
Abstract
Autism spectrum disorder (ASD) is a prevalent and complex neurodevelopmental disorder which has strong genetic basis. Despite the rapidly rising incidence of autism, little is known about its aetiology, risk factors, and disease progression. There are currently neither validated biomarkers for diagnostic screening nor specific medication for autism. Over the last two decades, there have been remarkable advances in genetics, with hundreds of genes identified and validated as being associated with a high risk for autism. The convergence of neuroscience methods is becoming more widely recognized for its significance in elucidating the pathological mechanisms of autism. Efforts have been devoted to exploring the behavioural functions, key pathological mechanisms and potential treatments of autism. Here, as we highlight in this review, emerging evidence shows that signal transduction molecular events are involved in pathological processes such as transcription, translation, synaptic transmission, epigenetics and immunoinflammatory responses. This involvement has important implications for the discovery of precise molecular targets for autism. Moreover, we review recent insights into the mechanisms and clinical implications of signal transduction in autism from molecular, cellular, neural circuit, and neurobehavioural aspects. Finally, the challenges and future perspectives are discussed with regard to novel strategies predicated on the biological features of autism.
Similar content being viewed by others

Introduction
Autism spectrum disorder (ASD), a group of early developmental disorders, is characterized by deficits in social communication and repetitive stereotyped behaviours. Over the past 80 years, risk factors, diagnostic criteria, clinical treatment options, and societal implications of ASD have attracted the concerns of neuroscientists and clinicians (Fig. 1).

The milestone events associated with autism. Original description of autism was in 1940s, subsequently leading to a series of studies on the definition, diagnosis and treatment of autism in 1960s and 1970s. From the first twin study in 1977, people began to realize that autism as a common highly heritable neurodevelopmental disorder. Up to now, advances in WGS and WES have revealed patterns of inheritance and the types of genetic variation that result in ASD and studies in models have identified a mountain of evidence for molecular mechanisms for ASD. PDD pervasive developmental disorder, EEG electroencephalography, WGS whole gene sequencing, WES whole-exome sequencing
In 1943, Leo Kanner of Johns Hopkins University published “Autistic disturbances of affect contact” in the special issue of the journal The Nervous Child, which systemically examined 11 cases of autism and named it “early infantile autism”.1 Kanner used the term ‘infantile autism’ to describe the children with symptoms of social isolation and linguistic disorders. However, some aspects of Kanner’s views also called the origin of early confusion in the field, such as the vague definition between schizophrenia and autism.2 In 1944, Hans Asperger identified a group children have severe social abnormalities and motor disorders but with very high intellectual functioning.3 This led to the diagnosis of high-functioning autism, that has been incorporated into the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) and the 10th edition of the World Health Organization’s International Statistical Classification of Diseases and Related Health Problems (ICD-10) and named “Asperger’s Syndrome”.4,5,6
In the 1960s and 1970s, early pioneering works on the diagnosis and treatment of autism were initiated. In 1964, Bernard Rimland first began to investigate new approaches to the objective diagnosis of autism.7 In 1972, based on studies of clinical phenomenology, Rutter made clear that autism has significant differences from schizophrenia in terms of onset, clinical symptoms, and family history.8 Rutter’s research also suggested that it would be more plausible to attribute autistic behaviours to developmental disorders from birth to early childhood. By the late 1970s, a consensus emerged about the importance of studying autism independently of schizophrenia, which promoted the updating of diagnostic criteria.9,10 In 1978, Rutter proposed new diagnostic criteria for autism emphasizing social skill dysfunction, language and communication impairment, and repetitive behaviours as three aspects of the basic criteria, abandoning the “special skills and attractive appearance” of Kanner’s criteria.9 The diagnostic approach provided by Rutter directly influenced the revision of DSM-III. In 1980, DSM-III first regarded “infantile autism” as a pervasive developmental disorder (PDD) and focused on early development. Over the same period, studies on intervention and treatment also greatly improved. In 1973, Bartak and Rutter recommended the importance of a structured, behavioural improvement-focused treatment plan.11 Subsequently, an increasing number of behavioural intervention studies have supported the notion that behavioural psychology and special education can be applied to inform autism therapy.
In the 1980s, autism research entered a new era, especially in terms of mechanisms. Autism gradually began to be viewed as a somatic developmental disorder unrelated to parenting styles. Researchers began exploring the aetiology of autism from a biological perspective and completely distinguished autism from schizophrenia on account of clinical symptom recognition and clinical diagnosis. In 1977, Folstein and Rutter’s first study on twins revealed the high heritability of autism.12 Subsequently, with the in-depth understanding of autism, people gradually realized that autism is a developmental disorder under the influence of certain genetic factors.13,14 On this foundation, substantial research into the genesis of autism has been conducted, including molecular genetics, neuroimmunity, functional imaging, neuroanatomy, and neurochemistry research.
ASD is considered to be the result of complex interactions among genetic, environmental, and immunological factors.15,16,17 There have been incredible improvements in the investigation of genetic correlations with autism over the past two decades, ranging from monoclonal gene studies18 to contemporary large-scale studies using whole-genome sequencing (WGS).19 A number of highly reliable and repetitive risk genes have been discovered.20,21 Based on studies of genetically modified mice, considerable progress has been made in illustrating the functions of genes such as Mecp2 (Rett syndrome), Tsc1/2 (tuberous sclerosis), Fmrp (fragile X syndrome), Pten and Shank3 (Phelan–McDermid syndrome) in several monogenetic diseases. These advances in disease mechanism research provide the basis for the design of drugs such as rapamycin (mTOR) inhibitors (tuberous sclerosis22 and fragile X syndrome,23,24) metabolic glutamate receptor (mGluR) antagonists (fragile X syndrome25 and 16p11.2 deletion26), and insulin growth factor (IGF-1) (Rett syndrome27 and Phelan–McDermid syndrome28,29).
In addition to the downregulation of synapse-related genes, microglia and immune-related genes were increased in the brains of autistic patients.30,31,32 The correlations among astrocytes, microglial activation, neuroinflammation caused by gut microbiota and immune dysregulation in ASD patients are also involved in the pathological mechanism.17,33,34,35,36 In particular, infection during pregnancy has been established to induce maternal immune activation that affects the offspring nervous system.37,38
Another pathological mechanism of ASD that has garnered much attention is the functional impairment of brain regions and neural circuits. Autopsies of patients with ASD have revealed significant structural changes in their brains, including altered grey/white matter ratios, increased neuronal numbers, decreased neuronal body volume, increased numbers of glia, and changes in dendritic spines and cerebral blood vessels.39 Additionally, there is established evidence of alterations in glutamate circuits and GABAergic circuits in ASD patients, as manifested by increased numbers of excitatory synapses and spine densities, significantly reduced levels of glutamic acid decarboxylase, and GABAA and GABAB receptor alterations in the postmortem brains of patients with autism.40,41
In this review, we integrate recent advances from genetic, neuropathological, and neurobiochemical studies on ASD to further elucidate the pathogenetic mechanism at the molecular, cellular, and neural circuit levels.
Clinical overview and genetic features
Definition and diagnosis of ASD
Since autism was discovered 80 years ago, its clinical definition and diagnostic criteria have undergone several iterations. In 1980, the DSM-III classified “infantile autism” as one of the generic “PDDs”.42 In 1994, five PDDs were included in the DSM-IV: autism disorder, Asperger’s syndrome, PDD-not otherwise specified (PDD-NOS), Rett syndrome and childhood disintegrative disorder.5 Given the large variability in symptom severity across disease groups, it is difficult to effectively distinguish diseases. To remove this uncertainty, the DSM-5 classifies autism, Asperger’s syndrome, and PDD-NOS as ASD.43 With this revision, the diagnostic criteria have changed as well. ASD is characterized by two main symptoms: deficits in social interaction/communication, as well as repetitive stereotyped behaviours that first occur in early developmental stages and cause clinically substantial impairment.44 Aside from the core features above, individuals with ASD are frequently associated with co-occurring symptoms, including dyskinesia (hypotonia, bradykinesia), speech delay, sleep disorder, gastrointestinal problems, anxiety and epilepsy, which are the most common symptoms in preschool children, while in adolescents and adults, the proportion of depressive symptoms is higher.45,46,47 These comorbidities also pose challenges to disease modelling of ASD, as they may complicate the evaluation of ASD core behaviours in animal models.
The diagnosis of autism is based on thorough consideration of medical history, physical and neurological examination, psychiatric examination, and auxiliary examinations.48 A comprehensive review of the family history of ASD or other neurological disorders should also be included. Autism diagnoses from preschool to mid-childhood are highly stable.49 Due to the complexity, severity, and overlap of ASD features, the correct diagnosis of ASD with instruments and scales is essential for improving the clinical management of patients. Several scales have been suggested that can be helpful for identifying ASD.50
Epidemiology of ASD
Over the past two decades, the prevalence of ASD reported worldwide has been steadily increasing. In 2000, according to the Autism and Developmental Disabilities Monitoring (ADDM), the incidence of ASD was estimated to be 1 in 150 children. In 2006, the incidence was 1 in 110 children, and by 2008, the incidence had increased to 1 in 88 children.50 According to recent estimates, more than 70 million people worldwide have suffered from autism, and the overall estimated prevalence is between 1.5% and 2%.51,52 Modifications in diagnostic criteria and increased awareness of autism in people might be responsible for the surge in autism. Estimates of autism prevalence in different populations and settings vary by definition, sampling, and assessment of independent population cases among studies.
Notably, there is a prominent sex difference in the prevalence of ASD, with prevalences of 2.8% in males and 0.65% in females and a male-to-female ratio of 4.3:1.51,52 This suggests that unknown biological factors may play a role.53,54,55,56 Moreover, a recent study showed an increased female-to-male odds ratio for ASD comorbidities and showed that comorbidity occurrence was associated with the age at first autism diagnosis.57 There may be differences in gene expression induced by gonadal hormones or sex chromosomes in mammals.58 In the brain, more genes are expressed from the X chromosome than from the Y chromosome. The mutations in the X chromosome are generally associated with intellectual disability syndrome, which is more prevalent in males than in females.59,60 The earliest studies on the rare variant of ASD have also tended to focus on the contributions of chromosomal abnormalities in girls. A rare LGD mutation has been found in the NLGN4 and NLGN3 genes, both of which are located on the X chromosome.61 As an X-linked neurodevelopment disorder, Rett syndrome almost exclusively influences females. One possibility is that mutations in Rett syndrome occur almost exclusively on the paternally derived X chromosome and are lethal in male embryos.62 In general, the contribution of gender aetiology to autism remains largely unexplained. Human studies have only identified minor sex variations in cerebral cortical gene expression.63,64,65,66 Resolving sex differences is a significant aspect in the process of ASD and shows great potential for the development of widely applicable therapies. Many psychiatric disorders, including ASD, will probably be better understood if key sex differences in cellular and molecular events during brain differentiation can be identified.
Genetic architecture of ASD
Twin and family studies have consistently suggested that autism have a strong heritability.14,67,68 Recent advances in genetic technology, microarrays, WGS, and whole-exome sequencing (WES) have revealed patterns of genetic variation that result in ASD.19,69,70 Here, we highlight the contributions of inheritance patterns, variation types and epidemic rates to ASD (Fig. 2). Heritability measurements have been derived from investigations on identical twins, fraternal twins and sibling concordance, including a survey of more than 2 million Swedish households in 2014,71 which is the largest human-based ASD study to date, eventually estimating the heritability of ASD as ranging from 52% to 90%.68,72,73 Moreover, the epidemiological and molecular data suggest that the genetic contribution of ASD results from the combination of rare deleterious variants and a large number of low-risk alleles.74 Therefore, different phenotypes can arise because prevalent low-risk alleles buffer the effects of detrimental variantion.74,75,76

Genetic architecture of autism spectrum disorder (ASD). a The inheritance patterns of high-risk gene and syndromes associated with ASD. Major gene model includes autosomal recessive, autosomal dominant and X-linked inheritance patterns. The red stars indicate a causal allele. b The shown types of genetic variation including SNP and CNVs. Genes and syndrome that have been associated with ASD are also indicated. SNP single-nucleotide polymorphisms, CNV copy number variation. (Adapted with permission from reference15)
The genetic structure of ASD is extremely complex. Approximately 600–1200 genes and genomes have been identified that associated with autism.77 At least 5% of ASD cases are caused by single-nucleotide polymorphisms (SNPs) in genes such as NLGN3, NLGN4, NRXN1, MECP2, SHANK3, FMR1, TSC1/2 and UBE3A.78,79 In addition, rare de novo mutations of CHD8, SCN1A, SCN2A, SYNGAP1, ARID1B, GRIN2B, DSCAM, TBR1, KATNAL2, LAMC3 and NTNG1 have been identified, with strong evidence for their association with ASD.78,80,81,82 Approximately 10% of them are copy number variations (CNVs) that disrupt protein coding, including chromosomal duplications, large deletions, inversions, and translocations, such as 1q21.1 duplications or deletions, 3q29 deletions, 7q11.23 duplications, 15q11-q13 deletions, 15q13.3 microdeletions, 15q11-13 duplications, 17q12 deletions, 22q11.2 deletions and 22q13.33 duplications or deletions.78,83,84 Mutations located in intronic and intergenic regions are the third variation type of ASD.85
ASD is thought to contain two subtypes: syndromic and non-syndromic forms. Syndromic generally refers to mutations in a specific gene or genome, manidesting as neurological syndromes (such as fragile X syndrome, tuberous sclerosis, Rett syndrome, Phelan–McDermid syndrome and Angelman syndrome).79,85 Non-syndromic, also regarded as idiopathic, which accounts for the vast majority, is not associated with other neurological disorders (or syndromes) but is related to some genes associated with autism.85 In heterogeneous genetic structures, syndromic ASD caused by high-penetrance single-gene mutations represent only a minority of ASD cases, the majority of cases are idiopathic.86 In fact, due to the overlap of phenotypes and growing understanding of intersecting biology, it remains controversial that the definition and boundary between syndromic and idiopathic ASD. With the advance of genetics, more efforts have been invested in identifying individuals with rare mutations of same gene and the convergence among them. Some retrospective analysis of gene fragments (for example, CDH8 and ADNP) from individuals with typical idiopathic ASD has revealed different clinical phenotypic features.87,88 This suggests significant variability in the symptoms, as well as the persistence of previously overlooked syndromes in idiopathic ASD. Therefore, continuous and holistic analysis rather than isolated studies may help us better comprehend ASD.
Neurobiological mechanisms of ASD
Due to the above unknown factors and challenges, many genetic variations associated with ASD have been suggested to be possibly concentrated on common molecular or cellular pathways. Key literature from recent years has suggested that ASD-associated genes enriched in aspect of transcription and translation, synapse, epigenetics, immunity and inflammation. These are closely related to the occurrence, development and outcome of autism. The first category is the dysregulation of important transcripts and translational signalling pathways.15,89,90 The second category involves synaptic proteins, including cell adhesion, scaffolding, and signalling molecules, which can affect synapse structure and function during different processes of synapse formation, elimination, transmission, and plasticity.89,91,92 The third category is the overtranslation of certain transcripts, which can lead to widespread epigenetic dysregulation, creating a positive feedback loop between translation and transcription processes that exacerbates neuronal dysfunction in ASD.93 The immunoinflammatory response caused by the activation of reactive glial cell proliferation and intestinal flora dysbiosis can be classified into the fourth type of abnormal signal transduction.94,95 These types of signalling pathways can interact or participate in the pathophysiology of ASD in a cascading manner rather than acting independently. For example, alterations in Wnt signalling, alterations in neuronal translation and defects in synaptogenesis or synaptic function during brain development can all affect the formation and activity of neural circuits.96,97 In turn, altered neural activity can further influence transcription factors or chromatin remodelling by transmitting action potential cascades that trigger signals and initiate specific transcriptional programmes.89,98
Numerous animal genetic models of autism have been developed and characterised as a result of genetic advances, allowing relevant phenotypes and mechanisms to be discovered and further studied (Table 1). Mouse models have provided a mountain of evidence for molecular pathways in autism, especially in translation and synaptic function.15 Manipulation of individual risk genes in model systems may lead to identification of important phenotypes. Although they cannot completely simulate the pathological process of human beings, these techniques still help us to understand the occurrence and development of autism. Stem cell models have also demonstrated that abnormalities in specific molecular processes contribute to the pathogenesis of ASD (Table 2), including chromatin remodelling, Ca2+ and Wnt signalling.99,100 In recent years, accumulated evidence from modelling studies has identified many specific types of viable mutations, which may paint a bright picture for elucidation of the underlying pathogenesis of ASD.
Activity-dependent gene transcription and mRNA translation
Neuronal activity regulates gene transcription and mRNA translation in a dynamic manner.101,102,103 Many transcription factors and de novo mutations associated with ASD are thought to regulate or engage in cross-talk with canonical Wnt signalling, such as CHD8 and CTNNB1. Disorders in several upstream signalling pathways of translation, including mTOR, Ras and MAPK pathways, contribute to increased protein synthesis and therefore to altered synaptic plasticity (Fig. 3).

