–359 (2004).
Quann, E. J., Liu, X., Altan-Bonnet, G. & Huse, M. A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat. Immunol. 12, 647–654 (2011).
Fu, G. et al. Protein kinase C eta is required for T cell activation and homeostatic proliferation. Sci. Signal. 4, ra84 (2011).
Singleton, K. L. et al. Spatiotemporal patterning during T cell activation is highly diverse. Sci. Signal. 2, ra15 (2009).
Weil, R. & Israel, A. T-cell-receptor- and B-cell-receptor-mediated activation of NF-kappaB in lymphocytes. Curr. Opin. Immunol. 16, 374–381 (2004).
Lin, X. & Wang, D. The roles of CARMA1, Bcl10, and MALT1 in antigen receptor signaling. Semin. Immunol. 16, 429–435 (2004).
Arendt, C. W., Albrecht, B., Soos, T. J. & Littman, D. R. Protein kinase C-theta: signaling from the center of the T-cell synapse. Curr. Opin. Immunol. 14, 323–330 (2002).
Liu, Y. et al. Regulation of protein kinase Ctheta function during T cell activation by Lck-mediated tyrosine phosphorylation. J. Biol. Chem. 275, 3603–3609 (2000).
Park, S. G. et al. The kinase PDK1 is essential for B-cell receptor mediated survival signaling. PLoS ONE 8, e55378 (2013).
Park, S. G. et al. T regulatory cells maintain intestinal homeostasis by suppressing gammadelta T cells. Immunity 33, 791–803 (2010).
Park, S. G. et al. The kinase PDK1 integrates T cell antigen receptor and CD28 coreceptor signaling to induce NF-kappaB and activate T cells. Nat. Immunol. 10, 158–166 (2009).
Hinton, H. J., Alessi, D. R. & Cantrell, D. A. The serine kinase phosphoinositide-dependent kinase 1 (PDK1) regulates T cell development. Nat. Immunol. 5, 539–545 (2004).
Kang, J. A. et al. PKCtheta-mediated PDK1 phosphorylation enhances T cell activation by increasing PDK1 stability. Mol. Cells 40, 37–44 (2017).
Park, S. H., Cho, G. & Park, S. G. NF-kappaB activation in T helper 17 cell differentiation. Immune Netw. 14, 14–20 (2014).
Kang, J. A. et al. Transition from heterotypic to homotypic PDK1 homodimerization is essential for TCR-mediated NF-kappaB activation. J. Immunol. 190, 4508–4515 (2013).
Lin, X., O’Mahony, A., Mu, Y., Geleziunas, R. & Greene, W. C. Protein kinase C-theta participates in NF-kappaB activation induced by CD3-CD28 costimulation through selective activation of IkappaB kinase beta. Mol. Cell. Biol. 20, 2933–2940 (2000).
Lee, K. Y., D’Acquisto, F., Hayden, M. S., Shim, J. H. & Ghosh, S. PDK1 nucleates T cell receptor-induced signaling complex for NF-kappaB activation. Science 308, 114–118 (2005).
Matsumoto, R. et al. Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity 23, 575–585 (2005).
Blonska, M. & Lin, X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 21, 55–70 (2011).
So, T. & Croft, M. Regulation of the PKCtheta-NF-kappaB axis in T lymphocytes by the tumor necrosis factor receptor family member OX40. Front. Immunol. 3, 133 (2012).
Pfeifhofer, C. et al. Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J. Exp. Med. 197, 1525–1535 (2003).
Sun, Z. et al. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 404, 402–407 (2000).
Baier-Bitterlich, G. et al. Protein kinase C-theta isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell. Biol. 16, 1842–1850 (1996).
Gaide, O. et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat. Immunol. 3, 836–843 (2002).
van Slegtenhorst, M. et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 7, 1053–1057 (1998).
Pollizzi, K. N. & Powell, J. D. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol. 36, 13–20 (2015).
Battaglia, M., Stabilini, A. & Roncarolo, M. G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 105, 4743–4748 (2005).
Peterson, T. R. et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 (2009).
Facchinetti, V. et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 27, 1932–1943 (2008).
Chiang, G. G. & Sefton, B. M. Specific dephosphorylation of the Lck tyrosine protein kinase at Tyr-394 by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 276, 23173–23178 (2001).
Lorenz, U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol. Rev. 228, 342–359 (2009).
Kammerer, R., Hahn, S., Singer, B. B., Luo, J. S. & von Kleist, S. Biliary glycoprotein (CD66a), a cell adhesion molecule of the immunoglobulin superfamily, on human lymphocytes: structure, expression and involvement in T cell activation. Eur. J. Immunol. 28, 3664–3674 (1998).