Transcription factors and translation mechanism associated with ASD. Activity-regulated translational pathways including the Ras/ERK and PI3K/mTOR. Both of them could be activated upon the stimulation of TrKB. Activation of L-type voltage-sensitive calcium channels (L-VSCCs) triggers calcium influx, induction of calcium-dependent signalling molecules and Ras/ERK pathways, involving in transcriptional regulation. These signalling cascades transcription regulators in the nucleus lead to the expression of transcription factors, thereby contributing to the regulation of activity-dependent gene transcription. Mutations of proteins involved in transcriptional regulation are associated with some syndromes of ASD, including L-VSCC in Timothy syndrome, MeCP2 in Rett syndrome and UBE3A in Angleman syndrome. Mutations of proteins involved in translation regulation including PTEN, ADNP, EN2, TSC1/TSC2 (tuberous sclerosis) and FMRP (fragile X syndrome). These genes have been highlighted in red
Activity-dependent gene transcription
Neuronal activity regulates programmes of gene expression in the nucleus, and disruption of activity-dependent transcriptional regulators or their targets is associated with ASD. Such disruption includes mutations in methyl-CpG-binding protein 2 (MeCP2),104,105 activity-dependent neuroprotective protein (ADNP),106 engrailed 2 (EN2),107 voltage-dependent calcium channel subunit α1C (CACNA1C),108 T-box brain 1 (TBR1),109,110 myocyte enhancer factor 2C (MEF2C)111 and de novo deletions or duplications in 15q11-q13 (which cover ubiquitin-protein ligase E3A (UBE3A)).112
MeCP2 deletions or point mutations on the X chromosome in females manifest as Rett syndrome, a serious neurological disorder with autism-like symptoms.104 This is consistent with observations in model mice. Mecp2308/Y mutant mice exhibit ASD-like deficits in social behaviour and learning.105,113 MeCP2 is a transcriptional repressor which covers almost the whole genome, and its deletion raises overall transcriptional levels and accompanies with modification of the entire chromatin structure.114,115 Neuronal activity, brain-derived neurotrophic factor (BDNF), or drugs that increase intracellular 3’,5’-cyclic AMP (cAMP) levels induce MeCP2 phosphorylation and dissociation of the nuclear receptor corepressor (NCOR) complex, thereby enabling transcription.116,117,118 Notably, several studies have shown that MeCP2 binds with chromatin and transcriptional activators at the promoter of an activated target to activate gene expression, which means that MeCP2 can operate as both an activator and a repressor of transcription.119,120
Common genetic variations and rare mutations in genes encoding calcium channel subunits have extensive impact on the risk of ASD. For example, mutations in the L-type calcium channel Ca(v)1.2 generate Timothy syndrome, a monogenic disorder with a high penetrance for ASD.108 Transcriptional changes regulated by a series of calcium-dependent transcriptional regulators, including NFAT, MEF2, CREB, and FOXO, are found in Timothy syndrome.99 ADNP directly encodes a transcription factor and can bind and regulate ZFP161, which serves as a transcriptional activator of dopamine transporter (DAT; SLC6A3), interleukin 6 (IL-6), and leukaemia inhibitory factor (LIF) and a transcriptional repressor of FMR1.121 MEF2 is an activity-regulated transcription factor that regulates genes implicated in ASD, such as ARC, PCDH10, UBE3A and BDNF.111,122,123 The gene encoding the UBE3A is mutated in Angelman syndrome patients and duplicated on the maternal chromosome 15q11 in some ASD patients.124 Neuronal activity can promote the translation of UBE3A through the MEF2 complex.125 TBR1 is a neuron-specific transcription factor required for activity-dependent Grin2b expression, loss of a copy of which alters the expression of Ntng1, Cntn2 and Cdh8.109,110
Notably, the majority of the targets of the above-discussed transcription factors also show crucial effects in synaptic transmission and plasticity, which may explain why transcription and translation can modulate synaptic function in the aetiology of ASD.110,126,127,128
Wnt signalling pathway
The Wnt signalling pathway has long been implicated in neuronal overgrowth, and its alterations are thought to be pleiotropic in the aetiology of autism.129 Molecular, cellular, electrophysiological, and behavioural abnormalities in accordance with autism-like phenotypes in several Wnt signalling-related knockout mouse models.130,131 In the brain, there are two primary pathways for Wnt signalling: (1) β-catenin-dependent stabilized “canonical” signalling and (2) β-catenin-independent “noncanonical” signalling.96 Notably, many key proteins in both signalling pathways are localized at synapses and play key roles in synaptic growth and maturation.132,133,134 Canonical Wnt signalling acts indirectly on β-catenin to enhance its stability, allowing it to translocate from the cell surface to the nucleus, thereby linking extracellular signalling to nuclear gene expression regulation through downstream transcriptional machinery (Fig. 3).72 On the one hand, ASD-associated MET tyrosine kinases (such as CDH8) release β-catenin to bind to surface calcium.135 On the other hand, free cytoplasmic β-catenin is phosphorylated by GSK3β to reflect the level of proteasomal degradation.129 Multiple Wnt molecules, including Wnt2, transmit signals at the surface membrane by interacting with frizzled receptors and LRP5/6 coreceptors.136
It is noteworthy that the gene CTNNB1, which encodes β-catenin, has been identified among ASD risk variation.137 CDH8 is one of the best examples of an autism-related chromatin modifier that regulates the expression of other autism risk genes.130,138 As a negative regulator, CDH8 participates in the canonical Wnt signalling pathway by directly binding to β-catenin or being recruited to the promoter regions of β-catenin-responsive genes.139 This is consistent with the hypothesis that elevated canonical Wnt signalling contributes to the hyperproliferation of embryonic neural progenitor cells (NPCs) in the brain, which may partially explain the macrocephaly observed in individuals with autism.88,100,140,141 However, some studies have also found that CHD8 is a positive regulator of the Wnt/β-catenin signalling pathway in NPCs and negatively regulates this pathway in nonneuronal cell lines, suggesting that CHD8 may regulate Wnt signalling in a cell-specific manner.130
In addition, PTEN participates in Wnt signalling by working with β-catenin to regulate normal brain growth.142 A dynamic trajectory of brain overgrowth and elevated β-catenin signalling has been reported in the developing cerebral cortex in Pten-haploinsufficient mice, highlighting the roles of Pten and β-catenin signalling in regulating normal brain growth.142
Activity-dependent mRNA translation and protein synthesis
Several activity-regulated translational control pathways have been demonstrated to participate in pathologies of autism, such as the ERK/MAPK (mitogen-activated protein kinase)143 and PI3K/mTOR (mammalian target of rapamycin) pathways.144,145 Mutations in several genes, such as TSC1, TSC2, PTEN and FMR1, are canonical components involved in the mTOR pathways and play crucial roles in mRNA translation and protein synthesis.146,147,148
Tuberous sclerosis is an autosomal dominant disorder arising from heterozygous mutations in the TSC1 and TSC2 genes that is commonly associated with deficits in long-term and working memory, intellectual disability, and ASD.22,149,150 TSC1 acts as a regulator of the stability of TSC2, preventing the degradation of TSC2, while TSC2 is a GTPase activating protein (GAP) that inactivates Rheb, a GTPase of the Ras family, and other small G proteins.151 Activated AKT can phosphorylate and inhibit TSC2, which regulates translation, transcription, and other cellular processes by removing the inhibition of mTORC1 by the TSC1/2 complex and promoting mTORC1 activity.151 In the absence of a functioning TSC1/2 complex, overactive mTORC1 leads to unrepressed protein synthesis and subsequent cell growth.152,153 It is worth mentioning that a major activator of TSC1/2 signalling is BDNF, a secreted protein that binds to the receptor tyrosine factor TrKB and is thereby involved in the PI3K/mTOR pathway.154,155 PTEN is an ASD risk gene located on chromosome 10q23 that encodes a lipid specific for phosphatidylinositol (3,4,5)-triphosphate (PIP3), which is a negative regulator of PI3K/AKT/mTORC1 signalling upstream of TSC1/TSC2, resulting in symptoms of ASD. Mutations that inactivate PTEN lead to a constitutively active PI3K/AKT/mTOR signalling pathway and ultimately may result in abnormal protein synthesis.156
FMRP loss of function causes fragile X syndrome and autistic features, which is the most commonly known single-gene cause of ASD.157 FMRP is an RNA-binding protein whose target mRNAs encode transcription factors, and chromatin modifiers have been identified by high-throughput sequencing of RNA isolated with cross-linking immunoprecipitation (HITS-CLIP).148,158,159,160,161 The target genes of the mRNAs include several well-studied autism candidate genes, such as ARC, NLGN3, NRXN1, SHANK3, PTEN, TSC2 and NF1.23,148,162,163,164,165 Notably, the proteins encoded by FMRP target mRNAs regulate the balance of activity-dependent translation in synaptic plasticity.148 The proteins include mGluR5 and the NMDAR subunits, consistent with findings of altered mGluR5 and NMDAR-dependent synaptic plasticity in fragile X syndrome mouse models.166 Moreover, mGluR activation regulates FMRP-mediated translational repression, whereas FMRP regulates AMPAR trafficking and mGluR-mediated LTD.167 Regarding the link between translation initiation and autism, FMRP interacts with cytoplasmic FMRP-interacting protein 1 (CYFIP1), which binds to the cap-binding protein eukaryotic initiation factor 4E (eIF4E) to form a protein complex that inhibits mRNA translation initiation and acts on the RAS-ERK pathway.168,169 Notably, the FMRP-eIF4E-CYFIP1 complex regulates the translation of more than 1000 genes, many of which are ASD risk genes.170,171,172,173 In addition, several transcriptional regulators, such as ADNP and ENP, also impact translation by interacting with eIF4E.121,174
In summary, current evidence suggests that there is a complex level of dynamic regulation between translation and transcription that likely contributes to ASD pathophysiology. Interestingly, most mutations in translation pathways such as mTOR, ERK, and FMRP-eIF4E-CYFIP lead to abnormally high levels of synaptic translation and synaptic proteins. This is one of the few convergences seen in the heterogeneous context of autism and provides a good foundation for pharmacological target development. Moreover, determining the dynamics of spatio-temporal relationship between transcription and translation will help us to link the molecular dysfunction to the complex behavioural characteristics of ASD patients.
Synaptic function
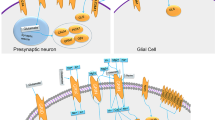
A growing number of genes that have been associated with ASD seem to play roles in synaptic structure and function by directly encoding synaptic scaffold proteins, neurotransmitter receptors, cell adhesion molecules, and actin cytoskeletal dynamics-related proteins (Fig. 4).74,175 Therefore, abnormalities in synaptic proteins might be some of the mechanisms that increase the risk of developing ASD. Among the synaptic proteins, cell adhesion molecules (neuroligins (NLGNs)176 and neurexins (NRXNs)61), postsynaptic scaffolding proteins (SH3 and multiple ankyrin repeat domains protein (SHANK),177 glutamate receptors (NMDAR subunit, GluN2B),178 inhibitory GABAA receptor subunits α3 and β3 (GABRA3 and GABRB3, respectively)179 and permeable ion channels (voltage-dependent calcium channel subunit α1C (CACNA1C)180 and sodium channel protein type 1 subunit-α (SCN1A)181) are reported to be important signal transduction molecules associated with ASD. Signalling changes in these proteins can modulate the strength or number of synapses and ultimately alter the structure and functional connectivity of neuronal networks in the brain.

Molecular pathways implicated in synaptic function for ASD. At the excitatory synapse, encoded proteins including synaptic scaffold proteins (for example, SHANKs), neurotransmitter receptors (for example, NMDARs, AMPARs and mGluRs) and cell adhesion molecules (NRXNs and NLGNs) associated with autism risk genes. Activation of cell surface receptors is closely linked to activation of the Ras/ERK and PI3K/AKT/mTOR pathways. In addition, mutations in ion channels, such as L-VSCCs and sodium channel protein type 1 subunit-α (SCN1A), both of which have been illuminated result in synaptic dysfunction and autism-like behaviour
Synaptic structure and homoeostasis
Intact synaptic structure and homoeostasis are fundamental for the normal function of the brain. Neuropathological studies have provided evidence of increased dendritic spine density and aberrant dendritic spine morphology in individuals with ASD.182,183 Moreover, reduced developmental synaptic pruning in layer V pyramidal neurons in the postmortem ASD temporal lobe has been shown to hyperactive mTOR and defective autophagy.146 At excitatory synapses, the molecular diversity of surface receptors impacts proper synapse formation, maturation and transmission by organizing clustering of interaction partners at postsynaptic regions. For example, the intracellular carboxy-terminal portions of cell adhesion molecules (NLGNs) can bind to several scaffolding proteins of the postsynaptic density, such as postsynaptic density protein 95 (PSD95) and SHANKs.184,185 SHANK3 can interact with PSD95, AMPA receptor and glutamate receptor 1 (GluR1), which is critical for dendritic spine formation and synaptic transmission.186,187
NRXNs and NLGNs are presynaptic and postsynaptic binding partners that cooperate to form transsynaptic complexes that directly mediate synapse formation and stabilization but are abnormally manifested during autism pathology.61,176,188 Whereas NLGN-1, NLGN-3 and NLGN-4 localize to the glutamate postsynaptic membrane, NLGN-2 localizes primarily to GABA synapses.189,190 NLGNs can participate in the formation of glutamatergic and GABAergic synapses in an activity-dependent manner.189 Specifically, inhibition of NMDARs or the downstream protein CaMKII suppresses the formation of glutamatergic synapses through the activity of NLGN1, whereas inhibition of NLGN2 activity suppresses the formation of GABAergic synapses.189,191,192 Various combinations of these cell adhesion molecules have been linked to the differentiation of glutamatergic or GABAergic synapses in Nlgn-3 and Nlgn-4 mutant mice.193,194,195,196,197 In addition to alterations in NLGNs, mutations in NRXNs result in extensive changes in synaptic structure and plasticity.198,199 Moreover, NRXNs are critical for Ca2+-triggered neurotransmitter release but are not required for synapse formation, which has also been demonstrated in knockout mice.198,199
SHANK genes, including SHANK1, SHANK2 and SHANK3, directly encode the proteins in the postsynaptic scaffolding protein family, which are located in the PSDs of excitatory synapses.177 SHANKs were first implicated in ASD by studies on Phelan–McDermid syndrome,200,201 a neurodevelopmental disorder caused by 22q13.3 deletion, and are deleted in almost all reported Phelan–McDermid syndrome cases. Consistent with studies in humans, different studies on Shank mutation sites in mice have also confirmed the strong genetic associations between Shank genes and ASD, especially Shank3.202,203,204,205,206,207,208 Individuals with ASD with SHANK3 mutation exhibit defects in dendrite development and morphology and axonal growth cone motility.209,210 Shank3-knockout mice showed a decrease in the number of corticostriatal connections,202,211 whereas defects in NMDAR-dependent excitatory neurotransmission and synaptic plasticity have been observed in Shank2-knockout mice.207
In addition, recent genome-wide association studies have linked polymorphisms and rare variations in ion channels and their subunits to ASD susceptibility. Haploinsufficiency of SCN1A encoding the voltage-gated sodium channel Na(V)1.1 causes Dravet’s syndrome, which has been proven to result in the display of autism-like behaviour.181 The Na(V)1.1 channel is the major Na+ channel expressed in the somata and axon initiation segments of excitatory and inhibitory neurons in the brain.212,213,214 In GABAergic interneurons, Na currents and action potential firing are harmed when Na(V)1.1 is deleted.181,215 Calcium channels act as sensors electrical activity sensors, converting membrane potential changes into protein conformational changes and transmitting information about neuronal activity to downstream effector systems.
There is clear evidence to illuminate that defective Ca2+ channel function can lead to ASD with penetrance as high as 60-80%.216 Mutations relevant to ASD typically sensitize voltage-dependent Ca2+ channel gating, shifting their activation to more hyperpolarized potentials of ~10 mV.217,218 CACNA1C and CACNA1D encode the Ca(V)1.2 and Ca(V)1.3 proteins, respectively, which localize to the postsynaptic membrane and signal to the nucleus.99,219 In excitatory neurons, CaMKII functions as a shuttle molecule to collect Ca2+/Calmodulin from the cytoplasm and transport it to the nucleus, where Ca2+/Calmodulin release activates CaMKK and its substrate CaMKIV to further phosphorylate CREB, thereby participating in the regulation of transcription and translation.72,220,221
Synaptic signalling pathways
Neuronal activity-dependent synaptic mRNA translation pathways can directly influence the levels of synaptic proteins, thereby controlling synaptic strength and number.102 The extracellular mTOR and FMRP-eIF4E-CYFIP1 signalling pathways are the two primary regulators of mRNA translation.15 Interestingly, the majority of ASD-related gene mutations (such as MEF2C, FMR1, PTEN, TSC1, TSC2 mutations) result in enhanced gene transcription and mRNA translation, ultimately leading to an aberrant increase in the strength or number of synapses within certain neural networks. In fact, glutamate and BDNF can also induce a cascade of mTOR and FMRP pathways, resulting in an increase in mRNA translation.74 Consistently, increased glutamate and BDNF levels have been found in the blood of ASD patients.222,223
Moreover, activation of cell surface receptors such as NMDARs, AMPARs, mGluR, IGFR and TrKB is closely linked to activation of the ERK/MAPK and PI3K/mTOR pathways (Fig. 4). Among them, mGluRs are located in the perisynaptic zone of excitatory synapses, ideally contributing to orchestrating AMPARs and NMDARs.224 Mechanistically, mGluRs can directly regulate glutamatergic signalling by anchoring in complexes with SHANK and HOMER proteins and further control the synthesis of synaptic proteins via activation of the PI3K/AKT/mTOR pathways.225 In addition to being involved in dendritic protein synthesis, activation of mGluRs can also stimulate long-term depression (LTD), which is accompanied by rapid loss of both AMPA and NMDA receptors.72 Interestingly, several ASD animal models, including Fmrp-mutant,167 Mecp2-mutant,113 Tsc1/2-mutant,226 Pten-mutant,227 Shank3-knockout,211,228 Nlgn3-knockout229 and 16p11.2-knockout models,26 have shown dysregulation of mGluRs and abnormal mGluR-dependent LTD. There are encouraging signs that some pharmacological manipulations of mGluR have shown initial success in restoring impaired LTD and improving ASD-related behaviours in mouse models.211,228 These will be detailed in the section “THERAPEUTIC STRATEGIES”.
In addition, proteinases play posttranslational roles by regulating the activity-dependent cleavage of postsynaptic adhesion molecules at glutamatergic synapses. For example, the cleavage of NLGNs is triggered by NMDA receptor activation and is mediated by the proteolytic activity of matrix metalloprotease 9 (MMP9).230 The ubiquitin–proteasome system is required for the degradation of AMPA receptors, which influence synaptic elimination and plasticity.231 UBE3A modulates excitatory synapse development by regulating the degradation of ARC, which reduces LTP by promoting the internalization of AMPA receptors.232 Several studies have demonstrated that loss of function of UBE3A leads to increased ARC expression and subsequently decreases the number of AMPARs, ultimately impairing synaptic plasticity at excitatory synapses.232,233
Epigenetic factors
Increasing evidence indicates that ASD is the result of a complicated interaction between genes and the environment.234 Epigenetic factors are ideally positioned at the genome-environment interface, allowing for steady gene expression regulation without alterations to the underlying DNA sequence.93,235,236 Epigenetic mechanisms, including DNA methylation, histone modification, chromatin remodelling, and non-coding RNA activity, are involved in the regulation of social behaviour in autism.93,237,238,239 Together, these mechanisms form an epigenetic network that integrates transient social experiences into the genome to regulate social–emotional dispositions in mammals (Fig. 5).

The epigenetic network associated with ASD pathophysiology. a Despite the exceptions, DNA methylation usually leads to transcriptional repression or even silencing of the affected gene. MeCP2 binds to methylated CpG sites in gene promoters and associates with chromatin silencing complexes, thereby suppressing gene expression. b Histone modification and chromatin remodelling cause transcriptional activation or inactivation, and chromatin packaging. c Non-coding RNAs control the expression of genes at the level of post-transcription by blocking protein synesis or inducing mRNA degradation
DNA methylation
Many epigenetic researches have focused on DNA methylation with consideration of the contact between genes and environmental factors.240,241,242 Early studies on ASD-associated DNA methylation focused on several candidate genes, such as MECP2, glutamate decarboxylase 65 (GAD65), reelin (RELN), oxytocin receptor (OXTR), SHANK3 and UBE3A.
MeCP2 is a chromatin architectural regulator and a reader of epigenetic information contained in methylation (or hydroxymethylated) DNA that has been well studied.243 Decreased MeCP2 expression in the PFC in ASD patients is associated with aberrant hypermethylation of its promoter.244,245 MeCP2 binds to methylated CpG sites in gene promoters and associates with chromatin silencing complexes, thereby suppressing gene expression.246,247,248 GAD1 and RELN are downregulated in postmortem ASD and are selectively expressed in GABAergic neurons.249 Enhanced binding of MeCP2 to GAD1 and GAD2 promoters, which leads to reduced expression of RELN and mRNA, has been found in the cerebellum and frontal cortex in ASD patients.249,250 While the methylation rate of CpG islands is elevated during mouse brain development, SHANK3 is upregulated two weeks postnatal, suggesting that methylation of CpG islands is a strong regulator of SHANK3 expression.251 The neuropeptide oxytocin (peptide: OT, gene: OXT) sends signals via its receptor OXTR, which is a highly conserved G protein-coupled receptor. Both genetic and epigenetic changes in OXTR have been identified to be related to ASD.252,253,254,255 OXTR mRNA expression is affected by methylation of promoter, and high levels of methylation have been associated with ASD.252,256 Consistent with this, a study on siblings and adults with ASD found increased OXTR promoter methylation.257,258
Taken together, the findings indicate that DNA methylation status may serve as a potential biomarker for risk prediction, diagnosis, and targeting, as well as provide information for the study of ASD pathological mechanisms. Highly specific DNA methylation has been identified that may help predict transcriptional regulation in autism.93
Histone modification and chromatin remodelling
Recent studies have revealed a characteristic histone acetylation signature in the brains of ASD patients, providing strong evidence that histone modifications, especially acetylation, lead to ASD-like behaviours.259 A cross-generational study has confirmed that children exposed to prenatal anticonvulsants and the mood stabilizer valproate, a well-known histone deacetylase (HDAC) inhibitor, are at increased risk of being diagnosed with autism, providing insights into the involvement of histone modifications in ASD.260,261 Furthermore, treatment with a histone deacetylase inhibitor in Shank3-knockout mice significantly improves the behavioural phenotype of the mice, suggesting that abnormal histone modification is a potential mechanism of ASD.262 Trimethylation of the fourth lysine residue of histone H3 (H3K4me3) is essential for chromatin formation and gene activation, regulating hippocampal plasticity by recruiting chromatin remodellers to gene transcription initiation sites.263,264 H3K4me3-ChIP deep sequencing of the prefrontal cortex in postmortem tissue from patients aged 6 months to 70 years has revealed that alterations of H3K4me3 levels in neurons are associated with autism.265 Mutations in the lysine-specific demethylase 5 C (KDM5C) gene damage its function of transcriptional regulation, resulting in reduced H3K4me3 methyl group removal and suppressed gene expression in ASD patients.266,267,268
Chromatin remodelling is mediated via ATP-dependent enzymes or chromatin remodelling complexes.269 The chromatin structure or proteins that bind to DNA are altered when nucleosomes positioned differently, causing gene expression to shift. Chromatin remodelling genes (including CHD8, ARID1B, BCL11A and ADNP) have been identified to be linked to autism.106 De novo mutations in the autism-related chromatin modifier CHD8 are well studied,88,270 with multiple de novo, truncating, or missense mutations observed in ASD patients.81,82,88,130 CHD8 is located at active transcription sites with the histone modification H3K4me3 or H3K27ac and recruits histone H1 to target genes by remodelling the chromatin structure.141,270 ARID1B is a component of SWI/SNF (or BAF), an ATP-dependent human chromatin remodelling complex that is frequently mutated in ASD.89,271 Proteins encoded by BCL11A and ADNP can also interact directly with members of the SWI/SNF complex, which is related to alternative splicing of tau and prediction of tauopathy.106,272
Non-coding RNAs
The majority of genome-wide association studies have concentrated on protein-coding regions, disregarding non-coding RNA. Because non-coding RNAs primarily target transcripts and rarely interact directly with DNA, they are considered nonclassical epigenetic pathways.93,273 Posttranscriptional regulation by non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), is associated with ASD. miRNAs are short non-coding RNA molecules that regulate the expression of most genes by blocking protein synthesis or increasing mRNA degradation at the posttranscriptional level. A preliminary assessment suggested that autism does not induce global dysfunction of miRNA expression, as only 28 of 466 miRNAs were significantly altered in postmortem cerebellar cortex tissue of ASD patients.274 Interestingly, the predicted targets of the differentially expressed miRNAs were enriched with genes related to neurobiology, the cell cycle, and cell signalling and largely overlapped with genes previously identified via differential mRNA expression analysis of ASD patients.30,275 Considering that miRNAs can be delivered into cells without being integrated into the host genome, miRNA-based therapy is a prospect strategy for the treatment of ASD.237 Highly expressed miRNAs in ASD patients can be downregulated by miRNA antagonist treatment (i.e., miRNA-inhibitory therapy), while miRNA mimic replacement therapy can compensate for weakly expressed miRNAs.276 Compared with mRNAs, lncRNAs exhibited higher tissue-specific expression, and a considerable number of lncRNAs were confined to the brain.277 The evolution of lncRNA-specific and synaptic function-enriched gene expression in primates suggests that this category of RNAs may have a broad range of roles in the brain and may help to elucidate the aetiology of ASD.31,278,279
In animal studies, mice with heterozygous knockout of miR-137 show repetitive behaviours and social behavioural deficits.280 Another example of the use of miRNA profile screens in a genetic model of ASD comes from a study on Mecp2-knockout mice. Expression profiling of miRNAs in the cerebella of Mecp2-knockout mice revealed the downregulation of a subset of miRNAs.280 Moreover, some of these miRNAs targeted BDNF, which is consistent with the finding that miR-132 targets MeCP2 and BDNF in vitro and is downregulated in the cortices of Mecp2-knockout mice.281,282 Therefore, the regulatory loop including BDNF, miR-132 and MeCP2 may be involved in ASD.237,282 The deletions in regions of differentially expressed lncRNAs are similar to those reported for miRNAs and mRNAs.30 BC1 is an lncRNA whose deletion in the mouse cortex can cause social dysfunction. The underlying mechanism is that BC1 tends to increase the affinity of FMRP and CYFIPI, both of which are ASD risk genes.168,283,284
In general, many differentially expressed and functionally significant non-coding RNA genes and overall epigenetic disorders have been identified in ASD patients and animal models. Preliminary evidence for a relationship between epigenetic regulation and social behaviour has been obtained at the animal level. Nevertheless, the epigenetic network is intricate, and the recently discovered genes with differential expression may be just the tip of the iceberg in the context of ASD. The important topic is how social stress induces temporary changes in the epigenetic network and whether gene expression might contribute to long-term social–behavioural adaptations. Future studies need to further identify more brain-specific epigenetic regulatory genes and clarify their practical functional significance.
Immunology and neuroinflammation
Immune dysfunction is another factor attributed to gene–environment interactions in the context of ASD. Persistent immune dysregulation has been identified in ASD patients and animal models.37,94,285,286 An earlier study identified 150 differentially expressed genes in ASD patients compared to controls, 85% of which were upregulated and involved in immune response pathways.275 Inflammatory molecular signalling pathways in both the central nervous system and the periphery can affect brain connections and synaptic function by affecting components including microglia, complement factors, cytokines and their receptors, MET receptors, and major histocompatibility complex class I molecules (MHCI) (Fig. 6).36

Mechanisms underlying the effects of microbiota, immunology and neuroinflammation on ASD. In periphery, microbiome and immune disorders in individuals with autism can lead to the change of peripheral immune environment. In the brain, abnormal proliferation and activation of glial cells can induce the secretion of cytokines and may cause vascular-endothelial dysfunction. Disorders in the periphery and brain all can affect brain functional connections and density of dendritic spines. Alterations in expression of immune mediators in the brain and at synapse, including cytokines and MHCI molecules. Notably, glutamate and cytokine receptors downstream signalling may converge upon the mTORC1 pathway, further regulating translation, synapse formation and plasticity. MHCI major histocompatibility complex class I molecules, mTORC1 mammalian target of rapamycin complex 1
Alterations of immune mediators in the central and periphery
In the brains of ASD patients, the numbers and activation of reactive microglia and astrocytes are increased in multiple brain regions.30,287,288,289,290,291 A cascade of cytokines and chemokines can be released by reactive microglia and astrocytes, which can signal across cells. Dysregulation of cytokines in ASD has also been associated with symptom severity and presentation on diagnostic tests for ASD.292 Therefore, abnormal cytokine profiles may be sensitive biomarkers indicative of immune system disturbances and abnormal neuroinflammation in autism. Some studies have found increases in GM-CSF, IL-6, IL-8, TNF-α, TGFβ, CCL2 and IFNγ levels in the brains of individuals with ASD, which supports this theory.287,293 Paralleling findings in humans, findings from several established animal models of ASD, including offspring with maternal immune activation (MIA) (IL2, IL6 and IL17)294,295,296 and offspring of VPA-treated rodents (TNF-α and IL-6),297 and the naturally occurring BTBR strain (IL-33, IL-18, IL-1β and CXCL7)298,299 have also shown alterations in the secretion of cytokines and chemokines. Due to the secretion of signalling molecules and cytokines, the cross-talk between microglia and astrocytes is enhanced, which can lead to vascular-endothelial dysfunction and damage to blood–brain barrier (BBB) permeability.94,300 Some cytokines, such as IL-1α, IL-1β, IL-6 and TNF-α, can migrate from the periphery into the brain via the BBB transport systems.301
Moreover, multiple studies have indicated different expressions of cytokine and chemokine in the periphery in autism patients.94 The results of cerebrospinal fluid and blood tests of ASD samples are similar, and cytokine changes in the blood can potentially provide information on inflammation and alterations in synapse connectivity in the brain. The levels of proinflammatory cytokines (such as IL-1β, IL-6, IL-8, IL-12p40, IFN-γ, TNF-α and GM-CSF) are increased, while those of anti-inflammatory cytokines (such as IL-10 and TGF-β) are decreased, in the blood of ASD patients.302,303,304 However, some alterations in cytokines are different between the central and peripheral regions, including IL-1β and TGF-β. In the CNS, IL-1β levels appear to be unchanged, but they have increased in the periphery.293 TGF-β1 levels have been reported to be rising in one study, while the vast majority of data point to a decline in TGF-β1 levels in peripheral blood.287 Hence, additional studies with persuasive datasets are warranted to confirm whether higher blood IL-1β levels influence CNS levels and whether TGF-β1 has dual roles in the brain and periphery in autism.
Notably, maternal autoimmune disorders, including autoimmune disorders (such as fever, allergies and asthma) and external exposures (such as mercury, lead, air pollutant, pesticide, and PCB exposures) can lead to elevated immune responses and increase ASD risk in offspring.36,294,305,306 The MIA model is an appropriate model for researching related mechanisms between maternal infection and ASD phenotypes. This model is created with influenza, viral infection molecules (poly(I:C)), bacterial mimics (lipopolysaccharide) and specific cytokines (such as IL-2 and IL-6).37,38,307,308 Poly(I:C) injection at midgestation generates offspring that display three core behavioural symptoms of ASD in all mice and some nonhuman primates.37,309 Changes in maternal cytokines such as IL-2, IL-6 and IL-10 levels, which may explain the MIA-induced ASD-like behaviours.296,310
Gut–brain axis of microbial–immune–neuronal communication
Recently, the gut gained attention as a key connection in the microbial–immune–neuronal system interplay. In addition to symptoms of inflammatory dysregulation, people with autism also experience gastrointestinal symptoms, including constipation, diarrhoea, and inflammatory bowel disease.311,312 The abundance of gut microbes in ASD patients, including Clostridium, Desulfovibrio, Bifidobacterium and Bacteroides, is significantly different from that in healthy controls.313,314,315,316,317 Consistently, several established animal models of ASD, including the naturally occurring BTBR strain (Bifidobacterium and Blautia flora), MIA model offspring (Clostridium),318,319 VPA-treated rodents (Desulfovibrionales)320,321 and mice lacking the synaptic adhesion protein SHANK3 (Lactobacillus reuteri),322,323 all show disturbance of the intestinal flora. Indeed, studies in animals and people with ASD have revealed that intestinal imbalance can affect peripheral immunological responses and contribute to immune cell dysfunction. For example, certain microbiota in the gut influence T-cell populations, and administration of Bacteroides fragilis restores the proper balance of T-cell populations in mice.324 Moreover, gut dysfunction affects brain function through neural, hormonal, and immune signalling.95 Interestingly, the gut microbiota is essential for microglial morphological and functional maturation, and microglial damage can be corrected to some extent by a complex microbiota.325 Therefore, microglia and inflammation alterations in the CNS may be at least partially attributable to microbial dysregulation.
Potential mechanisms of neuroimmune cross-talk
With the growing recognition and understanding of neuroimmune cross-talk, there is growing interest in how immune dysregulation affects brain functional connectivity. Most cytokines and their receptors are expressed by neurons and glial cells throughout development, and several studies have revealed that cytokines play important roles in neurogenesis, synapse formation, and plasticity, including IL-1β, IL-6, TNF-α, TGF-β1 and IFNγ326,327,328,329,330,331 Cytokines activate several signal transduction pathways, including the Janus kinase-signal transducer and activation of transcription (JAK-STAT) and PI3K/AKT/mTOR pathways, which regulate numerous cellular responses.36,286,332
In addition to participating in inflammatory responses, microglia and astrocytes also play key roles in maintaining brain homoeostasis by regulating synaptic morphology and plasticity.333,334,335,336 Specifically, glial cells engage in cross-talk with synapses through surface-expressed ion channels, receptors and transporters.333,334,335,336,337 Microglia regulate neuronal developmental remodelling and synaptic transmission by regulating the release of cytokines and chemokines in the adult brain.334,336,338 Consistently, significant impairments in synaptic pruning and synaptic transmission and ASD-like behaviours have been observed in CX3C chemokine receptor 1 (Cx3cr1)-knockout mice.335,339 These deficits may be attributable to increased signalling by IL-1β secreted from microglia.339 The engulfment of microglia is dependent upon the microglia-specific phagocytic signalling pathway via complement receptor 3 (CR3)/C3.340 This process is disrupted in mice with autism: increased C1q expression and enhanced phagocytic capacity have been found in the microcytes of Pten-mutant mice.337 Astrocytes affect synaptic transmission via glutamate uptake by the glutamate transporters GLAST and GLT1 and via regulation of synaptic function and plasticity mediated by calcium signalling.341,342,343,344 Correspondingly, astroglial GLT1 and glutamate uptake is significantly reduced in the cortex in fmr1−/− mice, which may explain the enhanced neuronal excitability observed in mice with fragile X syndrome.345
On the other hand, immune molecules and their receptors, such as MET and MHCI, are involved in a wide range of physiological events during brain development.36 MET is an immune gene encoding hepatocyte growth factor (HGF), mutations in which induce disruption of multiple downstream targets in signalling cascades, resulting in critical functional deficits in brain development.346,347 Decreases in MET expression have been observed in ASD postmortem tissues.348,349 MET can indirectly lead to changes in neural circuits and functions by negatively regulating immune responses and gastrointestinal homoeostasis, which is a putative hallmark of ASD pathophysiology.350,351 In addition to mediating the adaptive and innate immune responses, MHCI molecules contribute to controlling axonal and synaptic growth and participate in the regulation of synaptic plasticity and synaptic homeostasis in the presynaptic and postsynaptic regions associated with glutamate.352,353,354,355,356 Cortical neurons from offspring of MIA exhibit increased expression of MHCI molecules and its downstream effect factors MEF2. Remarkably, normalizing the MHCI-MEF2 signalling pathway in cultured MIA neurons prevents the MIA-induced decrease in synapse density.353 Notably, despite recent advances, most of the details of when, where and how immune molecules function in the brain remain unknown.
In summary, dysregulation of immunoregulatory signalling molecules, including cytokines, microglial complement, MET, and MHCI, is an important link in the pathological process of ASD that possibly regulates synaptic morphology and plasticity in the CNS through common downstream pathways. Among them, mTOR serves as a focal point for integrating immunological signalling in the brain, cytokine signalling, perinatal environmental exposures, and chronic immune disorders. Determining whether and how immune contributions concentrate on the common mTOR pathway in future studies will be critical for our understanding of the importance of mTOR in different aspects, not just from an immune perspective, as well as for future targeted drug development.
Brain functional connectivity and the neurotransmitter system
Early brain development in people with ASD is accelerated, which leads to changes in brain connectivity, including physical and functional connectivity between different regions and concomitant neurotransmitter changes. Different types of genetic variants may disrupt the circuits of social interactions and repetitive behaviours, resulting in a complex matrix of genes, synapses, circuits, and behaviours. Here, we summarize and review these topics on three levels. We first describe abnormal functional connectivity in the brains of ASD patients at a macroscopic scale. We then summarize the results of recent animal studies at the level of neural circuits, providing insights into the mechanisms of multiple types of specific neuronal and molecular regulation of circuit networks (Fig. 7). Finally, we summarize the relevant signal transduction pathways that regulate neurotransmitters in ASD patients.

Social behaviour-related neural circuits, neurotransmitter system and E/I balance in the rodent brain associated with ASD. a A sagittal view of the rodent brain used to illustrate the local and distal circuits implicated in social behaviours. Recent studies use behavioural neuroscience, optogenetics, chemical genetics and electrophysiology have illuminated the relationships between various social behaviour and the activity of specific neural circuits. Alterations in brain connectivity usually accompany changes of neurotransmitter, including glutamate, GABA, oxytocin, serotonin and dopamine. b In addition, the hypothesis of disruption of cortical “E/I imbalance” in autism is widely accepted, which has also been highlighted in the figure. AMY amygdala, AOB olfactory bulb, BNST bed nucleus of the stria terminalis, DRN dorsal raphe nucleus, LS lateral septum, MOB main olfactory bulb, MOE main olfactory epithelium, NAc nucleus accumbens, PFC prefrontal cortex, PVN paraventricular nucleus, RCrusl right Crus I, VNO vomeronasal organ, VTA ventral tegmental area
Brain regions and neural circuits
According to human neuroimaging and neuropathological investigations, global brain developmental anomalies in children with ASD emerge in the cerebral cortex, striatum, cerebellum, brainstem, and other subcortical structures.357,358,359,360,361,362,363 Recent studies have identified that the medial prefrontal cortex (mPFC) integrates social and spatial information through neuronal coding. The mPFC is one of the best-studied brain regions related to social behaviour.364,365 In both mice and humans, several pieces of evidence imply that striatal dysfunction is a neurological substrate for repetitive behaviours.366,367,368 For example, Nlgn1-knockout mice exhibit ASD-like repetitive behaviours and corticostriatal synaptic abnormalities,369 whereas mice lacking Nlgn3 exhibit similar behavioural changes caused by neuronal inhibitory transmission from D1-MSN in the nucleus accumbens (NAc).370 Mice lacking Shank3 exhibit striatal hypertrophy and decreased corticostriatal excitatory synaptic transmission, as well as repetitive behaviours.202 In early assessments of autism, the amygdala exhibits reduced volume and increased neuronal density in the medial, central and lateral nuclei, which play critical roles in modulating fear conditioning, anxiety and social behaviour.357,361,371,372,373 Consistently, amygdalar axonal projections and neuronal activation are defective in Tbr1(+/−) mice, but these defects are ameliorated by infusion of an NMDA receptor agonist (D-cycloserine).110 The cerebellum is best known for its role in controlling motor behaviours, and most individuals with ASD have comorbidities associated with movement disorders such as ADHD. Histopathological changes in cerebellar neuronal structure, such as loss of Purkinje cells (PCs), have been discovered in the postmortem brains of many ASD patients.357,374,375 Validation data on key signalling molecules suggest that cerebellar PC-specific knockout of Tsc1, Tsc2 and Bmal1 is sufficient to induce core ASD-like behaviour.376,377,378 Notably, a growing number of studies have found that the cerebellum is involved in the pathophysiology of autism in the form of nonmotor regulation.379,380,381
Rodents and humans share similar brain regions and neural circuits, facilitating our investigation of social behaviour and related signalling mechanisms.382 Currently, rodents and nonhuman primates, such as chimpanzees, are accepted models for identifying social behavioural changes in autism. Numerous studies have shown that mice exhibit unique social behaviours, such as territorial aggression and mating, interpret olfactory traits as social information, and transmit and interpret emotional contagion and empathic responses.383,384,385 Novel approaches in optogenetics, chemical genetics, electrophysiology and behavioural neuroscience have helped to construct the links between various social behaviours and brain circuit activity (Fig. 7).386,387,388,389 In the huge and complex neural network involving social behaviour, the PFC and its massive reciprocal loop connections constitute a top-down social behaviour regulation system. Various subcortical networks communicate with the mPFC, including the amygdala (responsible for emotional processing), the NAc (responsible for social incentive), and the hypothalamus (responsible for stress regulation).390,391,392,393 Recently, the right crus I (RCrusI) of the cerebellum was identified as a key brain region for social interaction in mice that can project to the cortex to modulate social interaction and repetitive behaviours in mice.394,395 In addition, oxytocinergic, serotonergic and dopaminergic-related circuits also play critical roles in social regulation, which will be discussed below.
Neurotransmitter system
From a neurobiochemical perspective, the activity of brain structures and neural circuits is coordinated by multiple neurotransmitters and neuromodulators. Therefore, dynamic changes in neurotransmitter concentration, release, and receptor density may directly affect neural circuit function and thus behavioural performance.396 Increasing evidence shows that disturbances in neurotransmitter systems, including the glutamate, GABA, serotonin (5-hydroxytryptamine, 5-HT),397,398 melatonin,397,399 dopamine (DA),396,400,401 OT and arginine vasopressin (AVP) systems, are associated with autism (Fig. 7).
Classic neurotransmitters. glutamate and GABA:
An appropriate balance between excitation and inhibition (E/I) in synaptic transmission and neural circuits is essential for appropriate brain functioning. In 2011, Yizhar et al. used optogenetics to study excitatory projection neurons and inhibitory PV neurons of the mPFC and subsequently found that an increase in the cellular E/I ratio leads to severe impairments in information processing and behaviour.402 Currently, the hypothesis of cortical “E/I imbalance” in autism is widely accepted (Fig. 7).403,404,405,406
E/I balance is controlled by the ratio of excitatory to inhibitory cells, as well as their activity. Plasma levels of GABA and glutamate are changed in autistic children, who exhibit significantly increased GABA levels and decreased glutamate/GABA ratios.223 Previous findings have highlighted the importance of glutamate dysfunction in contributing to the aetiology of autism.407,408,409,410,411 In addition to the above mentioned changes in glutamatergic neurons in ASD, the functional role of GABAergic inhibitory neurons is becoming increasingly clear. Neuropathological studies have provided evidence of reduced GABAR levels in the cortex and hippocampus, aberrant GAD1 and GAD2 mRNA expression in the postmortem cortex and cerebellum, and the interneuron markers parvalbumin (PV) and somatostatin (SST) are downregulated.412,413,414,415,416,417 Loss of inhibitory neurons and impairment of inhibitory neurotransmission are also observed in ASD mouse models as a result of mutations in genes such as Pten, Mecp2, Cntnap2, Shank3 and BTBR mice, which may directly lead to alterations in the balance of excitation and inhibition.418,419,420,421,422,423 It is worth noting, however, that investigations on E/I imbalance have primarily been carried out using animal models, therefore a detailed assessment of the pathophysiology of E/I imbalance contributing to human ASD is warranted.
Biogenic amines. 5-HT and DA:
5-HT has long been suggested to be related to social behaviour. Early researches suggested increased 5-HT levels in the blood of children with autism. According to data from neuroimaging and neurobiochemical analyses, up to 45% of individuals with autism have hyperserotonaemia.398 Abnormal 5-HT neurotransmission and social behavioural deficits have been reported in SERT and MAOA mutant animal models.398 Serotonergic neurons are mainly located in the dorsal raphe nuclei (DRNs), which can project to the PVN of the hypothalamus and modulate OT release.424 Moreover, other brain areas, such as the NAc, can also receive projections from the DRNs and display OXTR. A study in mice has elucidated that the coordinated activity of OT and 5-HT inside the NAc is essential for social reward.425 These studies have highlighted the synergistic effects of 5-HT and OT in ASD.