Nagaishi, T. et al. SHP1 phosphatase-dependent T cell inhibition by CEACAM1 adhesion molecule isoforms. Immunity 25, 769–781 (2006).
Stefanova, I. et al. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 4, 248–254 (2003).
Mikhailik, A. et al. A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol. Cell 27, 486–497 (2007).
Carpino, N. et al. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity 20, 37–46 (2004).
Caunt, C. J., Armstrong, S. P., Rivers, C. A., Norman, M. R. & McArdle, C. A. Spatiotemporal regulation of ERK2 by dual specificity phosphatases. J. Biol. Chem. 283, 26612–26623 (2008).
Stambolic, V. et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 (1998).
Hershko, A. & Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998).
Pickart, C. M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 (2001).
Deshaies, R. J. & Joazeiro, C. A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 (2009).
Hu, H. & Sun, S. C. Ubiquitin signaling in immune responses. Cell Res. 26, 457–483 (2016).
Park, Y., Jin, H. S., Aki, D., Lee, J. & Liu, Y. C. The ubiquitin system in immune regulation. Adv. Immunol. 124, 17–66 (2014).
Hsu, T. S., Hsiao, H. W., Wu, P. J., Liu, W. H. & Lai, M. Z. Deltex1 promotes protein kinase Ctheta degradation and sustains Casitas B-lineage lymphoma expression. J. Immunol. 193, 1672–1680 (2014).
Huang, F. & Gu, H. Negative regulation of lymphocyte development and function by the Cbl family of proteins. Immunol. Rev. 224, 229–238 (2008).
Heissmeyer, V. et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol. 5, 255–265 (2004).
Gao, M. et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 306, 271–275 (2004).
Wang, H. Y. et al. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. J. Biol. Chem. 276, 26004–26011 (2001).
Huang, H. et al. K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity 33, 60–70 (2010).
Mueller, D. L. E3 ubiquitin ligases as T cell anergy factors. Nat. Immunol. 5, 883–890 (2004).
Zhang, Q. et al. New insights into the RNA-binding and E3 ubiquitin ligase activities of Roquins. Sci. Rep. 5, 15660 (2015).
Vogel, K. U. et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity 38, 655–668 (2013).
Chen, Z. et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 9, 1586–1597 (1995).
Brown, K., Park, S., Kanno, T., Franzoso, G. & Siebenlist, U. Mutual regulation of the transcriptional activator NF-kappa B and its inhibitor, I kappa B-alpha. Proc. Natl Acad. Sci. USA 90, 2532–2536 (1993).
Zhou, H. et al. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature 427, 167–171 (2004).
Sun, L., Deng, L., Ea, C. K., Xia, Z. P. & Chen, Z. J. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 (2004).
Deng, L. et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 (2000).
Merida, I., Avila-Flores, A. & Merino, E. Diacylglycerol kinases: at the hub of cell signalling. Biochem. J. 409, 1–18 (2008).
Riese, M. J., Moon, E. K., Johnson, B. D. & Albelda, S. M. Diacylglycerol kinases (DGKs): novel targets for improving T cell activity in cancer. Front. Cell Dev. Biol. 4, 108 (2016).
Shirai, Y. & Saito, N. Diacylglycerol kinase as a possible therapeutic target for neuronal diseases. J. Biomed. Sci. 21, 28 (2014).
Zhong, X. P. et al. Regulation of T cell receptor-induced activation of the Ras-ERK pathway by diacylglycerol kinase zeta. J. Biol. Chem. 277, 31089–31098 (2002).
Shin, J., O’Brien, T. F., Grayson, J. M. & Zhong, X. P. Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J. Immunol. 188, 2111–2117 (2012).
Zhong, X. P., Guo, R., Zhou, H., Liu, C. & Wan, C. K. Diacylglycerol kinases in immune cell function and self-tolerance. Immunol. Rev. 224, 249–264 (2008).
Zhong, X. P., Olenchock, B. A. & Koretzky, G. A. The role of diacylglycerol kinases in T cell anergy. Ernst Schering Found. Symp. Proc. 139–149 (2007).
Zha, Y. et al. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat. Immunol. 7, 1166–1173 (2006).
Olenchock, B. A. et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 7, 1174–1181 (2006).
Zhong, X. P. et al. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat. Immunol. 4, 882–890 (2003).
Guo, R. et al. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc. Natl Acad. Sci. USA 105, 11909–11914 (2008).