The DA system is also involved in ASD, and an early study identified elevations in HVA (a DA metabolite) in the cerebrospinal fluid of patients.426 Children with autism have defects in mesolimbic dopaminergic signalling, such as decreased dopamine release in the prefrontal cortex and decreased NAc neural responses.427,428 The majority of DA-producing neurons are located in two primary regions, the substantia nigra (SN) and VTA, in the brain.429 VTA dopaminergic neurons project to various brain structures, such as the NAc, involved in the control of social cognition.388,430 Although DA release has long been linked to reward, there is growing evidence that DA is released in response to aversive behaviour.431,432,433,434 The NAc has been well studied for its role in reward processing behaviour, which is predominantly composed of inhibitory MSNs that differ in the type of DA receptor they express, D1R or D2R.388 Notably, the two subtypes of neurons may play different roles in social and repetitive behaviours.435,436
Neuropeptides. OT and AVP:
The neuropeptide hormones OT and AVP belong to the same superfamily, and genetic variants in OXT, OXTR, arginine vasopressin receptor 1a (AVPR1a) and CD38 (lately demonstrated as essentiall for social behaviour because it mediates oxytocin secretion) have been verified to be associated with autism.437,438,439,440 Compared to neurotransmitters (approximately 5 ms), neuropeptides (approximately 20 min) display a substantially longer half-life and are stored in dense core vesicles, which are much larger in size and scope than synaptic vesicles.441,442 Hence, OT and AVP have much broader neuromodulatory roles and less spatial/temporal specificity than classical neurotransmitters.442,443 The changes in OT and AVP levels in autistic patients’ plasma are often associated with abnormal functional connectivity.444 For example, OT administration increases the connectivity of brain regions critical for processing socioemotional information, such as the NAc, amygdala and PFC.445 Studies in animals have implicated OT and AVP in mammalian sexual, territorial, attachment and social behaviours.442 Moreover, OT also plays a recognized role in anxiety, which is common a comorbid symptom of ASD.446
OT is mainly produced by neurons located in the paraventricular nucleus (PVN) and supraventricular nucleus (SON) of the hypothalamic–neurohypophysial system. Social cues induce OT release from the PVN; the OT acts on downstream structures such as the LS, amygdala, VTA and NAc.425,447,448,449 OT release from oxytocinergic neuron axon terminals in the VTA drives the excitability of dopaminergic neurons in the NAc, and eventual activation of the PVN–VTA circuit enhances social behaviour.448
For nearly two decades, an increasing number of studies on the modulation of circuits and neurotransmitter systems have gained insight into different brain areas and circuits involved in particular behavioural states. Nevertheless, it is unclear to what extent the mouse phenotypes recapitulate the relationships among neural circuits in autism. It should be noted that the human brain with its multimodal structure has undergone dramatic changes in brain regions such as the frontal and temporal lobes during evolution. Therefore, more comparative studies between primate and mouse models are required to precisely correlate neuroanatomical features with candidate brain circuits involved in ASD pathogenesis. More importantly, identification of molecular mechanisms that are specific to social behaviours and circuits is needed. Such information will be essential for developing targeted treatments aimed at ASD.
Therapeutic strategies
The current treatment strategies for autism are divided into nonpharmacological treatment and pharmacological treatment approaches. Combining pharmacotherapy with behavioural psychosocial learning interventions may have significant impacts on long-term outcomes for people with autism. However, based on the complex mechanism of the superposition of multiple aetiologies of autism, there is still a lack of clinical cures for core symptoms. In any case, the lack of molecular targets is the rate-limiting barrier for new drug research for autism. Innovative drug development for autism is currently the most challenging work in the field. The development of strategies to intervene in or block the transduction of key signalling molecules involved in the pathogenesis of autism is a primary research direction. In this section, we mainly review and discuss pharmacotherapies based on pathological features and signal transduction mechanisms (Fig. 8).

Potential novel therapeutic strategies and target of ASD. Abundant basic research on mouse and iPSC models exploited potential treatments to be used in ASD patients.It is noteworthy that emerging treatments including brain stimulation, gene therapy and exosome modulators are also been indicated
Nonpharmacological therapies
Nonpharmacological treatment mainly refers to educational interventions and behaviour modification but also includes adjunctive treatments such as music and art therapy. The main purpose of nonpharmacological treatment is to develop children’s self-care and social skills, thus improving their quality of life. With advances in neuroscience, brain stimulation has also gradually attracted clinicians’ attention and has shown potential to improve the symptoms of ASD patients.450,451
Behavioural and psychological intervention
Physical intervention is usually considered as a priority because many young autistic children have difficulty communicating and interacting with others. Music therapy, cognitive behavioural therapy (CBT) and social behavioural therapy (SBT) have all showed promise in helping autistic patients improve their social interaction and verbal communication.50,452 One potential pathway by which music therapy affects ASD is by changing the structural and functional connectivity of the cortex to achieve a greater degree of multisensory integration across cortical and subcortical regeions during early development.453 CBT is a commonly used psychotherapeutic intervention and can both target core symptoms and treat comorbid anxiety and depression symptoms of ASD.454,455 SBT targets emotional regulation, social skills and functional communication, with an emphasis on independence and quality of life. Considering that the behavioural symptoms of ASD appear at a fairly early stage of development, intervening before symptoms appear may lead to better outcomes. Although treatments vary widely around the world, they generally follow a typical developmental psychology sequence that emphasizes play, social interaction, and communication with children. It is worth noting that clinical services should not be solely diagnosis oriented but should provide step-by-step specific interventions.175
Brain stimulation
Non-invasive brain stimulation is a relatively recent treatment option that has shown hope in the treatment of ASD. The molecular mechanisms underlying brain stimulation-dependent neuronal excitability and synaptic plasticity have been well elucidated with extensive preclinical animal models.456,457,458 Neuroimaging studies have demonstrated structural and functional imaging abnormalities in several brain regions of ASD patients. There have been more than a dozen trials of brain stimulation techniques, including transcranial direct current stimulation (tDCS) and transcranial magnetic stimulation (TMS), in the ASD population. tDCS is primarily conducted in the brain via a constant current through scalp electrodes. In contrast, in TMS, intracranial currents are induced in the cortex by fluctuating extracranial magnetic fields. Both techniques modulate regional cortical excitability and are well tolerated in children and adults.459,460 Neural stimulation has been reported to modify cortical excitability by affecting GABAergic function and causing LTP or LTD-like excitatory synaptic strength.461,462,463,464,465,466 tDCS has been shown to improve autism symptoms and language in several small clinical trials.467,468 Recent studies examining executive function in the dorsolateral prefrontal cortex (DLPFC) after TMS and improvements in social behaviour and social cognition in the posterior superior temporal sulcus and DLPFC in autistic patients after tDCS have shown preliminary therapeutic effects.469,470,471,472
Together, nonpharmacological therapies can partially alleviate autism symptoms. Although sufficient evidence is still lacking, the therapeutic effects of behavioural and psychological interventions and brain stimulation on autistic patients must have a theoretical basis related to neurobiochemistry and signal transduction.
Drug targets and pharmacological therapies
Because the pathogenetic and pathological mechanisms are still unclear, there is no effective treatment drug for the eradication of autism that has been officially approved. Several drugs targeting autism are under study (Table 3) and clinical trials (Table 4). At present, clinical drug treatment of autism generally involves appropriate amounts of atypical antipsychotics, antidepressants, and sleep disorder-improving drugs according to the core symptoms of children.50
Atypical antipsychotics, including risperidone (a dopamine antagonist) and aripiprazole (a dopamine agonist), are FDA-approved drugs that have been shown to relieve irritability symptoms such as aggression and self-mutilation in adolescent autistic patients in several large clinical trials.473,474,475,476,477 α-Adrenergic drugs such as guanfacine are used for ADHD and disruptive behaviour.478 Antidepressants such as SSRIs improve the symptoms of emotional instability, anxiety, and stereotyped repetitive behaviours in patients with ASD by blocking the reuptake of 5-HT and increasing the concentration of 5-HT in the synaptic cleft.479 Fluoxetine, sertraline, citalopram, escitalopram, and fluvoxamine are SSRIs widely used in ASD. However, SSRIs are not suitable for everyone and should be used with caution, especially in people with autism with anxiety or obsessive–compulsive disorder.480
Notably, nearly 40–86% of children with autism have sleep–wake rhythm disturbances.481,482 Clinical drugs that can treat ASD by improving sleep include melatonin, ramelteon, niperrazine, and clonidine.483,484 It is worth mentioning that many investigations have reported aberrant melatonin secretion in autistic patients, particularly decreased melatonin and metabolite secretion at night, and altered circadian rhythms of melatonin.481,484,485 Several clinical trials have shown that melatonin reduces sleep latency and improves sleep duration and nighttime arousal, suggesting that it is an effective treatment for sleep disturbances in children with ASD. In addition, a meta-analysis and some placebo-controlled studies have suggested that melatonin supplementation may also have positive effects on autistic behavioural disorders.481,486 One study on VPA-treated rats has proven that melatonin treatment significantly improves social behavioural deficits through CaMKII/PKA/PKC signalling.487,488 Therefore, melatonin or novel analogues may be promising drug therapies for improving behavioural disorders in autism. In the future, it will be necessary to study the regulatory mechanism of melatonin-related signal transduction and to verify the dose–response relationship in the improvement of behavioural disorders in clinical trials to test the therapeutic benefits of melatonin.
In addition, the development of other ASD-targeted drugs has been promoted due to in-depth basic scientific research on the pathogenesis of ASD in the past decade. Clinical trials targeting E/I balance, transcriptional and epigenetic regulation, immune regulation, biological peptides and intestinal flora are advancing in an orderly manner (Table 3).
Targeting E/I balance
The cortical E/I imbalance hypothesis in ASD patients highlights the potential of glutamate and GABAergic receptor modulators as therapeutic agents.402 Different pharmacological methods have been applied to restore E/I imbalance, such as mGluR5 antagonist treatment, NMDAR agonist treatment and GABAR agonist treatment.489,490,491 Extensive preclinical data demonstrate that overactivity of mGluR5 is central to the pathogenesis of fragile X syndrome.25,211,492 In addition to targeting fragile X syndrome, mGluR5 inhibition has been shown to salvage many phenotypes, including learning and memory deficits, social deficits, repetitive behaviours, hyperactivity, and dendritic spine dysmorphogenesis, in 16p11.2 deletion mice, BTBR mice and Shank3-knockout mice.26,228,493 Unfortunately, mGluR5 inhibitors developed by two companies have exhibited negative effects in large-scale patient trials targeting fragile X syndrome.494,495 Further reasons should be sought for the discrepancies in preclinical and clinical outcomes. In addition to expanding and refining the preclinical analyses of new drugs, it will also be necessary to scientifically stratify patients enrolled in clinical trials in order to increase the expected efficacy in patients.
NMDA receptors and mGluRs show positive reciprocal regulation. NMDA receptor agonist (d-cycloserine) intervention attenuates impaired sociability in Shank2-transgenic mice, highlighting the need for accurate signalling at excitatory synapses.207 The spatial and temporal selectivity offered by subtype-selective positive allosteric modulators of the NR2 receptor make these agents promising candidates for the treatment of ASD.496 Drugs targeting the NMDA receptor, such as memantine, have been demonstrated to alleviate core symptoms of ASD in early open-label trials.497,498,499,500 Although subsequent RCTs have shown no differences in primary and secondary indicators, memantine improves symptoms of ASD such as stereotyped behaviours, and social communication/interaction impairment as an adjuvant therapy.501,502,503 The results from the memantine trial have been mixed, suggesting that further research is needed, and a large randomized controlled trial is currently being conducted on the therapy of social impairment in adolescents. Several trials on other NMDA-modulating drugs, including ketamine,504 riluzole,44,505 and d-cycloserine,506 have been negative for the primary endpoint, indicating that further studies with increased sample sizes are required.506
Evidence from fragile X syndrome mice has indicated that alterations in GABA-mediated synaptic transmission are present in the mice, suggesting that there is potential therapeutic benefit of GABA receptor agonism.423 Arbaclofen, a GABA-B agonist, regulates glutamatergic activity through presynaptic action to reduce glutamate release. In Fmr1-knockout mice, arbaclofen reverses protein synthesis, synaptic abnormalities and dendritic spine density phenotypes.507 Consistently, two clinical studies have suggested that arbaclofen has the potential to improve symptoms of ASD.508,509 Bumetanide, an NKCC1 (Na+-K+-2Cl− cotransporter) chloride-importer inhibitor that reduces (Cl−)i levels, enhances GABAergic inhibition, which improves the behavioural symptoms of individuals with ASD.510,511,512 Data from three follow-up studies have been obtained: two studies showed improvement in the primary endpoint (the Childhood Autism Rating Scale),513 while the other study showed no difference in the primary endpoint (the Social Responsiveness Rating Scale).514
Targeting translation and epigenetic regulation
Transcriptional and translational studies have provided a scientific foundation for the discovery of drug targets for underlying mechanisms, such as PI3K/mTOR pathways.491 mTOR inhibitors, such as rapamycin and everolimus, have been utilized to cure behavioural and molecular abnormalities in TSC-deficient mice.22 Unfortunately, chemotherapeutic agents acting on the mTOR pathway have not been discovered to improve social interaction of children with tuberous sclerosis.515 Preliminary data have shown that the pharmacological effects of IGF-1 affect synaptic development primarily by modulating the MAPK and mTOR pathways, as validated in Phelan–McDermid syndrome and Rett syndrome.28,29,516 Specifically, IGF-1 treatment results in increases in synaptic protein levels and activation of signalling pathway proteins and enhances cortical excitatory synaptic transmission and dendritic spine density. Trials of the effects of IGF-1 on social interactions in individuals with ASD have shown positive results, but larger trials will provide more definitive information on efficacy.517,518,519
In terms of epigenetic regulation, many autism risk genes are involved in histone modification and chromatin remodelling, and disruption of this process has been observed in individuals with autism. Treatment strategies with epigenetic enzymes, primarily targeting histone modifiers (such as histone deacetylase,520 histone demethylase521 and histone methyltransferase,522) show therapeutic potential in animal models. The Shank3-mutant mouse model is one of the most commonly used models to study epigenetic enzymes, and it was found that using histone methyltransferase inhibitors and histone acetylase inhibitors alone520,521,522 or in combination523 can both significantly improve NMDA dysfunction and social interactions in Shank3-mutant mice. In a recent small randomized controlled trial, dietary supplementation with methylation-modifying leucovorin/folate improved core symptoms of ASD.524 Folate is crucial to normal neurodevelopment. Abnormal folate metabolism has been identified in patients with ASD.525 Three randomized double-blind placebo-controlled trials evaluated the effect of folic acid on verbal communication in patients with ASD.524,526,527 Encouragingly, compared to placebo, folic acid improved scores in communication and social interaction, providing promising preliminary evidence for language impairment in children with autism.
Other biological targets: biological peptides, neuroinflammation and the intestinal flora
The neuropeptide theory of autism is backed up by evidence from animal research.528,529 OT has been discovered to play an important role in relationship formation and social functioning.530 Dozens of clinical trials have studied the effects of intranasal oxytocin on ASD.531,532,533,534 Although there is no substantial treatment-specific improvement in core social symptoms, recent findings on the long-term beneficial effects on repeated behaviours and feelings of avoidance are encouraging and suggest that OT may have therapeutic promise in the treatment of ASD. Given the difficulty of exogenous drug interventions in penetrating the blood–brain barrier, several trials on strategies to promote endogenous OT production are underway. AVP is a neuropeptide primarily used to regulate renal water reabsorption and increase perivascular resistance that has been detected at lower levels in the cerebrospinal fluid of ASD children than in controls and has also been studied as a target for ASD drug therapy.535,536 A randomized double-blind controlled trial of intranasal AVP in children showed a beneficial effect on sociability deficits.537 Combined with evidence from preclinical studies, this evidence indicates that V1a receptor antagonists may exert prosocial, antidepressant, and anxiolytic effects in disorders of social and emotional dysfunction. In a large trial conducted in adult men, balovaptan, an orally administered selective vasopressin V1a receptor antagonist, showed promise in terms of improving social interaction and communication among people with ASD.538
Findings of elevated levels of inflammatory factors and altered gut bacterial stages in children with ASD underscore the importance of ASD immune mechanisms.539,540,541,542 Peroxisome proliferator-activated receptor (PPAR-ϒ) is a nuclear hormone receptor, and its anti-inflammatory function has received attention. Pioglitazone belongs to the thiazolidinediones drug class (TZDs) and acts on PPAR-ϒ. In addition, pioglitazone has been identified to reduce NMDA-mediated Ca2+ currents and transients.543 Two clinical trials have suggested that pioglitazone has the potential to improve behavioural symptoms of ASD.544,545 Basic and clinical data have emphasized the role of gut microbes in the regulation of brain immune function.546 Modulating the microbiome has been shown to improve social core symptoms and synaptic dysfunction in animal models.322,547,548,549 Clinical trials have demonstrated that children with ASD treated with microbiota transfer have significantly reduced abdominal pain, indigestion, diarrhoea and constipation. In addition, the abundance of Bifidobacterium, Prevotella and Desulfovibrio is significantly increased, and the increases are correlated with improved symptoms.546,550,551,552 A recent study has also shown that Lactobacillus plantarum intervention in children with ASD reduces common abnormal behaviours and social impairments in ASD patients.553 Multimodal interventions are aimed at achieving clinical maximal therapeutic effects. It is expected that drugs targeting specific facets of autism will be developed to improve the core symptoms of patients. New drugs that affect synaptic plasticity, social learning or neuroinflammation must be combined with psychological interventions to achieve complementary synergies that ultimately have a major impact on the long-term outcomes of individuals with autism.
Conclusion and perspectives
In conclusion, ASD is a complex disease caused by a series of combinations of different aetiological factors, including genetic factors, environmental and immune activation, etc., and ultimately manifests as abnormal changes in molecular signalling pathways, neuronal synapses, immune environment and brain functional connections. Animal models provide an opportunity to identify potential changes in circuit levels and their relation to behaviour regulation. Frustratingly, present medication only target concomitant symptoms rather than the core symptoms of autism, and the development of key molecular targets for signal transduction pathways is still in the basic research. To date, few trials have reached their primary endpoints, and little evidence has promoted the approval of drug administration agencies or the use of the tested treatments in clinical practice. For example, the efficacy of several small molecular targets has been well demonstrated in animal models, such as mGluR5 inhibitors, OT, Memantine, and mTOR inhibitors, but is still unsatisfactory in clinical trials. A serious challenge is how ASD can bridge the vast gap between molecular, cellular, and circuit convergence mechanisms to the heterogeneity of clinical manifestations. Therefore, basic research to clinical transformation remains the rate-limiting step in the development of treatment strategies for ASD, and the degree of heterogeneity may be considered, which may obscure the effect of experimental treatments. Conducting in-depth mechanistic studies using models such as nonhuman primates that can truly simulate human pathological processes would be crucial. The development of methods for manipulating nonhuman primate genomes may provide key insights for translation from model system experiments to human studies.
Despite these challenges, new therapies based on elucidated genes have been developed in recent years, such as gene replacement, gene editing and translating oligonucleotides.554 Relatively modest manipulation of gene expression using normal alleles may be sufficient to mitigate the effects of deleterious mutations. The development of technologies such as CRISPR–Cas9, which is based on targeted DNA editing, has facilitated rapid progress in gene therapy, and these technologies have also shown therapeutic effects in mice with fragile X syndrome. Thus, gene editing provides a new personalized medicine approach for the treatment of autism.555,556
To optimize and change the treatment strategy for autism, it is necessary to bridge biochemical molecular events, electrical oscillations and information processing and to explore the pathological mechanism of autism from a new systemic perspective. The coexistence of many clinical disorders in autism is quite common, but this autism comorbidity has not received enough attention thus far. Studies exploring potential biomarkers should design laboratory tests related to specific clinical syndromes based on the presence or absence of some specific comorbidities. Such research will require large-scale clinical cohort studies involving the same population, as well as focusing on spatiotemporal dynamics such as behaviour, development, and types of comorbidities. In conclusion, research on ASD is still challenging. ‘Bench to bedside’ progress will depend on integrative multidisciplinary approaches between basic scientists and clinical investigators to reveal the pathological mechanism of autism.
References
Kanner, L. Autistic disturbances of affect contact. Nerv. Child 2, 217–250 (1943).
Volkmar, F. R. & McPartland, J. C. From Kanner to DSM-5: autism as an evolving diagnostic concept. Annu Rev. Clin. Psychol. 10, 193–212 (2014).
Asperger, H. Die “autistichen Psychopathen” im Kindersalter. Arch. Psychiatr. Nervenkrankheiten 117, 76–136 (1944).
Hippler, K. & Klicpera, C. A retrospective analysis of the clinical case records of ‘autistic psychopaths’ diagnosed by Hans Asperger and his team at the University Children’s Hospital, Vienna. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 291–301 (2003).
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edn (American Psychiatric Association, 1994).
World Health Organ. International Classification of Diseases (Draft Version: Diagnostic Criteria for Research, 1990).
Rimland, B. Infantile Autism: The Syndrome and Its Implications for a Neural Theory of Behavior (Appleton-Century-Crofts, 1964).
Rutter, M. Childhood schizophrenia reconsidered. J. Autism Child Schizophr. 2, 315–337 (1972).
Rutter, M. Diagnosis and definition of childhood autism. J. Autism Child Schizophr. 8, 139–161 (1978).
Ritvo, E. R. & Freeman, B. J. Current research on the syndrome of autism: introduction. The National Society for Autistic Children’s definition of the syndrome of autism. J. Am. Acad. Child Psychiatry 17, 565–575 (1978).
Bartak, L. & Rutter, M. Special educational treatment of autistic children: A comparative study–II. Follow‐up findings and implications for services. J. Child Psychol. Psychiatry 14, 161–179 (1973).
Folstein, S. & Rutter, M. Genetic influences and infantile autism. Nature 265, 726–728 (1977).
Baron-Cohen, S., Leslie, A. M. & Frith, U. Does the autistic child have a “theory of mind”? Cognition 21, 37–46 (1985).
Zwaigenbaum, L. et al. Studying the emergence of autism spectrum disorders in high-risk infants: methodological and practical issues. J. Autism Dev. Disord. 37, 466–480 (2007).
de la Torre-Ubieta, L., Won, H., Stein, J. L. & Geschwind, D. H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 22, 345–361 (2016).
Mandy, W. & Lai, M. C. Annual Research Review: The role of the environment in the developmental psychopathology of autism spectrum condition. J. Child Psychol. Psychiatry 57, 271–292 (2016).
Onore, C., Careaga, M. & Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 26, 383–392 (2012).
European Chromosome 16 Tuberous Sclerosis, C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315 (1993).
Werling, D. M. et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 50, 727–736 (2018).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012).
Doan, R. N. et al. Recessive gene disruptions in autism spectrum disorder. Nat. Genet. 51, 1092–1098 (2019).
Tsai, P. & Sahin, M. Mechanisms of neurocognitive dysfunction and therapeutic considerations in tuberous sclerosis complex. Curr. Opin. Neurol. 24, 106–113 (2011).
Bhattacharya, A. et al. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337 (2012).
Troca-Marin, J. A., Alves-Sampaio, A. & Montesinos, M. L. Deregulated mTOR-mediated translation in intellectual disability. Prog. Neurobiol. 96, 268–282 (2012).
Michalon, A. et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56 (2012).
Tian, D. et al. Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat. Neurosci. 18, 182–184 (2015).
Castro, J. et al. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl Acad. Sci. USA 111, 9941–9946 (2014).
Shcheglovitov, A. et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271 (2013).
Bozdagi, O., Tavassoli, T. & Buxbaum, J. D. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol. Autism 4, 9 (2013).
Voineagu, I. et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011).
Parikshak, N. N. et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427 (2016).
Gupta, S. et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 5, 5748 (2014).
Takano, T. Role of Microglia in Autism: Recent Advances. Dev. Neurosci. 37, 195–202 (2015).
Chernikova, M. A. et al. The brain-gut-microbiome system: pathways and implications for autism spectrum disorder. Nutrients 13, 4497 (2021).
Zantomio, D. et al. Convergent evidence for mGluR5 in synaptic and neuroinflammatory pathways implicated in ASD. Neurosci. Biobehav Rev. 52, 172–177 (2015).
Estes, M. L. & McAllister, A. K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 16, 469–486 (2015).
Malkova, N. V., Yu, C. Z., Hsiao, E. Y., Moore, M. J. & Patterson, P. H. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 26, 607–616 (2012).
Patel, S. et al. Social impairments in autism spectrum disorder are related to maternal immune history profile. Mol. Psychiatry 23, 1794–1797 (2018).
Casanova, M. F. Neuropathological and genetic findings in autism: the significance of a putative minicolumnopathy. Neuroscientist 12, 435–441 (2006).
Volk, L., Chiu, S. L., Sharma, K. & Huganir, R. L. Glutamate synapses in human cognitive disorders. Annu Rev. Neurosci. 38, 127–149 (2015).
Gao, R. & Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167 (2015).
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-3 3rd (1980).
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-5 5th (2013).
Ajram, L. A. et al. Shifting brain inhibitory balance and connectivity of the prefrontal cortex of adults with autism spectrum disorder. Transl. Psychiatry 7, e1137 (2017).
Lai, M. C. et al. Prevalence of co-occurring mental health diagnoses in the autism population: a systematic review and meta-analysis. Lancet Psychiatry 6, 819–829 (2019).
Soke, G. N., Maenner, M. J., Christensen, D., Kurzius-Spencer, M. & Schieve, L. A. Prevalence of co-occurring medical and behavioral conditions/symptoms among 4- and 8-year-old children with autism spectrum disorder in selected areas of the United States in 2010. J. Autism Dev. Disord. 48, 2663–2676 (2018).
Pezzimenti, F., Han, G. T., Vasa, R. A. & Gotham, K. Depression in youth with autism spectrum disorder. Child Adolesc. Psychiatr. Clin. N. Am. 28, 397–409 (2019).
Constantino, J. N. & Charman, T. Diagnosis of autism spectrum disorder: reconciling the syndrome, its diverse origins, and variation in expression. Lancet Neurol. 15, 279–291 (2016).
Lord, C. et al. Autism from 2 to 9 years of age. Arch. Gen. Psychiatry 63, 694–701 (2006).
Sharma, S. R., Gonda, X. & Tarazi, F. I. Autism spectrum disorder: classification, diagnosis and therapy. Pharm. Ther. 190, 91–104 (2018).
Roman-Urrestarazu, A. et al. Association of race/ethnicity and social disadvantage with autism prevalence in 7 million school children in England. JAMA Pediatr. 175, e210054 (2021).
Morales Hidalgo, P., Voltas Moreso, N. & Canals Sans, J. Autism spectrum disorder prevalence and associated sociodemographic factors in the school population: EPINED study. Autism 25, 1999–2011 (2021).
Loomes, R., Hull, L. & Mandy, W. P. L. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 56, 466–474 (2017).
Werling, D. M. & Geschwind, D. H. Understanding sex bias in autism spectrum disorder. Proc. Natl Acad. Sci. USA 110, 4868–4869 (2013).
Robinson, E. B., Lichtenstein, P., Anckarsater, H., Happe, F. & Ronald, A. Examining and interpreting the female protective effect against autistic behavior. Proc. Natl Acad. Sci. USA 110, 5258–5262 (2013).
Brugha, T. S. et al. Epidemiology of autism in adults across age groups and ability levels. Br. J. Psychiatry 209, 498–503 (2016).
Rodgaard, E. M., Jensen, K., Miskowiak, K. W. & Mottron, L. Autism comorbidities show elevated female-to-male odds ratios and are associated with the age of first autism diagnosis. Acta Psychiatr. Scand. 144, 475–486 (2021).
Manoli, D. S. & Tollkuhn, J. Gene regulatory mechanisms underlying sex differences in brain development and psychiatric disease. Ann. N. Y Acad. Sci. 1420, 26–45 (2018).
Nguyen, D. K. & Disteche, C. M. Dosage compensation of the active X chromosome in mammals. Nat. Genet 38, 47–53 (2006).
Nguyen, D. K. & Disteche, C. M. High expression of the mammalian X chromosome in brain. Brain Res 1126, 46–49 (2006).
Jamain, S. et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29 (2003).
Trappe, R. et al. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 68, 1093–1101 (2001).
Kang, H. J. et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011).
Werling, D. M. et al. Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep. 31, 107489 (2020).
Werling, D. M. The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex. Differ. 7, 58 (2016).
Werling, D. M., Parikshak, N. N. & Geschwind, D. H. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat. Commun. 7, 10717 (2016).
Rosenberg, R. E. et al. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch. Pediatr. Adolesc. Med. 163, 907–914 (2009).
Klei, L. et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 3, 9 (2012).
Cross-Disorder Group of the Psychiatric Genomics, C. et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994 (2013).
Satterstrom, F. K. et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584 e23 (2020).
Sandin, S. et al. The familial risk of autism. JAMA 311, 1770–1777 (2014).
Mullins, C., Fishell, G. & Tsien, R. W. Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89, 1131–1156 (2016).
Ruzzo, E. K. et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell 178, 850–866.e26 (2019).
Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 16, 551–563 (2015).
Rutherford, S. L. From genotype to phenotype: buffering mechanisms and the storage of genetic information. Bioessays 22, 1095–1105 (2000).
Hartman, J. L. T., Garvik, B. & Hartwell, L. Principles for the buffering of genetic variation. Science 291, 1001–1004 (2001).
De Rubeis, S. & Buxbaum, J. D. Genetics and genomics of autism spectrum disorder: embracing complexity. Hum. Mol. Genet. 24, R24–R31 (2015).
Varghese, M. et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. 134, 537–566 (2017).
Quesnel-Vallieres, M., Weatheritt, R. J., Cordes, S. P. & Blencowe, B. J. Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Nat. Rev. Genet. 20, 51–63 (2019).
Mitra, I. et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature 589, 246–250 (2021).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012).
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012).
Sanders, S. J. et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015).
Girirajan, S. et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 92, 221–237 (2013).
Longo, F. & Klann, E. Reciprocal control of translation and transcription in autism spectrum disorder. EMBO Rep. 22, e52110 (2021).
Gaugler, T. et al. Most genetic risk for autism resides with common variation. Nat. Genet 46, 881–885 (2014).
Van Dijck, A. et al. Clinical presentation of a complex neurodevelopmental disorder caused by mutations in ADNP. Biol. Psychiatry 85, 287–297 (2019).
Bernier, R. et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276 (2014).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Krumm, N., O’Roak, B. J., Shendure, J. & Eichler, E. E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105 (2014).
Ebrahimi-Fakhari, D. & Sahin, M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr. Opin. Neurol. 28, 91–102 (2015).
Zoghbi, H. Y. & Bear, M. F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 4, a009886 (2012).
Bludau, A., Royer, M., Meister, G., Neumann, I. D. & Menon, R. Epigenetic regulation of the social brain. Trends Neurosci. 42, 471–484 (2019).
Matta, S. M., Hill-Yardin, E. L. & Crack, P. J. The influence of neuroinflammation in autism spectrum disorder. Brain Behav. Immun. 79, 75–90 (2019).
Luna, R. A. et al. Distinct microbiome-neuroimmune signatures correlate with functional abdominal pain in children with autism spectrum disorder. Cell Mol. Gastroenterol. Hepatol. 3, 218–230 (2017).
Salinas, P. C. & Zou, Y. Wnt signaling in neural circuit assembly. Annu. Rev. Neurosci. 31, 339–358 (2008).
Yap, E. L. & Greenberg, M. E. Activity-regulated transcription: bridging the gap between neural activity and behavior. Neuron 100, 330–348 (2018).
West, A. E. & Greenberg, M. E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3, a005744 (2011).
Tian, Y. et al. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 6, 75 (2014).
Sugathan, A. et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl Acad. Sci. USA 111, E4468–E4477 (2014).
Greer, P. L. & Greenberg, M. E. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860 (2008).
Buffington, S. A., Huang, W. & Costa-Mattioli, M. Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci. 37, 17–38 (2014).
Quesnel-Vallieres, M. et al. Misregulation of an activity-dependent splicing network as a common mechanism underlying autism spectrum disorders. Mol. Cell 64, 1023–1034 (2016).
Amir, R. E. et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188 (1999).
Moretti, P., Bouwknecht, J. A., Teague, R., Paylor, R. & Zoghbi, H. Y. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 14, 205–220 (2005).
Helsmoortel, C. et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 46, 380–384 (2014).
Hnoonual, A., Sripo, T. & Limprasert, P. Whole-exome sequencing identifies a novel heterozygous missense variant of the EN2 gene in two unrelated patients with autism spectrum disorder. Psychiatr. Genet. 26, 297–301 (2016).
Pasca, S. P. et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657–1662 (2011).
Chuang, H. C., Huang, T. N. & Hsueh, Y. P. Neuronal excitation upregulates Tbr1, a high-confidence risk gene of autism, mediating Grin2b expression in the adult brain. Front. Cell Neurosci. 8, 280 (2014).
Huang, T. N. et al. Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat. Neurosci. 17, 240–247 (2014).
Flavell, S. W. et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60, 1022–1038 (2008).
Nicholls, R. D. & Knepper, J. L. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu. Rev. Genomics Hum. Genet 2, 153–175 (2001).
Moretti, P. et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327 (2006).
Yazdani, M. et al. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells 30, 2128–2139 (2012).
Guy, J., Cheval, H., Selfridge, J. & Bird, A. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652 (2011).
Cohen, S. et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72, 72–85 (2011).
Ebert, D. H. et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499, 341–345 (2013).
Gabel, H. W. et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93 (2015).
Chahrour, M. et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229 (2008).
Ben-Shachar, S., Chahrour, M., Thaller, C., Shaw, C. A. & Zoghbi, H. Y. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 18, 2431–2442 (2009).
Malishkevich, A. et al. Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl. Psychiatry 5, e501 (2015).
Wilkerson, J. R. et al. A role for dendritic mGluR5-mediated local translation of Arc/Arg3.1 in MEF2-dependent synapse elimination. Cell Rep. 7, 1589–1600 (2014).
Tsai, N. P. et al. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594 (2012).
Vatsa, N. & Jana, N. R. UBE3A and its link with autism. Front. Mol. Neurosci. 11, 448 (2018).
Straub, J. et al. Genetic interaction screen for severe neurodevelopmental disorders reveals a functional link between Ube3a and Mef2 in Drosophila melanogaster. Sci. Rep. 10, 1204 (2020).
Flavell, S. W. et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012 (2006).
Zhou, Z. et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269 (2006).
Barbosa, A. C. et al. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl Acad. Sci. USA 105, 9391–9396 (2008).
Kwan, V., Unda, B. K. & Singh, K. K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 8, 45 (2016).
Durak, O. et al. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 19, 1477–1488 (2016).
Katayama, Y. et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679 (2016).
Caracci, M. O., Avila, M. E. & De Ferrari, G. V. Synaptic Wnt/GSK3beta signaling hub in autism. Neural Plast. 2016, 9603751 (2016).
Oliva, C. A., Vargas, J. Y. & Inestrosa, N. C. Wnts in adult brain: from synaptic plasticity to cognitive deficiencies. Front. Cell Neurosci. 7, 224 (2013).
Stamatakou, E. & Salinas, P. C. Postsynaptic assembly: a role for Wnt signaling. Dev. Neurobiol. 74, 818–827 (2014).
Judson, M. C., Eagleson, K. L. & Levitt, P. A new synaptic player leading to autism risk: Met receptor tyrosine kinase. J. Neurodev. Disord. 3, 282–292 (2011).
MacDonald, B. T. & He, X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb. Perspect. Biol. 4, a007880 (2012).
de Ligt, J. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 (2012).
Cotney, J. et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 6, 6404 (2015).
Thompson, B. A., Tremblay, V., Lin, G. & Bochar, D. A. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell Biol. 28, 3894–3904 (2008).
O’Roak, B. J. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012).
Nishiyama, M., Skoultchi, A. I. & Nakayama, K. I. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-beta-catenin signaling pathway. Mol. Cell Biol. 32, 501–512 (2012).
Chen, Y., Huang, W. C., Sejourne, J., Clipperton-Allen, A. E. & Page, D. T. Pten mutations alter brain growth trajectory and allocation of cell types through elevated beta-catenin signaling. J. Neurosci. 35, 10252–10267 (2015).
Xing, L. et al. Layer specific and general requirements for ERK/MAPK signaling in the developing neocortex. Elife 5, e11123 (2016).
Winden, K. D., Ebrahimi-Fakhari, D. & Sahin, M. Abnormal mTOR activation in autism. Annu. Rev. Neurosci. 41, 1–23 (2018).
Lipton, J. O. & Sahin, M. The neurology of mTOR. Neuron 84, 275–291 (2014).
Tang, G. et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 (2014).
Zhou, J. et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 29, 1773–1783 (2009).
Darnell, J. C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011).
Uysal, S. P. & Sahin, M. Tuberous sclerosis: a review of the past, present, and future. Turk. J. Med. Sci. 50, 1665–1676 (2020).
Ehninger, D. & Silva, A. J. Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol. Med. 17, 78–87 (2011).
Kwiatkowski, D. J. & Manning, B. D. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 14 Spec, R251–R258 (2005).
Ruvinsky, I. & Meyuhas, O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 31, 342–348 (2006).
Wullschleger, S., Loewith, R. & Hall, M. N. TOR signaling in growth and metabolism. Cell 124, 471–484 (2006).
Hong, E. J., McCord, A. E. & Greenberg, M. E. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron 60, 610–624 (2008).
Ebert, D. H. & Greenberg, M. E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337 (2013).
Tan, M. H. et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 88, 42–56 (2011).
Kidd, S. A. et al. Fragile X syndrome: a review of associated medical problems. Pediatrics 134, 995–1005 (2014).
Korb, E. et al. Excess TRanslation of Epigenetic Regulators Contributes to Fragile X syndrome and is alleviated by Brd4 inhibition. Cell 170, 1209–1223 e20 (2017).
Contractor, A., Klyachko, V. A. & Portera-Cailliau, C. Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715 (2015).
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H. & Bassell, G. J. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939 (2008).
Bassell, G. J. & Warren, S. T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214 (2008).
Niere, F., Wilkerson, J. R. & Huber, K. M. Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J. Neurosci. 32, 5924–5936 (2012).
Udagawa, T. et al. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat. Med. 19, 1473–1477 (2013).
Gross, C. et al. Selective role of the catalytic PI3K subunit p110beta in impaired higher order cognition in fragile X syndrome. Cell Rep. 11, 681–688 (2015).
Gross, C. et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 11, 727–736 (2015).
Darnell, J. C. & Klann, E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci. 16, 1530–1536 (2013).
Huber, K. M., Gallagher, S. M., Warren, S. T. & Bear, M. F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl Acad. Sci. USA 99, 7746–7750 (2002).
Napoli, I. et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054 (2008).
Budimirovic, D. B. & Kaufmann, W. E. What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 33, 379–394 (2011).
Fernandez, E., Rajan, N. & Bagni, C. The FMRP regulon: from targets to disease convergence. Front Neurosci. 7, 191 (2013).
De Rubeis, S. et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79, 1169–1182 (2013).
Santini, E. et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415 (2013).
Gkogkas, C. G. et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377 (2013).
Rossman, I. T. et al. Engrailed2 modulates cerebellar granule neuron precursor proliferation, differentiation and insulin-like growth factor 1 signaling during postnatal development. Mol. Autism 5, 9 (2014).
Lord, C. et al. Autism spectrum disorder. Nat. Rev. Dis. Prim. 6, 5 (2020).
Vaags, A. K. et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 90, 133–141 (2012).
Monteiro, P. & Feng, G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157 (2017).
Yang, Q. et al. Hippocampal synaptic metaplasticity requires the activation of NR2B-containing NMDA receptors. Brain Res. Bull. 84, 137–143 (2011).
Chevaleyre, V. & Castillo, P. E. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron 43, 871–881 (2004).
Splawski, I. et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004).
Han, S. et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390 (2012).
Hutsler, J. J. & Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94 (2010).
Lo, L. H. & Lai, K. O. Dysregulation of protein synthesis and dendritic spine morphogenesis in ASD: studies in human pluripotent stem cells. Mol. Autism 11, 40 (2020).
Grabrucker, A. M., Schmeisser, M. J., Schoen, M. & Boeckers, T. M. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21, 594–603 (2011).
Ting, J. T., Peca, J. & Feng, G. Functional consequences of mutations in postsynaptic scaffolding proteins and relevance to psychiatric disorders. Annu. Rev. Neurosci. 35, 49–71 (2012).
Naisbitt, S. et al. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582 (1999).
Uchino, S. et al. Direct interaction of post-synaptic density-95/Dlg/ZO-1 domain-containing synaptic molecule Shank3 with GluR1 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor. J. Neurochem. 97, 1203–1214 (2006).
Sudhof, T. C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 (2008).
Graf, E. R., Zhang, X., Jin, S. X., Linhoff, M. W. & Craig, A. M. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026 (2004).
Kwon, H. B. et al. Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat. Neurosci. 15, 1667–1674 (2012).
Chih, B., Engelman, H. & Scheiffele, P. Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328 (2005).
Chubykin, A. A. et al. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931 (2007).
Tabuchi, K. et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76 (2007).
Yan, J. et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 10, 329–332 (2005).
Etherton, M. et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl Acad. Sci. USA 108, 13764–13769 (2011).
Etherton, M. R., Tabuchi, K., Sharma, M., Ko, J. & Sudhof, T. C. An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 30, 2908–2919 (2011).
Hammer, M. et al. Perturbed hippocampal synaptic inhibition and gamma-oscillations in a neuroligin-4 knockout mouse model of autism. Cell Rep. 13, 516–523 (2015).
Dudanova, I., Tabuchi, K., Rohlmann, A., Sudhof, T. C. & Missler, M. Deletion of alpha-neurexins does not cause a major impairment of axonal pathfinding or synapse formation. J. Comp. Neurol. 502, 261–274 (2007).
Missler, M. et al. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948 (2003).
Boeckers, T. M., Bockmann, J., Kreutz, M. R. & Gundelfinger, E. D. ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem 81, 903–910 (2002).
Phelan, M. C. et al. 22q13 deletion syndrome. Am. J. Med. Genet. 101, 91–99 (2001).
Peca, J. et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442 (2011).
Schmeisser, M. J. et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260 (2012).
Wang, X. et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108 (2011).
Bozdagi, O. et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1, 15 (2010).
Zhou, Y. et al. Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162 (2016).
Won, H. et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265 (2012).
Peter, S. et al. Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 7, 12627 (2016).
Durand, C. M. et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 17, 71–84 (2012).
Mei, Y. et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484 (2016).
Wang, X. et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7, 11459 (2016).
Westenbroek, R. E., Merrick, D. K. & Catterall, W. A. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3, 695–704 (1989).
Van Wart, A., Trimmer, J. S. & Matthews, G. Polarized distribution of ion channels within microdomains of the axon initial segment. J. Comp. Neurol. 500, 339–352 (2007).