Hahm, K. et al. Helios, a T cell-restricted Ikaros family member that quantitatively associates with Ikaros at centromeric heterochromatin. Genes Dev. 12, 782–796 (1998).
Morgan, B. et al. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiation. EMBO J. 16, 2004–2013 (1997).
Molnar, A. & Georgopoulos, K. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol. Cell. Biol. 14, 8292–8303 (1994).
Georgopoulos, K., Moore, D. D. & Derfler, B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science 258, 808–812 (1992).
Georgopoulos, K. et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell 79, 143–156 (1994).
Wang, J. H. et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity 5, 537–549 (1996).
Winandy, S., Wu, L., Wang, J. H. & Georgopoulos, K. Pre-T cell receptor (TCR) and TCR-controlled checkpoints in T cell differentiation are set by Ikaros. J. Exp. Med. 190, 1039–1048 (1999).
Merika, M. & Orkin, S. H. DNA-binding specificity of GATA family transcription factors. Mol. Cell. Biol. 13, 3999–4010 (1993).
Ko, L. J. & Engel, J. D. DNA-binding specificities of the GATA transcription factor family. Mol. Cell. Biol. 13, 4011–4022 (1993).
Ho, I. C. et al. Human GATA-3: a lineage-restricted transcription factor that regulates the expression of the T cell receptor alpha gene. EMBO J. 10, 1187–1192 (1991).
Zheng, W. & Flavell, R. A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596 (1997).
Zhang, D. H., Cohn, L., Ray, P., Bottomly, K. & Ray, A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J. Biol. Chem. 272, 21597–21603 (1997).
Samson, S. I. et al. GATA-3 promotes maturation, IFN-gamma production, and liver-specific homing of NK cells. Immunity 19, 701–711 (2003).
Oosterwegel, M., Timmerman, J., Leiden, J. & Clevers, H. Expression of GATA-3 during lymphocyte differentiation and mouse embryogenesis. Dev. Immunol. 3, 1–11 (1992).
Ting, C. N., Olson, M. C., Barton, K. P. & Leiden, J. M. Transcription factor GATA-3 is required for development of the T-cell lineage. Nature 384, 474–478 (1996).
Pai, S. Y. et al. Critical roles for transcription factor GATA-3 in thymocyte development. Immunity 19, 863–875 (2003).
Wang, L. et al. Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4(+) T cells. Nat. Immunol. 9, 1122–1130 (2008).
Wharton, K. A., Johansen, K. M., Xu, T. & Artavanis-Tsakonas, S. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 43, 567–581 (1985).
Milner, L. A. & Bigas, A. Notch as a mediator of cell fate determination in hematopoiesis: evidence and speculation. Blood 93, 2431–2448 (1999).
Struhl, G. & Adachi, A. Nuclear access and action of notch in vivo. Cell 93, 649–660 (1998).
Schroeter, E. H., Kisslinger, J. A. & Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 (1998).
Pui, J. C. et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity 11, 299–308 (1999).
Izon, D. J. et al. Deltex1 redirects lymphoid progenitors to the B cell lineage by antagonizing Notch1. Immunity 16, 231–243 (2002).
Wilson, A., MacDonald, H. R. & Radtke, F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J. Exp. Med. 194, 1003–1012 (2001).
Koch, U. et al. Subversion of the T/B lineage decision in the thymus by lunatic fringe-mediated inhibition of Notch-1. Immunity 15, 225–236 (2001).
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (grants NRF-2016R1A5A1007318 and NRF-2017R1E1A1A01074299) and by a grant from the GIST Research Institute (GRI).
Author information
Authors and Affiliations
Contributions
All authors wrote the paper, and S.-G.P. oversaw the drafting of and reviewed the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hwang, JR., Byeon, Y., Kim, D. et al. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med 52, 750–761 (2020). https://doi.org/10.1038/s12276-020-0435-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0435-8
This article is cited by
-
Getting more bang for their buck: BCL2 inhibitors boost dendritic-cell function to enhance anti-cancer immune surveillance
Journal of Translational Medicine (2024)
-
Genome editing iPSC to purposing enhancement of induce CD8 killer T cell function for regenerative immunotherapy
Inflammation and Regeneration (2024)
-
Tumor heterogeneity and immune-evasive T follicular cell lymphoma phenotypes at single-cell resolution
Leukemia (2024)
-
Role of Sam68 as an adaptor protein in inflammatory signaling
Cellular and Molecular Life Sciences (2024)
-
Adoptive cellular immunotherapy for solid neoplasms beyond CAR-T
Molecular Cancer (2023)