Ogiwara, I. et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914 (2007).
Yu, F. H. et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149 (2006).
Splawski, I. et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl Acad. Sci. USA 102, 8089–8096 (2005).
Frohler, S. et al. Exome sequencing helped the fine diagnosis of two siblings afflicted with atypical Timothy syndrome (TS2). BMC Med. Genet 15, 48 (2014).
Hiippala, A., Tallila, J., Myllykangas, S., Koskenvuo, J. W. & Alastalo, T. P. Expanding the phenotype of Timothy syndrome type 2: an adolescent with ventricular fibrillation but normal development. Am. J. Med. Genet. A 167A, 629–634 (2015).
Ma, H. et al. gammaCaMKII shuttles Ca(2)(+)/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159, 281–294 (2014).
Impey, S. et al. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 34, 235–244 (2002).
Kwok, R. P. et al. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 370, 223–226 (1994).
Kasarpalkar, N. J., Kothari, S. T. & Dave, U. P. Brain-derived neurotrophic factor in children with autism spectrum disorder. Ann. Neurosci. 21, 129–133 (2014).
Al-Otaish, H. et al. Relationship between absolute and relative ratios of glutamate, glutamine and GABA and severity of autism spectrum disorder. Metab. Brain Dis. 33, 843–854 (2018).
Lujan, R., Nusser, Z., Roberts, J. D., Shigemoto, R. & Somogyi, P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur. J. Neurosci. 8, 1488–1500 (1996).
Tu, J. C. et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592 (1999).
Bateup, H. S., Takasaki, K. T., Saulnier, J. L., Denefrio, C. L. & Sabatini, B. L. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J. Neurosci. 31, 8862–8869 (2011).
Takeuchi, K. et al. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc. Natl Acad. Sci. USA 110, 4738–4743 (2013).
Vicidomini, C. et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 22, 689–702 (2017).
Baudouin, S. J. et al. Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338, 128–132 (2012).
Peixoto, R. T. et al. Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 76, 396–409 (2012).
Mabb, A. M. & Ehlers, M. D. Ubiquitination in postsynaptic function and plasticity. Annu. Rev. Cell Dev. Biol. 26, 179–210 (2010).
Greer, P. L. et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140, 704–716 (2010).
Yashiro, K. et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783 (2009).
Huang, J. Y. et al. Functional genomic analyses identify pathways dysregulated in animal model of autism. CNS Neurosci. Ther. 22, 845–853 (2016).
Campbell, R. R. & Wood, M. A. How the epigenome integrates information and reshapes the synapse. Nat. Rev. Neurosci. 20, 133–147 (2019).
Lv, J., Xin, Y., Zhou, W. & Qiu, Z. The epigenetic switches for neural development and psychiatric disorders. J. Genet. Genomics 40, 339–346 (2013).
Issler, O. & Chen, A. Determining the role of microRNAs in psychiatric disorders. Nat. Rev. Neurosci. 16, 201–212 (2015).
Gudenas, B. L., Srivastava, A. K. & Wang, L. Integrative genomic analyses for identification and prioritization of long non-coding RNAs associated with autism. PLoS ONE 12, e0178532 (2017).
Spadaro, P. A. et al. Long Noncoding RNA-Directed Epigenetic Regulation of Gene Expression Is Associated With Anxiety-like Behavior in Mice. Biol. Psychiatry 78, 848–859 (2015).
Jang, H. S., Shin, W. J., Lee, J. E. & Do, J. T. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes 8, 148 (2017).
Dolinoy, D. C., Weidman, J. R. & Jirtle, R. L. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod. Toxicol. 23, 297–307 (2007).
Tremblay, M. W. & Jiang, Y. H. DNA methylation and susceptibility to autism spectrum disorder. Annu Rev. Med. 70, 151–166 (2019).
Della Ragione, F., Vacca, M., Fioriniello, S., Pepe, G. & D’Esposito, M. MECP2, a multi-talented modulator of chromatin architecture. Brief. Funct. Genomics 15, 420–431 (2016).
Nagarajan, R. P., Hogart, A. R., Gwye, Y., Martin, M. R. & LaSalle, J. M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 1, e1–e11 (2006).
Kuwano, Y. et al. Autism-associated gene expression in peripheral leucocytes commonly observed between subjects with autism and healthy women having autistic children. PLoS ONE 6, e24723 (2011).
Nan, X., Campoy, F. J. & Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481 (1997).
Meehan, R. R., Lewis, J. D. & Bird, A. P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 20, 5085–5092 (1992).
Jones, P. L. et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191 (1998).
Zhubi, A., Chen, Y., Guidotti, A. & Grayson, D. R. Epigenetic regulation of RELN and GAD1 in the frontal cortex (FC) of autism spectrum disorder (ASD) subjects. Int. J. Dev. Neurosci. 62, 63–72 (2017).
Zhubi, A. et al. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 4, e349 (2014).
Waga, C. et al. Identification of two novel Shank3 transcripts in the developing mouse neocortex. J. Neurochem. 128, 280–293 (2014).
Jack, A., Connelly, J. J. & Morris, J. P. DNA methylation of the oxytocin receptor gene predicts neural response to ambiguous social stimuli. Front. Hum. Neurosci. 6, 280 (2012).
Baribeau, D. A. et al. Oxytocin receptor polymorphisms are differentially associated with social abilities across neurodevelopmental disorders. Sci. Rep. 7, 11618 (2017).
Jacob, S. et al. Association of the oxytocin receptor gene (OXTR) in Caucasian children and adolescents with autism. Neurosci. Lett. 417, 6–9 (2007).
Kosaka, H. et al. Oxytocin efficacy is modulated by dosage and oxytocin receptor genotype in young adults with high-functioning autism: a 24-week randomized clinical trial. Transl. Psychiatry 6, e872 (2016).
Mamrut, S. et al. DNA methylation of specific CpG sites in the promoter region regulates the transcription of the mouse oxytocin receptor. PLoS ONE 8, e56869 (2013).
Gregory, S. G. et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 7, 62 (2009).
Andari, E. et al. Epigenetic modification of the oxytocin receptor gene: implications for autism symptom severity and brain functional connectivity. Neuropsychopharmacology 45, 1150–1158 (2020).
Sun, W. et al. Histone acetylome-wide association study of autism spectrum disorder. Cell 167, 1385–1397.e11 (2016).
Phiel, C. J. et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276, 36734–36741 (2001).
Christensen, J. et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309, 1696–1703 (2013).
Qin, L. et al. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 21, 564–575 (2018).
Shilatifard, A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 20, 341–348 (2008).
Gupta, S. et al. Histone methylation regulates memory formation. J. Neurosci. 30, 3589–3599 (2010).
Shulha, H. P. et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch. Gen. Psychiatry 69, 314–324 (2012).
Vallianatos, C. N. et al. Altered gene-regulatory function of KDM5C by a novel mutation associated with autism and intellectual disability. Front. Mol. Neurosci. 11, 104 (2018).
Adegbola, A., Gao, H., Sommer, S. & Browning, M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD). Am. J. Med. Genet. A 146A, 505–511 (2008).
Goncalves, T. F. et al. KDM5C mutational screening among males with intellectual disability suggestive of X-Linked inheritance and review of the literature. Eur. J. Med. Genet. 57, 138–144 (2014).
Vogel-Ciernia, A. & Wood, M. A. Neuron-specific chromatin remodeling: a missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 80, 18–27 (2014).
Barnard, R. A., Pomaville, M. B. & O’Roak, B. J. Mutations and modeling of the chromatin remodeler CHD8 define an emerging autism etiology. Front. Neurosci. 9, 477 (2015).
Hamdan, F. F. et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 10, e1004772 (2014).
Gozes, I. The cytoskeleton as a drug target for neuroprotection: the case of the autism-mutated ADNP. Biol. Chem. 397, 177–184 (2016).
Qureshi, I. A. & Mehler, M. F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 13, 528–541 (2012).
Abu-Elneel, K. et al. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics 9, 153–161 (2008).
Garbett, K. et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol. Dis. 30, 303–311 (2008).
Zhang, Y., Wang, Z. & Gemeinhart, R. A. Progress in microRNA delivery. J. Control Release 172, 962–974 (2013).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012).
Ng, S. Y., Johnson, R. & Stanton, L. W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31, 522–533 (2012).
Ramos, A. D. et al. The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 16, 439–447 (2015).
Cheng, Y. et al. Partial loss of psychiatric risk gene Mir137 in mice causes repetitive behavior and impairs sociability and learning via increased Pde10a. Nat. Neurosci. 21, 1689–1703 (2018).
Wu, H. et al. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl Acad. Sci. USA 107, 18161–18166 (2010).
Klein, M. E. et al. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 10, 1513–1514 (2007).
Briz, V. et al. The non-coding RNA BC1 regulates experience-dependent structural plasticity and learning. Nat. Commun. 8, 293 (2017).
Zalfa, F. et al. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327 (2003).
Noriega, D. B. & Savelkoul, H. F. Immune dysregulation in autism spectrum disorder. Eur. J. Pediatr. 173, 33–43 (2014).
Estes, M. L. & McAllister, A. K. Maternal immune activation: implications for neuropsychiatric disorders. Science 353, 772–777 (2016).
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W. & Pardo, C. A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81 (2005).
Morgan, J. T. et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 68, 368–376 (2010).
Suzuki, K. et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 70, 49–58 (2013).
Laurence, J. A. & Fatemi, S. H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 4, 206–210 (2005).
Edmonson, C., Ziats, M. N. & Rennert, O. M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism 5, 3 (2014).
Masi, A., Glozier, N., Dale, R. & Guastella, A. J. The immune system, cytokines, and biomarkers in autism spectrum disorder. Neurosci. Bull. 33, 194–204 (2017).
Li, X. et al. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 207, 111–116 (2009).
Meyer, U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 75, 307–315 (2014).
Choi, G. B. et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939 (2016).
Ponzio, N. M., Servatius, R., Beck, K., Marzouk, A. & Kreider, T. Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann. N. Y Acad. Sci. 1107, 118–128 (2007).
Lucchina, L. & Depino, A. M. Altered peripheral and central inflammatory responses in a mouse model of autism. Autism Res. 7, 273–289 (2014).
Heo, Y., Zhang, Y., Gao, D., Miller, V. M. & Lawrence, D. A. Aberrant immune responses in a mouse with behavioral disorders. PLoS ONE 6, e20912 (2011).
Wang, H. et al. Cathepsin B inhibition ameliorates leukocyte-endothelial adhesion in the BTBR mouse model of autism. CNS Neurosci. Ther. 25, 476–485 (2019).
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P. & Malik, A. B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal 20, 1126–1167 (2014).
Banks, W. A., Kastin, A. J. & Broadwell, R. D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 2, 241–248 (1995).
Ashwood, P. et al. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 25, 40–45 (2011).
Krakowiak, P. et al. Neonatal cytokine profiles associated with autism spectrum disorder. Biol. Psychiatry 81, 442–451 (2017).
Masi, A. et al. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Mol. Psychiatry 20, 440–446 (2015).
Knuesel, I. et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 10, 643–660 (2014).
Garay, P. A. & McAllister, A. K. Novel roles for immune molecules in neural development: implications for neurodevelopmental disorders. Front. Synaptic Neurosci. 2, 136 (2010).
Hsiao, E. Y., McBride, S. W., Chow, J., Mazmanian, S. K. & Patterson, P. H. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl Acad. Sci. USA 109, 12776–12781 (2012).
Careaga, M., Murai, T. & Bauman, M. D. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol. Psychiatry 81, 391–401 (2017).
Bauman, M. D. et al. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol. Psychiatry 75, 332–341 (2014).
Meyer, U. et al. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol. Psychiatry 13, 208–221 (2008).
McElhanon, B. O., McCracken, C., Karpen, S. & Sharp, W. G. Gastrointestinal symptoms in autism spectrum disorder: a meta-analysis. Pediatrics 133, 872–883 (2014).
Lee, M. et al. Association of autism spectrum disorders and inflammatory Bowel disease. J. Autism Dev. Disord. 48, 1523–1529 (2018).
Wang, L. et al. Low relative abundances of the mucolytic bacterium Akkermansia muciniphila and Bifidobacterium spp. in feces of children with autism. Appl Environ. Microbiol. 77, 6718–6721 (2011).
Dan, Z. et al. Altered gut microbial profile is associated with abnormal metabolism activity of Autism Spectrum Disorder. Gut Microbes 11, 1246–1267 (2020).
Hughes, H. K., Rose, D. & Ashwood, P. The gut microbiota and dysbiosis in autism spectrum disorders. Curr. Neurol. Neurosci. Rep. 18, 81 (2018).
Needham, B. D. et al. Plasma and fecal metabolite profiles in autism spectrum disorder. Biol. Psychiatry 89, 451–462 (2021).
Levi Mortera, S. et al. A metaproteomic-based gut microbiota profiling in children affected by autism spectrum disorders. J. Proteom. 251, 104407 (2022).
Parracho, H. M., Bingham, M. O., Gibson, G. R. & McCartney, A. L. Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 54, 987–991 (2005).
Finegold, S. M., Downes, J. & Summanen, P. H. Microbiology of regressive autism. Anaerobe 18, 260–262 (2012).
de Theije, C. G. et al. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun. 37, 197–206 (2014).
Liu, F., Horton-Sparks, K., Hull, V., Li, R. W. & Martinez-Cerdeno, V. The valproic acid rat model of autism presents with gut bacterial dysbiosis similar to that in human autism. Mol. Autism 9, 61 (2018).
Sgritta, M. et al. Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron 101, 246–259 e6 (2019).
Tabouy, L. et al. Dysbiosis of microbiome and probiotic treatment in a genetic model of autism spectrum disorders. Brain Behav. Immun. 73, 310–319 (2018).
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O. & Kasper, D. L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118 (2005).
Erny, D. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977 (2015).
Goshen, I. et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 32, 1106–1115 (2007).
Pavlowsky, A. et al. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr. Biol. 20, 103–115 (2010).
Gruol, D. L. IL-6 regulation of synaptic function in the CNS. Neuropharmacology 96, 42–54 (2015).
Wei, H. et al. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim. Biophys. Acta 1822, 831–842 (2012).
Diniz, L. P., Matias, I. C., Garcia, M. N. & Gomes, F. C. Astrocytic control of neural circuit formation: highlights on TGF-beta signaling. Neurochem Int 78, 18–27 (2014).
Nagakura, I., Van Wart, A., Petravicz, J., Tropea, D. & Sur, M. STAT1 regulates the homeostatic component of visual cortical plasticity via an AMPA receptor-mediated mechanism. J. Neurosci. 34, 10256–10263 (2014).
Malemud, C. J. Negative regulators of JAK/STAT signaling in rheumatoid arthritis and osteoarthritis. Int. J. Mol. Sci. 18, 484 (2017).
Ben Achour, S. & Pascual, O. Glia: the many ways to modulate synaptic plasticity. Neurochem. Int. 57, 440–445 (2010).
Paolicelli, R. C. et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011).
Zhan, Y. et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406 (2014).
Schafer, D. P., Lehrman, E. K. & Stevens, B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61, 24–36 (2013).
Sarn, N. et al. Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol. Psychiatry 26, 1458–1471 (2021).
Xavier, A. L., Menezes, J. R., Goldman, S. A. & Nedergaard, M. Fine-tuning the central nervous system: microglial modelling of cells and synapses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130593 (2014).
Rogers, J. T. et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250 (2011).
Schafer, D. P. et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012).
Eroglu, C. & Barres, B. A. Regulation of synaptic connectivity by glia. Nature 468, 223–231 (2010).
Schummers, J., Yu, H. & Sur, M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320, 1638–1643 (2008).
Liu, X. X. et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 217, e20180992 (2020).
Haydon, P. G. & Nedergaard, M. How do astrocytes participate in neural plasticity? Cold Spring Harb. Perspect. Biol. 7, a020438 (2014).
Higashimori, H. et al. Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum. Mol. Genet. 22, 2041–2054 (2013).
Tyndall, S. J. & Walikonis, R. S. The receptor tyrosine kinase Met and its ligand hepatocyte growth factor are clustered at excitatory synapses and can enhance clustering of synaptic proteins. Cell Cycle 5, 1560–1568 (2006).
Nakano, M. et al. Hepatocyte growth factor promotes the number of PSD-95 clusters in young hippocampal neurons. Exp. Neurol. 207, 195–202 (2007).
Campbell, D. B. et al. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann. Neurol. 62, 243–250 (2007).
Rudie, J. D. et al. Autism-associated promoter variant in MET impacts functional and structural brain networks. Neuron 75, 904–915 (2012).
Okunishi, K. et al. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J. Immunol. 175, 4745–4753 (2005).
Ido, A., Numata, M., Kodama, M. & Tsubouchi, H. Mucosal repair and growth factors: recombinant human hepatocyte growth factor as an innovative therapy for inflammatory bowel disease. J. Gastroenterol. 40, 925–931 (2005).
Corriveau, R. A., Huh, G. S. & Shatz, C. J. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21, 505–520 (1998).
Elmer, B. M., Estes, M. L., Barrow, S. L. & McAllister, A. K. MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J. Neurosci. 33, 13791–13804 (2013).
Lee, H. et al. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 509, 195–200 (2014).
Goddard, C. A., Butts, D. A. & Shatz, C. J. Regulation of CNS synapses by neuronal MHC class I. Proc. Natl Acad. Sci. USA 104, 6828–6833 (2007).
Qiu, S., Anderson, C. T., Levitt, P. & Shepherd, G. M. Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated Met receptor tyrosine kinase. J. Neurosci. 31, 5855–5864 (2011).
Bauman, M. L. & Kemper, T. L. Neuroanatomic observations of the brain in autism: a review and future directions. Int. J. Dev. Neurosci. 23, 183–187 (2005).
Minshew, N. J. & Williams, D. L. The new neurobiology of autism: cortex, connectivity, and neuronal organization. Arch. Neurol. 64, 945–950 (2007).
Carper, R. A. & Courchesne, E. Localized enlargement of the frontal cortex in early autism. Biol. Psychiatry 57, 126–133 (2005).
Vissers, M. E., Cohen, M. X. & Geurts, H. M. Brain connectivity and high functioning autism: a promising path of research that needs refined models, methodological convergence, and stronger behavioral links. Neurosci. Biobehav. Rev. 36, 604–625 (2012).
Wegiel, J. et al. Brain-region-specific alterations of the trajectories of neuronal volume growth throughout the lifespan in autism. Acta Neuropathol. Commun. 2, 28 (2014).
Wegiel, J. et al. Neuronal nucleus and cytoplasm volume deficit in children with autism and volume increase in adolescents and adults. Acta Neuropathol. Commun. 3, 2 (2015).
Casanova, M. F. The neuropathology of autism. Brain Pathol. 17, 422–433 (2007).
Zhang, C. et al. Dynamics of a disinhibitory prefrontal microcircuit in controlling social competition. Neuron 110, 516–531.e6 (2022).
Zhou, T. et al. History of winning remodels thalamo-PFC circuit to reinforce social dominance. Science 357, 162–168 (2017).
Langen, M., Durston, S., Kas, M. J., van Engeland, H. & Staal, W. G. The neurobiology of repetitive behavior:…and men. Neurosci. Biobehav Rev. 35, 356–365 (2011).
Yu, X. et al. Reducing Astrocyte Calcium Signaling In Vivo Alters Striatal Microcircuits and Causes Repetitive Behavior. Neuron 99, 1170–1187 e9 (2018).
Platt, R. J. et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. 19, 335–350 (2017).
Blundell, J. et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 30, 2115–2129 (2010).
Rothwell, P. E. et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158, 198–212 (2014).
Sah, P. Fear, anxiety, and the amygdala. Neuron 96, 1–2 (2017).
Adhikari, A. et al. Basomedial amygdala mediates top-down control of anxiety and fear. Nature 527, 179–185 (2015).
Ferrara, N. C., Trask, S. & Rosenkranz, J. A. Maturation of amygdala inputs regulate shifts in social and fear behaviors: a substrate for developmental effects of stress. Neurosci. Biobehav. Rev. 125, 11–25 (2021).
Whitney, E. R., Kemper, T. L., Bauman, M. L., Rosene, D. L. & Blatt, G. J. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum 7, 406–416 (2008).
Wegiel, J. et al. Contribution of olivofloccular circuitry developmental defects to atypical gaze in autism. Brain Res. 1512, 106–122 (2013).
Tsai, P. T. et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651 (2012).
Reith, R. M. et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 51, 93–103 (2013).
Liu, D. et al. Autistic-like behavior and cerebellar dysfunction in Bmal1 mutant mice ameliorated by mTORC1 inhibition. Mol. Psychiatry (2022).
Miterko, L. N. et al. Consensus Paper: Experimental Neurostimulation of the Cerebellum. Cerebellum 18, 1064–1097 (2019).
Bruchhage, M. M. K., Bucci, M. P. & Becker, E. B. E. Cerebellar involvement in autism and ADHD. Handb. Clin. Neurol. 155, 61–72 (2018).
Wang, S. S., Kloth, A. D. & Badura, A. The cerebellum, sensitive periods, and autism. Neuron 83, 518–532 (2014).
Silverman, J. L., Yang, M., Lord, C. & Crawley, J. N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502 (2010).
Rennie, S. M., Moita, M. M. & Mainen, Z. F. Social cognition in the rodent: nothing to be sniffed at. Trends Cogn. Sci. 17, 306–307 (2013).
Smith, M. L., Asada, N. & Malenka, R. C. Anterior cingulate inputs to nucleus accumbens control the social transfer of pain and analgesia. Science 371, 153–159 (2021).
Burkett, J. P. et al. Oxytocin-dependent consolation behavior in rodents. Science 351, 375–378 (2016).
Insel, T. R. & Fernald, R. D. How the brain processes social information: searching for the social brain. Annu. Rev. Neurosci. 27, 697–722 (2004).
Stanley, D. A. & Adolphs, R. Toward a neural basis for social behavior. Neuron 80, 816–826 (2013).
Gunaydin, L. A. et al. Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551 (2014).
Lerner, T. N., Ye, L. & Deisseroth, K. Communication in neural circuits: tools, opportunities, and challenges. Cell 164, 1136–1150 (2016).
Gangopadhyay, P., Chawla, M., Dal Monte, O. & Chang, S. W. C. Prefrontal-amygdala circuits in social decision-making. Nat. Neurosci. 24, 5–18 (2021).
Huang, W. C., Zucca, A., Levy, J. & Page, D. T. Social behavior is modulated by valence-encoding mPFC-amygdala sub-circuitry. Cell Rep. 32, 107899 (2020).
Murugan, M. et al. Combined social and spatial coding in a descending projection from the prefrontal cortex. Cell 171, 1663–1677 e16 (2017).
Ferguson, B. R. & Gao, W. J. Thalamic control of cognition and social behavior via regulation of gamma-aminobutyric acidergic signaling and excitation/inhibition balance in the medial prefrontal cortex. Biol. Psychiatry 83, 657–669 (2018).
Stoodley, C. J. et al. Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat. Neurosci. 20, 1744–1751 (2017).
Kelly, E. et al. Regulation of autism-relevant behaviors by cerebellar-prefrontal cortical circuits. Nat. Neurosci. 23, 1102–1110 (2020).
Fernandez, M., Mollinedo-Gajate, I. & Penagarikano, O. Neural circuits for social cognition: implications for autism. Neuroscience 370, 148–162 (2018).
Pagan, C. et al. The serotonin-N-acetylserotonin-melatonin pathway as a biomarker for autism spectrum disorders. Transl. Psychiatry 4, e479 (2014).
Muller, C. L., Anacker, A. M. J. & Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 321, 24–41 (2016).
Melke, J. et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry 13, 90–98 (2008).
Farook, M. F. et al. Altered serotonin, dopamine and norepinepherine levels in 15q duplication and Angelman syndrome mouse models. PLoS One 7, e43030 (2012).
Marotta, R. et al. The neurochemistry of autism. Brain Sci. 10, 163 (2020).
Yizhar, O. et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178 (2011).
Antoine, M. W., Langberg, T., Schnepel, P. & Feldman, D. E. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101, 648–661.e4 (2019).
Citri, A. & Malenka, R. C. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41 (2008).
Rubenstein, J. L. & Merzenich, M. M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267 (2003).
Qin, L. et al. Deficiency of autism risk factor ASH1L in prefrontal cortex induces epigenetic aberrations and seizures. Nat. Commun. 12, 6589 (2021).
Siegel-Ramsay, J. E. et al. Glutamate and functional connectivity—support for the excitatory-inhibitory imbalance hypothesis in autism spectrum disorders. Psychiatry Res. Neuroimaging 313, 111302 (2021).
Zheng, Z., Zhu, T., Qu, Y. & Mu, D. Blood glutamate levels in autism spectrum disorder: a systematic review and meta-analysis. PLoS ONE 11, e0158688 (2016).
Chao, H. T., Zoghbi, H. Y. & Rosenmund, C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65 (2007).
Stoner, R. et al. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 370, 1209–1219 (2014).
Nisar, S. et al. Genetics of glutamate and its receptors in autism spectrum disorder. Mol. Psychiatry 27, 2380–2392 (2022).
Oblak, A., Gibbs, T. T. & Blatt, G. J. Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res. 2, 205–219 (2009).
Oblak, A. L., Gibbs, T. T. & Blatt, G. J. Decreased GABA(B) receptors in the cingulate cortex and fusiform gyrus in autism. J. Neurochem 114, 1414–1423 (2010).
Yip, J., Soghomonian, J. J. & Blatt, G. J. Increased GAD67 mRNA expression in cerebellar interneurons in autism: implications for Purkinje cell dysfunction. J. Neurosci. Res. 86, 525–530 (2008).
Yip, J., Soghomonian, J. J. & Blatt, G. J. Decreased GAD65 mRNA levels in select subpopulations of neurons in the cerebellar dentate nuclei in autism: an in situ hybridization study. Autism Res. 2, 50–59 (2009).
Yip, J., Soghomonian, J. J. & Blatt, G. J. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol. 113, 559–568 (2007).
Fatemi, S. H. et al. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 52, 805–810 (2002).
Vogt, D., Cho, K. K. A., Lee, A. T., Sohal, V. S. & Rubenstein, J. L. R. The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep. 11, 944–956 (2015).
Goffin, D., Brodkin, E. S., Blendy, J. A., Siegel, S. J. & Zhou, Z. Cellular origins of auditory event-related potential deficits in Rett syndrome. Nat. Neurosci. 17, 804–806 (2014).
Chao, H. T. et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269 (2010).
Penagarikano, O. et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246 (2011).
Filice, F., Vorckel, K. J., Sungur, A. O., Wohr, M. & Schwaller, B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 9, 10 (2016).
Paluszkiewicz, S. M., Martin, B. S. & Huntsman, M. M. Fragile X syndrome: the GABAergic system and circuit dysfunction. Dev. Neurosci. 33, 349–364 (2011).
Swanson, L. W. & Sawchenko, P. E. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu. Rev. Neurosci. 6, 269–324 (1983).
Dolen, G., Darvishzadeh, A., Huang, K. W. & Malenka, R. C. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 501, 179–184 (2013).
Gillberg, C. Not less likely than before that mean CSF HVA may be high in autism. Biol. Psychiatry 34, 746–747 (1993).
Ernst, M., Zametkin, A. J., Matochik, J. A., Pascualvaca, D. & Cohen, R. M. Low medial prefrontal dopaminergic activity in autistic children. Lancet 350, 638 (1997).
Scott-Van Zeeland, A. A., Dapretto, M., Ghahremani, D. G., Poldrack, R. A. & Bookheimer, S. Y. Reward processing in autism. Autism Res. 3, 53–67 (2010).
Bjorklund, A. & Dunnett, S. B. Dopamine neuron systems in the brain: an update. Trends Neurosci. 30, 194–202 (2007).
Yang, H. et al. Nucleus accumbens subnuclei regulate motivated behavior via direct inhibition and disinhibition of VTA dopamine subpopulations. Neuron 97, 434–449.e4 (2018).
Schultz, W. Updating dopamine reward signals. Curr. Opin. Neurobiol. 23, 229–238 (2013).
Hamid, A. A. et al. Mesolimbic dopamine signals the value of work. Nat. Neurosci. 19, 117–126 (2016).
Bromberg-Martin, E. S., Matsumoto, M. & Hikosaka, O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 68, 815–834 (2010).
Brooks, A. M. & Berns, G. S. Aversive stimuli and loss in the mesocorticolimbic dopamine system. Trends Cogn. Sci. 17, 281–286 (2013).
Shonesy, B. C. et al. Role of striatal direct pathway 2-arachidonoylglycerol signaling in sociability and repetitive behavior. Biol. Psychiatry 84, 304–315 (2018).
Wang, W. et al. Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Invest. 127, 1978–1990 (2017).
Ebstein, R. P. et al. Arginine vasopressin and oxytocin modulate human social behavior. Ann. N. Y Acad. Sci. 1167, 87–102 (2009).
LoParo, D. & Waldman, I. D. The oxytocin receptor gene (OXTR) is associated with autism spectrum disorder: a meta-analysis. Mol. Psychiatry 20, 640–646 (2015).
Munesue, T. et al. Two genetic variants of CD38 in subjects with autism spectrum disorder and controls. Neurosci. Res. 67, 181–191 (2010).
Francis, S. M. et al. Variants in adjacent oxytocin/vasopressin gene region and associations with ASD diagnosis and other autism related endophenotypes. Front. Neurosci. 10, 195 (2016).
Mens, W. B., Laczi, F., Tonnaer, J. A., de Kloet, E. R. & van Wimersma Greidanus, T. B. Vasopressin and oxytocin content in cerebrospinal fluid and in various brain areas after administration of histamine and pentylenetetrazol. Pharm. Biochem. Behav. 19, 587–591 (1983).
Romano, A., Tempesta, B., Micioni Di Bonaventura, M. V. & Gaetani, S. From autism to eating disorders and more: the role of oxytocin in neuropsychiatric disorders. Front. Neurosci. 9, 497 (2015).
Morris, J. F. & Pow, D. V. Widespread release of peptides in the central nervous system: quantitation of tannic acid-captured exocytoses. Anat. Rec. 231, 437–445 (1991).
Husarova, V. M. et al. Plasma oxytocin in children with autism and its correlations with behavioral parameters in children and parents. Psychiatry Investig. 13, 174–183 (2016).
Gordon, I. et al. Intranasal oxytocin enhances connectivity in the neural circuitry supporting social motivation and social perception in children with autism. Sci. Rep. 6, 35054 (2016).
Amico, J. A., Mantella, R. C., Vollmer, R. R. & Li, X. Anxiety and stress responses in female oxytocin deficient mice. J. Neuroendocrinol. 16, 319–324 (2004).
Menon, R. et al. Oxytocin signaling in the lateral septum prevents social fear during lactation. Curr. Biol. 28, 1066–1078.e6 (2018).
Hung, L. W. et al. Gating of social reward by oxytocin in the ventral tegmental area. Science 357, 1406–1411 (2017).
Knobloch, H. S. et al. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 73, 553–566 (2012).
Baribeau, D., Vorstman, J. & Anagnostou, E. Novel treatments in autism spectrum disorder. Curr. Opin. Psychiatry 35, 101–110 (2022).
Liu, C., Li, T., Wang, Z., Zhou, R. & Zhuang, L. Scalp acupuncture treatment for children’s autism spectrum disorders: A systematic review and meta-analysis. Medicine 98, e14880 (2019).
Tarver, J. et al. Child and parent outcomes following parent interventions for child emotional and behavioral problems in autism spectrum disorders: A systematic review and meta-analysis. Autism 23, 1630–1644 (2019).
Altenmuller, E. & Schlaug, G. Apollo’s gift: new aspects of neurologic music therapy. Prog. Brain Res. 217, 237–252 (2015).
Danial, J. T. & Wood, J. J. Cognitive behavioral therapy for children with autism: review and considerations for future research. J. Dev. Behav. Pediatr. 34, 702–715 (2013).
Hesselmark, E., Plenty, S. & Bejerot, S. Group cognitive behavioural therapy and group recreational activity for adults with autism spectrum disorders: a preliminary randomized controlled trial. Autism 18, 672–683 (2014).
Cao, G. & Harris, K. M. Developmental regulation of the late phase of long-term potentiation (L-LTP) and metaplasticity in hippocampal area CA1 of the rat. J. Neurophysiol. 107, 902–912 (2012).
Guerriero, R. M., Giza, C. C. & Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 15, 27 (2015).
Mix, A., Hoppenrath, K. & Funke, K. Reduction in cortical parvalbumin expression due to intermittent theta-burst stimulation correlates with maturation of the perineuronal nets in young rats. Dev. Neurobiol. 75, 1–11 (2015).
Rajapakse, T. & Kirton, A. Non-invasive brain stimulation in children: applications and future directions. Transl. Neurosci. 4, 217–233 (2013).
Palm, U. et al. Transcranial direct current stimulation in children and adolescents: a comprehensive review. J. Neural Transm. (Vienna) 123, 1219–1234 (2016).
Trippe, J., Mix, A., Aydin-Abidin, S., Funke, K. & Benali, A. theta burst and conventional low-frequency rTMS differentially affect GABAergic neurotransmission in the rat cortex. Exp. Brain Res 199, 411–421 (2009).
Ahmed, Z. & Wieraszko, A. Modulation of learning and hippocampal, neuronal plasticity by repetitive transcranial magnetic stimulation (rTMS). Bioelectromagnetics 27, 288–294 (2006).
Funke, K. & Benali, A. Cortical cellular actions of transcranial magnetic stimulation. Restor. Neurol. Neurosci. 28, 399–417 (2010).
Desarkar, P. et al. Assessing and stabilizing atypical plasticity in autism spectrum disorder using rTMS: results from a proof-of-principle study. Clin. Neurophysiol. (2021).
Masuda, F. et al. Motor cortex excitability and inhibitory imbalance in autism spectrum disorder assessed with transcranial magnetic stimulation: a systematic review. Transl. Psychiatry 9, 110 (2019).
Oberman, L. M. et al. Transcranial magnetic stimulation in autism spectrum disorder: challenges, promise, and roadmap for future research. Autism Res. 9, 184–203 (2016).
Amatachaya, A. et al. Effect of anodal transcranial direct current stimulation on autism: a randomized double-blind crossover trial. Behav. Neurol. 2014, 173073 (2014).
Schneider, H. D. & Hopp, J. P. The use of the Bilingual Aphasia Test for assessment and transcranial direct current stimulation to modulate language acquisition in minimally verbal children with autism. Clin. Linguist Phon. 25, 640–654 (2011).
Ameis, S. H. et al. Treatment of Executive Function Deficits in autism spectrum disorder with repetitive transcranial magnetic stimulation: A double-blind, sham-controlled, pilot trial. Brain Stimul. 13, 539–547 (2020).
Ni, H. C. et al. Intermittent theta burst stimulation over the posterior superior temporal sulcus for children with autism spectrum disorder: a 4-week randomized blinded controlled trial followed by another 4-week open-label intervention. Autism 25, 1279–1294 (2021).
Garcia-Gonzalez, S. et al. Transcranial direct current stimulation in autism spectrum disorder: a systematic review and meta-analysis. Eur. Neuropsychopharmacol. 48, 89–109 (2021).
Khaleghi, A., Zarafshan, H., Vand, S. R. & Mohammadi, M. R. Effects of non-invasive neurostimulation on autism spectrum disorder: a systematic review. Clin. Psychopharmacol. Neurosci. 18, 527–552 (2020).
Sandler, L. Risperidone in children with autism and serious behavioral problems. N. Engl. J. Med. 347, 1890–1891 (2002). author reply.
Shea, S. et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics 114, e634–e641 (2004).
Owen, R. et al. Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics 124, 1533–1540 (2009).
Marcus, R. N. et al. A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry 48, 1110–1119 (2009).
Kent, J. M. et al. Risperidone dosing in children and adolescents with autistic disorder: a double-blind, placebo-controlled study. J. Autism Dev. Disord. 43, 1773–1783 (2013).
Politte, L. C. et al. A randomized, placebo-controlled trial of extended-release guanfacine in children with autism spectrum disorder and ADHD symptoms: an analysis of secondary outcome measures. Neuropsychopharmacology 43, 1772–1778 (2018).
Baldwin, D. S. et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J. Psychopharmacol. 28, 403–439 (2014).
Nadeau, J. et al. Treatment of comorbid anxiety and autism spectrum disorders. Neuropsychiatry 1, 567–578 (2011).
Rossignol, D. A. & Frye, R. E. Melatonin in autism spectrum disorders: a systematic review and meta-analysis. Dev. Med. Child Neurol. 53, 783–792 (2011).
Takumi, T., Tamada, K., Hatanaka, F., Nakai, N. & Bolton, P. F. Behavioral neuroscience of autism. Neurosci. Biobehav. Rev. 110, 60–76 (2020).
Lalanne, S. et al. Melatonin: from pharmacokinetics to clinical use in autism spectrum disorder. Int. J. Mol. Sci. 22, 1490 (2021).
Wu, Z. Y. et al. Autism spectrum disorder (ASD): disturbance of the melatonin system and its implications. Biomed. Pharmacother. 130, 110496 (2020).
Tordjman, S. et al. Advances in the research of melatonin in autism spectrum disorders: literature review and new perspectives. Int. J. Mol. Sci. 14, 20508–20542 (2013).
Wright, B. et al. Melatonin versus placebo in children with autism spectrum conditions and severe sleep problems not amenable to behaviour management strategies: a randomised controlled crossover trial. J. Autism Dev. Disord. 41, 175–184 (2011).
Tian, Y. et al. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J. Pineal Res. 56, 1–11 (2014).
Taleb, A. et al. Emerging mechanisms of valproic acid-induced neurotoxic events in autism and its implications for pharmacological treatment. Biomed. Pharmacother. 137, 111322 (2021).
Lacivita, E., Perrone, R., Margari, L. & Leopoldo, M. Targets for drug therapy for autism spectrum disorder: challenges and future directions. J. Med. Chem. 60, 9114–9141 (2017).
Ghosh, A., Michalon, A., Lindemann, L., Fontoura, P. & Santarelli, L. Drug discovery for autism spectrum disorder: challenges and opportunities. Nat. Rev. Drug Disco. 12, 777–790 (2013).
Sahin, M. & Sur, M. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 350, aab3897 (2015).
Dolen, G. et al. Correction of fragile X syndrome in mice. Neuron 56, 955–962 (2007).
Silverman, J. L. et al. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med. 4, 131ra51 (2012).
Scharf, S. H., Jaeschke, G., Wettstein, J. G. & Lindemann, L. Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharm. 20, 124–134 (2015).
Diaz-Caneja, C. M. et al. A white paper on a neurodevelopmental framework for drug discovery in autism and other neurodevelopmental disorders. Eur. Neuropsychopharmacol. 48, 49–88 (2021).
Burket, J. A. & Deutsch, S. I. Metabotropic functions of the NMDA receptor and an evolving rationale for exploring NR2A-selective positive allosteric modulators for the treatment of autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 90, 142–160 (2019).
Joshi, G. et al. A prospective open-label trial of memantine hydrochloride for the treatment of social deficits in intellectually capable adults with autism spectrum disorder. J. Clin. Psychopharmacol. 36, 262–271 (2016).
Chez, M. G. et al. Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J. Child Neurol. 22, 574–579 (2007).
Owley, T. et al. A prospective, open-label trial of memantine in the treatment of cognitive, behavioral, and memory dysfunction in pervasive developmental disorders. J. Child Adolesc. Psychopharmacol. 16, 517–524 (2006).
Erickson, C. A., Mullett, J. E. & McDougle, C. J. Open-label memantine in fragile X syndrome. J. Autism Dev. Disord. 39, 1629–1635 (2009).
Aman, M. G. et al. Safety and efficacy of memantine in children with autism: randomized, placebo-controlled study and open-label extension. J. Child Adolesc. Psychopharmacol. 27, 403–412 (2017).
Hardan, A. Y. et al. Efficacy and safety of memantine in children with autism spectrum disorder: results from three phase 2 multicenter studies. Autism 23, 2096–2111 (2019).
Karahmadi, M., Tarrahi, M. J., Vatankhah Ardestani, S. S., Omranifard, V. & Farzaneh, B. Efficacy of memantine as adjunct therapy for autism spectrum disorder in children aged <14 years. Adv. Biomed. Res 7, 131 (2018).
Wink, L. K. et al. Brief Report: intranasal ketamine in adolescents and young adults with autism spectrum disorder-initial results of a randomized, controlled, crossover, pilot study. J. Autism Dev. Disord. 51, 1392–1399 (2021).
Wink, L. K. et al. A randomized placebo-controlled cross-over pilot study of riluzole for drug-refractory irritability in autism spectrum disorder. J. Autism Dev. Disord. 48, 3051–3060 (2018).
Minshawi, N. F. et al. A randomized, placebo-controlled trial of D-cycloserine for the enhancement of social skills training in autism spectrum disorders. Mol. Autism 7, 2 (2016).
Henderson, C. et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4, 152ra128 (2012).
Veenstra-VanderWeele, J. et al. Arbaclofen in children and adolescents with autism spectrum disorder: a randomized, controlled, phase 2 trial. Neuropsychopharmacology 42, 1390–1398 (2017).
Erickson, C. A. et al. STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J. Autism Dev. Disord. 44, 958–964 (2014).
Lemonnier, E. et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7, e1056 (2017).
Lemonnier, E. et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2, e202 (2012).
van Andel, D. M. et al. Effects of bumetanide on neurodevelopmental impairments in patients with tuberous sclerosis complex: an open-label pilot study. Mol. Autism 11, 30 (2020).
Zhang, L. et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 10, 9 (2020).
Sprengers, J. J. et al. Bumetanide for core symptoms of autism spectrum disorder (BAMBI): a single center, double-blinded, participant-randomized, placebo-controlled, phase-2 superiority trial. J. Am. Acad. Child Adolesc. Psychiatry 60, 865–876 (2021).
Overwater, I. E. et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 93, e200–e209 (2019).
Tropea, D. et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl Acad. Sci. USA 106, 2029–2034 (2009).
Kolevzon, A. et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol. Autism 5, 54 (2014).
Khwaja, O. S. et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl Acad. Sci. USA 111, 4596–4601 (2014).
Pini, G. et al. Illness severity, social and cognitive ability, and EEG analysis of ten patients with rett syndrome treated with mecasermin (recombinant human IGF-1). Autism Res. Treat. 2016, 5073078 (2016).
Ma, K. et al. Histone deacetylase inhibitor MS-275 restores social and synaptic function in a Shank3-deficient mouse model of autism. Neuropsychopharmacology 43, 1779–1788 (2018).
Rapanelli, M. et al. Targeting histone demethylase LSD1 for treatment of deficits in autism mouse models. Mol. Psychiatry (2022).
Wang, Z. J. et al. Amelioration of autism-like social deficits by targeting histone methyltransferases EHMT1/2 in Shank3-deficient mice. Mol. Psychiatry 25, 2517–2533 (2020).
Zhang, F. et al. Synergistic inhibition of histone modifiers produces therapeutic effects in adult Shank3-deficient mice. Transl. Psychiatry 11, 99 (2021).
Batebi, N. et al. Folinic acid as adjunctive therapy in treatment of inappropriate speech in children with autism: a double-blind and placebo-controlled randomized trial. Child Psychiatry Hum. Dev. 52, 928–938 (2021).
Frye, R. E. et al. Treatment of folate metabolism abnormalities in autism spectrum disorder. Semin Pediatr. Neurol. 35, 100835 (2020).
Frye, R. E. et al. Folinic acid improves verbal communication in children with autism and language impairment: a randomized double-blind placebo-controlled trial. Mol. Psychiatry 23, 247–256 (2018).
Renard, E. et al. Folinic acid improves the score of Autism in the EFFET placebo-controlled randomized trial. Biochimie 173, 57–61 (2020).
Shamay-Tsoory, S. G. & Abu-Akel, A. The social salience hypothesis of oxytocin. Biol. Psychiatry 79, 194–202 (2016).
Bertoni, A. et al. Oxytocin administration in neonates shapes hippocampal circuitry and restores social behavior in a mouse model of autism. Mol. Psychiatry 26, 7582–7595 (2021).
LeClerc, S. & Easley, D. Pharmacological therapies for autism spectrum disorder: a review. P T 40, 389–397 (2015).
Bernaerts, S., Boets, B., Bosmans, G., Steyaert, J. & Alaerts, K. Behavioral effects of multiple-dose oxytocin treatment in autism: a randomized, placebo-controlled trial with long-term follow-up. Mol. Autism 11, 6 (2020).
Yamasue, H. et al. Effect of intranasal oxytocin on the core social symptoms of autism spectrum disorder: a randomized clinical trial. Mol. Psychiatry 25, 1849–1858 (2020).
Watanabe, T. et al. Clinical and neural effects of six-week administration of oxytocin on core symptoms of autism. Brain 138, 3400–3412 (2015).
Ooi, Y. P., Weng, S. J., Kossowsky, J., Gerger, H. & Sung, M. Oxytocin and autism spectrum disorders: a systematic review and meta-analysis of randomized controlled trials. Pharmacopsychiatry 50, 5–13 (2017).
Oztan, O. et al. Cerebrospinal fluid vasopressin and symptom severity in children with autism. Ann. Neurol. 84, 611–615 (2018).
Baribeau, D. & Anagnostou, E. Novel treatments for autism spectrum disorder based on genomics and systems biology. Pharm. Ther. 230, 107939 (2022).
Parker, K. J. et al. A randomized placebo-controlled pilot trial shows that intranasal vasopressin improves social deficits in children with autism. Sci. Transl. Med. 11, eaau7356 (2019).
Bolognani, F. et al. A phase 2 clinical trial of a vasopressin V1a receptor antagonist shows improved adaptive behaviors in men with autism spectrum disorder. Sci. Transl. Med. 11, eaat7838 (2019).
Saghazadeh, A. et al. A meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: effects of age, gender, and latitude. J. Psychiatr. Res. 115, 90–102 (2019).
Heuer, L. S. et al. An exploratory examination of neonatal cytokines and chemokines as predictors of autism risk: the early markers for autism study. Biol. Psychiatry 86, 255–264 (2019).
Vuong, H. E. & Hsiao, E. Y. Emerging roles for the gut microbiome in autism spectrum disorder. Biol. Psychiatry 81, 411–423 (2017).
Desbonnet, L., Clarke, G., Shanahan, F., Dinan, T. G. & Cryan, J. F. Microbiota is essential for social development in the mouse. Mol. Psychiatry 19, 146–148 (2014).
Almasi-Nasrabadi, M. et al. Involvement of NMDA receptors in the beneficial effects of pioglitazone on scopolamine-induced memory impairment in mice. Behav. Brain Res. 231, 138–145 (2012).
Ghaleiha, A. et al. A pilot double-blind placebo-controlled trial of pioglitazone as adjunctive treatment to risperidone: Effects on aberrant behavior in children with autism. Psychiatry Res. 229, 181–187 (2015).
Capano, L. et al. A pilot dose finding study of pioglitazone in autistic children. Mol. Autism 9, 59 (2018).
Kang, D. W. et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome 5, 10 (2017).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013).
Needham, B. D., Tang, W. & Wu, W. L. Searching for the gut microbial contributing factors to social behavior in rodent models of autism spectrum disorder. Dev. Neurobiol. 78, 474–499 (2018).
Buffington, S. A. et al. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell 165, 1762–1775 (2016).
Isolauri, E., Salminen, S. & Rautava, S. Early microbe contact and obesity risk: evidence of causality? J. Pediatr. Gastroenterol. Nutr. 63, S3–S5 (2016).
Patusco, R. & Ziegler, J. Role of probiotics in managing gastrointestinal dysfunction in children with autism spectrum disorder: an update for practitioners. Adv. Nutr. 9, 637–650 (2018).
Kang, D. W. et al. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 9, 5821 (2019).
Liu, Y. W. et al. Effects of Lactobacillus plantarum PS128 on children with autism spectrum disorder in taiwan: a randomized, double-blind, placebo-controlled trial. Nutrients 11, 820 (2019).
Weuring, W., Geerligs, J. & Koeleman, B. P. C. Gene Therapies for Monogenic Autism Spectrum Disorders. Genes (Basel) 12, 1664 (2021).
Sandweiss, A. J., Brandt, V. L. & Zoghbi, H. Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: prospects for future therapies. Lancet Neurol. 19, 689–698 (2020).
Wykes, R. C. & Lignani, G. Gene therapy and editing: Novel potential treatments for neuronal channelopathies. Neuropharmacology 132, 108–117 (2018).
Radyushkin, K. et al. Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 8, 416–425 (2009).
Jamain, S. et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl Acad. Sci. USA 105, 1710–1715 (2008).
Zhang, B., Gokce, O., Hale, W. D., Brose, N. & Sudhof, T. C. Autism-associated neuroligin-4 mutation selectively impairs glycinergic synaptic transmission in mouse brainstem synapses. J. Exp. Med. 215, 1543–1553 (2018).
Grayton, H. M., Missler, M., Collier, D. A. & Fernandes, C. Altered social behaviours in neurexin 1alpha knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS ONE 8, e67114 (2013).
Etherton, M. R., Blaiss, C. A., Powell, C. M. & Sudhof, T. C. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl Acad. Sci. USA 106, 17998–18003 (2009).
Born, G. et al. Genetic targeting of NRXN2 in mice unveils role in excitatory cortical synapse function and social behaviors. Front Synaptic Neurosci. 7, 3 (2015).
Samaco, R. C. et al. Female Mecp2(+/-) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet 22, 96–109 (2013).
Samaco, R. C. et al. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat. Genet. 44, 206–211 (2012).
Jaramillo, T. C. et al. Altered striatal synaptic function and abnormal behaviour in Shank3 Exon4-9 deletion mouse model of autism. Autism Res 9, 350–375 (2016).
Duffney, L. J. et al. Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413 (2015).
Auerbach, B. D., Osterweil, E. K. & Bear, M. F. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68 (2011).
Gantois, I. et al. Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav. Brain Res. 239, 72–79 (2013).
Sato, A. et al. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 3, 1292 (2012).
Smith, S. E. et al. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3, 103ra97 (2011).
Guo, X. et al. Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of Schizophrenia. Neuropsychopharmacology 34, 1659–1672 (2009).
Clement, J. P. et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723 (2012).
Jung, E. M. et al. Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat. Neurosci. 20, 1694–1707 (2017).
Clipperton-Allen, A. E. & Page, D. T. Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum. Mol. Genet 23, 3490–3505 (2014).
Page, D. T., Kuti, O. J., Prestia, C. & Sur, M. Haploinsufficiency for Pten and Serotonin transporter cooperatively influences brain size and social behavior. Proc. Natl Acad. Sci. USA 106, 1989–1994 (2009).
Kwon, C. H. et al. Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388 (2006).
Ogawa, S. et al. A seizure-prone phenotype is associated with altered free-running rhythm in Pten mutant mice. Brain Res. 1168, 112–123 (2007).
Lugo, J. N. et al. Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front. Mol. Neurosci. 7, 27 (2014).
Amiri, A. et al. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J. Neurosci. 32, 5880–5890 (2012).
Brielmaier, J. et al. Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS ONE 7, e40914 (2012).
Phan, M. L. et al. Engrailed 2 deficiency and chronic stress alter avoidance and motivation behaviors. Behav. Brain Res. 413, 113466 (2021).
Nakatani, J. et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137, 1235–1246 (2009).
Kogan, J. H. et al. Mouse model of chromosome 15q13.3 microdeletion syndrome demonstrates features related to autism spectrum disorder. J. Neurosci. 35, 16282–16294 (2015).
Rees, K. A. et al. Molecular, physiological and behavioral characterization of the heterozygous Df[h15q13]/+ mouse model associated with the human 15q13.3 microdeletion syndrome. Brain Res. 1746, 147024 (2020).
Horev, G. et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl Acad. Sci. USA 108, 17076–17081 (2011).
Pucilowska, J. et al. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci. 35, 3190–3200 (2015).
Earls, L. R. et al. Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J. Neurosci. 30, 15843–15855 (2010).
Wong, C. T., Bestard-Lorigados, I. & Crawford, D. A. Autism-related behaviors in the cyclooxygenase-2-deficient mouse model. Genes Brain Behav. 18, e12506 (2019).
Mahmood, U. et al. Dendritic spine anomalies and PTEN alterations in a mouse model of VPA-induced autism spectrum disorder. Pharm. Res 128, 110–121 (2018).
McFarlane, H. G. et al. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. 7, 152–163 (2008).
Arakawa, H. Implication of the social function of excessive self-grooming behavior in BTBR T(+)ltpr3(tf)/J mice as an idiopathic model of autism. Physiol. Behav. 237, 113432 (2021).
Kim, J. E. et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc. Natl Acad. Sci. USA 108, 3005–3010 (2011).
Avazzadeh, S. et al. NRXN1alpha(+/−) is associated with increased excitability in ASD iPSC-derived neurons. BMC Neurosci. 22, 56 (2021).
Lam, M. et al. Single cell analysis of autism patient with bi-allelic NRXN1-alpha deletion reveals skewed fate choice in neural progenitors and impaired neuronal functionality. Exp. Cell Res. 383, 111469 (2019).
Marchetto, M. C. et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539 (2010).
Mellios, N. et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 23, 1051–1065 (2018).
Tang, X. et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl Acad. Sci. USA 113, 751–756 (2016).
Williams, E. C. et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum. Mol. Genet. 23, 2968–2980 (2014).
Nageshappa, S. et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 21, 178–188 (2016).
Huang, G. et al. Uncovering the functional link between SHANK3 deletions and deficiency in neurodevelopment using iPSC-Derived human neurons. Front Neuroanat. 13, 23 (2019).
Gouder, L. et al. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. Sci. Rep. 9, 94 (2019).
Kathuria, A. et al. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 23, 735–746 (2018).
Zaslavsky, K. et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 22, 556–564 (2019).
Liu, J. et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum. Mol. Genet 21, 3795–3805 (2012).
Zhang, Z. et al. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci. Transl. Med. 10, eaar4338 (2018).
Raj, N. et al. Cell-type-specific profiling of human cellular models of fragile X syndrome reveal PI3K-dependent defects in translation and neurogenesis. Cell Rep. 35, 108991 (2021).
Li, Y. et al. Abnormal neural progenitor cells differentiated from induced pluripotent stem cells partially mimicked development of TSC2 neurological abnormalities. Stem Cell Rep. 8, 883–893 (2017).
Zucco, A. J. et al. Neural progenitors derived from Tuberous Sclerosis Complex patients exhibit attenuated PI3K/AKT signaling and delayed neuronal differentiation. Mol. Cell Neurosci. 92, 149–163 (2018).
Winden, K. D. et al. Biallelic mutations in TSC2 lead to abnormalities associated with cortical tubers in human iPSC-derived neurons. J. Neurosci. 39, 9294–9305 (2019).
Fink, J. J. et al. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 8, 15038 (2017).
Wang, P. et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 8, 11 (2017).
Llamosas, N. et al. SYNGAP1 controls the maturation of dendrites, synaptic function, and network activity in developing human neurons. J. Neurosci. 40, 7980–7994 (2020).
Ricciardi, S. et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 14, 911–923 (2012).
Sanchez-Sanchez, S. M. et al. Rare RELN variants affect Reelin-DAB1 signal transduction in autism spectrum disorder. Hum. Mutat. 39, 1372–1383 (2018).
de Jong, J. O. et al. Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder. Nat. Commun. 12, 4087 (2021).
Mariani, J. et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162, 375–390 (2015).
Griesi-Oliveira, K. et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 20, 1350–1365 (2015).
Krey, J. F. et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 16, 201–209 (2013).
Birey, F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017).
Deneault, E. et al. CNTN5(−)(/+)or EHMT2(−)(/+)human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. Elife 8, e40092 (2019).
Fink, J. J. et al. Hyperexcitable phenotypes in induced pluripotent stem cell-derived neurons from patients with 15q11-q13 duplication syndrome, a genetic form of autism. Biol. Psychiatry 90, 756–765 (2021).
Germain, N. D. et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol. Autism 5, 44 (2014).
Meganathan, K. et al. Altered neuronal physiology, development, and function associated with a common chromosome 15 duplication involving CHRNA7. BMC Biol. 19, 147 (2021).
Deshpande, A. et al. Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Rep. 21, 2678–2687 (2017).
Khan, T. A. et al. Neuronal defects in a human cellular model of 22q11.2 deletion syndrome. Nat. Med. 26, 1888–1898 (2020).
Moore, D. et al. Downregulation of an evolutionary young miR-1290 in an iPSC-derived neural stem cell model of autism spectrum disorder. Stem Cells Int. 2019, 8710180 (2019).
Marchetto, M. C. et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 22, 820–835 (2017).
Russo, F. B. et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol. Psychiatry 83, 569–578 (2018).
Cortesi, F., Giannotti, F., Sebastiani, T., Panunzi, S. & Valente, D. Controlled-release melatonin, singly and combined with cognitive behavioural therapy, for persistent insomnia in children with autism spectrum disorders: a randomized placebo-controlled trial. J. Sleep. Res. 21, 700–709 (2012).
Ming, X., Gordon, E., Kang, N. & Wagner, G. C. Use of clonidine in children with autism spectrum disorders. Brain Dev. 30, 454–460 (2008).
Erickson, C. A. et al. A retrospective study of memantine in children and adolescents with pervasive developmental disorders. Psychopharmacol. (Berl.) 191, 141–147 (2007).
Posey, D. J. et al. A pilot study of D-cycloserine in subjects with autistic disorder. Am. J. Psychiatry 161, 2115–2117 (2004).
Mahdavinasab, S. M. et al. Baclofen as an adjuvant therapy for autism: a randomized, double-blind, placebo-controlled trial. Eur. Child Adolesc. Psychiatry 28, 1619–1628 (2019).
Hollander, E. et al. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology 28, 193–198 (2003).
Hollander, E. et al. Oxytocin increases retention of social cognition in autism. Biol. Psychiatry 61, 498–503 (2007).
Stigler, K. A., Mullett, J. E., Erickson, C. A., Posey, D. J. & McDougle, C. J. Paliperidone for irritability in adolescents and young adults with autistic disorder. Psychopharmacology 223, 237–245 (2012).
Hardan, A. Y. & Handen, B. L. A retrospective open trial of adjunctive donepezil in children and adolescents with autistic disorder. J. Child Adolesc. Psychopharmacol. 12, 237–241 (2002).
Arnold, L. E. et al. Placebo-controlled pilot trial of mecamylamine for treatment of autism spectrum disorders. J. Child Adolesc. Psychopharmacol. 22, 198–205 (2012).
Erickson, C. A. et al. An open-label naturalistic pilot study of acamprosate in youth with autistic disorder. J. Child Adolesc. Psychopharmacol. 21, 565–569 (2011).
King, B. H. et al. Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry 40, 658–665 (2001).
Hardan, A. Y. et al. A randomized controlled pilot trial of oral N-acetylcysteine in children with autism. Biol. Psychiatry 71, 956–961 (2012).
Kemner, C., Willemsen-Swinkels, S. H., de Jonge, M., Tuynman-Qua, H. & van Engeland, H. Open-label study of olanzapine in children with pervasive developmental disorder. J. Clin. Psychopharmacol. 22, 455–460 (2002).
Loebel, A. et al. Lurasidone for the treatment of irritability associated with autistic disorder. J. Autism Dev. Disord. 46, 1153–1163 (2016).
Niederhofer, H., Staffen, W. & Mair, A. Galantamine may be effective in treating autistic disorder. BMJ 325, 1422 (2002).
Gringras, P., Nir, T., Breddy, J., Frydman-Marom, A. & Findling, R. L. Efficacy and safety of pediatric prolonged-release melatonin for insomnia in children with autism spectrum disorder. J. Am. Acad. Child Adolesc. Psychiatry 56, 948–957 e4 (2017).
Malow, B. A. et al. Sleep, growth, and puberty after 2 years of prolonged-release melatonin in children with autism spectrum disorder. J. Am. Acad. Child Adolesc. Psychiatry 60, 252–261.e3 (2021).
Sikich, L. et al. Intranasal oxytocin in children and adolescents with autism spectrum disorder. N. Engl. J. Med. 385, 1462–1473 (2021).
Aye, S. Z. et al. The effectiveness and adverse effects of D-cycloserine compared with placebo on social and communication skills in individuals with autism spectrum disorder. Cochrane Database Syst. Rev. 2, CD013457 (2021).
Derks, M. et al. Bioavailability and pharmacokinetic profile of balovaptan, a selective, brain-penetrant vasopressin 1a receptor antagonist, in healthy volunteers. Expert Opin. Investig. Drugs 30, 893–901 (2021).
Jacob, S. et al. Efficacy and safety of balovaptan for socialisation and communication difficulties in autistic adults in North America and Europe: a phase 3, randomised, placebo-controlled trial. Lancet Psychiatry 9, 199–210 (2022).
McDougle, C. J. et al. A randomized double-blind, placebo-controlled pilot trial of mirtazapine for anxiety in children and adolescents with autism spectrum disorder. Neuropsychopharmacology 47, 1263–1270 (2022).
Acknowledgements
This work was supported by the National Innovation of Science and Technology-2030 (Program of Brain Science and Brain-Inspired Intelligence Technology, Grant 2021ZD0204002).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.-C.J., Y.-M.L. and F.H. Investigation: C.-CJ., L.-S.L., S.L. and X.-Y.K. Funding acquisition: C.-C.J., L.-S.L., Y.-M.L. and F.H. Project administration: C.-C.J., Y.-M.L. and F.H. Supervision: Y.-M.L. and F.H. Writing-original draft: C.-C.J., Y.-M.L. and F.H. Writing-review and editing: C.-C.J., L.-S.L., S.L., X.-Y.K., K.F., Y.-M.L. and F.H. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, CC., Lin, LS., Long, S. et al. Signalling pathways in autism spectrum disorder: mechanisms and therapeutic implications. Sig Transduct Target Ther 7, 229 (2022). https://doi.org/10.1038/s41392-022-01081-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01081-0
This article is cited by
-
Gephyrin phosphorylation facilitates sexually dimorphic development and function of parvalbumin interneurons in the mouse hippocampus
Molecular Psychiatry (2024)
-
Loss-of-function variant in spermidine/spermine N1-acetyl transferase like 1 (SATL1) gene as an underlying cause of autism spectrum disorder
Scientific Reports (2024)
-
Current and future applications of light-sheet imaging for identifying molecular and developmental processes in autism spectrum disorders
Molecular Psychiatry (2024)
-
Deficiency of FRMD5 results in neurodevelopmental dysfunction and autistic-like behavior in mice
Molecular Psychiatry (2024)
-
Structural insights into the functional mechanism of the ubiquitin ligase E6AP
Nature Communications (2024)
