
Abstract
Programmed death ligand 1 (PD-L1, CD274) is an essential immune checkpoint protein that binds to programmed death 1 (PD-1) on T-lymphocytes. T cell plays a critical role in killing cancer cells while the cancer cell exhibits immune escape by the expression of PD-L1. The binding of PD-L1 to PD-1 inhibits T cell proliferation and activity, leading to tumor immunosuppression. Increasing evidence shows that PD-L1 protein undergoes degradation in proteasomes or lysosomes by multiple pathways, leading to enhanced immunotherapy for cancer. Although some specific drugs induce PD-L1 degradation and increase antitumor activity, the combination of these drugs with PD-L1/PD-1 blockade significantly enhances cancer immunotherapy. In this review, we have discussed the interaction of PD-L1 degradation with cancer immunotherapy.
Similar content being viewed by others


Facts
-
1.
PD-L1 is an essential immune checkpoint protein that binds to PD-1 on T cells, which plays a critical role in killing cancer cells, while cancer cell exhibits immune escape by the expression of PD-L1.
-
2.
Increasing evidence shows that PD-L1 protein will be degraded in proteasomes or lysosomes, leading to enhanced immunotherapy for cancer.
-
3.
Some specific drugs or a combination of these drugs with PD-L1/PD-1 blockade inhibitors can effectively enhance antitumor immunotherapy.
Open questions
-
1.
How does GSK3β or AMPK induce the extracellular fragment of PD-L1 phosphorylation?
-
2.
It remains unclear that how membrane PD-L1 protein can be translocated into the cytoplasm and degraded. Is there any other E3 ligase or autophagy receptor for PD-L1 degradation by proteasomes or lysosomes?
-
3.
Does the FDA-approved agents that target PD-L1 (atezolizumab, etc.) or PD-1 (nivolumab, etc.) induce PD-L1 degradation?
-
4.
Although some specific drugs or a combination of these drugs with PD-L1/PD-1 blockade inhibitors can effectively enhance antitumor immunotherapy, the mechanism of PD-L1 degradation remains unclear.
Introduction
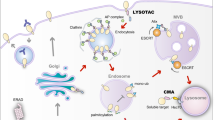
The host immune system exhibits the ability of antitumor activity by activation of the immune response1,2. As a “don’t find me” signal, the programmed death ligand 1 (PD-L1), a critical immune checkpoint protein, binds to programmed death 1 (PD-1) on T cells, leading to cancer immunosuppression3. The binding of PD-L1 to PD-1 on T cells results in the dephosphorylation of the T-cell receptor (SHP-1/2). It inhibits T cells from killing cancer cells by reducing T cell proliferation and activity4. In contrast, the immune checkpoint inhibitors such as PD-L1 or PD-1 monoclonal antibodies have been used for cancer treatment, including melanoma, non-small-cell lung cancer, gastric cancer, and breast cancer5. Although PD-1/PD-L1 blockade therapy exhibits significant clinical benefits for multiple types of cancer, the response rates of patients are less than 40% with an unclear mechanism6. The high expression of PD-L1 protein levels is observed in different types of cancers, which promotes cancer cell immune escape5,7. The expression of PD-L1 in cancer cells is regulated by multiple signaling pathways, including NFκB, MAPK, mTOR, STAT, and c-Myc8,9, while PD-L1 protein undergoes degradation in proteasomes or lysosomes by multiple pathways10,11,12,13,14,15,16, leading to increased effectiveness of cancer immunotherapy (Figs. 1 and 2 and Tables 1 and 2).

PD-L1 undergoes ubiquitination and degradation by E3 ubiquitin ligases, including STUB1, Cullin3SPOP, and β-TrCP, which is abolished by CMTM4/6, CSN5, and STT3. Although glycosylation of PD-L1 increases its protein stability, the AMPK agonist or EGFR inhibitor reverses this process and induces PD-L1 proteasome-dependent degradation. Moreover, in response to extracellular stimuli, PD-L1 protein triggers ubiquitination and degradation by multiple pathways.

HIP1R, PKCα/GSK3β/MITF, ADAM10/17, and endosomal sorting-signal induce PD-L1 protein degradation by autophagy, which is inhibited by CMTM6, DHHC3, and Sigma I. In response to extracellular stimuli or specific anti-PD-L1 antibody such as HA or STM108, PD-L1 protein is degraded via autophagy.
The pathways of PD-L1 ubiquitination and degradation
The ubiquitin-proteasome system plays an important role in the regulation of protein stability, which consists of ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) that delivers ubiquitin from E2 to the specific substrates17,18,19. PD-L1 undergoes ubiquitination and degradation by E3 ubiquitin ligases such as STUB110, Cullin3SPOP 11, and β-TrCP (β-transducin repeat-containing protein)12,13. Although STUB1 ubiquitin ligase destabilizes PD-L1 protein by inducing its lysosomal degradation in A375 melanoma cells10, the mechanism is still unclear. In contrast, Zhang et al.11 described the detailed mechanism of PD-L1 ubiquitination and degradation by the cyclinD-CDK4/SPOP/Cdh1 pathway. Mechanistically, cyclinD-CDK4 mainly induced SPOP phosphorylation at serine-6, resulting in the recruitment of 14-3-3γ to SPOP and thereby inhibiting APC/Cdh1-mediated SPOP degradation; consequently, this promoted PD-L1 ubiquitination and degradation by SPOP ubiquitin ligase. However, SPOP function loss by mutations enhanced PD-L1 protein stability, resulting in tumor immunosuppression. Since glycogen synthase kinase 3β (GSK3β) can induce phosphorylation and degradation of multiple substrates by proteasomes20, the interaction of GSK3β with PD-L1 induces its phosphorylation at tyrosine-180/serine-184, resulting in β-TrCP ubiquitin ligase-mediated PD-L1 ubiquitination and degradation13. In addition, activation of AMP-activated protein kinase (AMPK) induces PD-L1 phosphorylation at serine-195, leading to abnormal PD-L1 glycosylation and ER-associated protein degradation (ERAD)21.
Although PD-L1 undergoes ubiquitination and degradation, cancer cells exhibit the ability to inhibit this process. Mezzadra et al.10 reported that the cellular membrane protein CMTM4/6 interacted with PD-L1, leading to inhibition of PD-L1 ubiquitination and degradation, which consequently impaired T cell activity. In the tumor microenvironment, macrophage-secreted TNFα activates NFκB in cancer cells, leading to increased deubiquitinase CSN5 (COP9 signalosome 5) gene transcription and expression, and CSN5 stabilizes PD-L1 protein by inhibiting its ubiquitination and degradation, resulting in cancer cell immune escape22. In response to EGF, active EGFR induces GSK3β phosphorylation, leading to inhibition of the binding of GSK3β to PD-L1, and facilitates PD-L1 glycosylation; consequently, this inhibits PD-L1 degradation by β-TrCP ubiquitin ligase13. Since PD-L1 glycosylation enhances PD-L1 protein stability13, epithelial-mesenchymal transition (EMT) triggers β-catenin-induced STT3 (N-glycosyltransferase) gene transcription and expression, resulting in PD-L1 glycosylation, which subsequently inhibits PD-L1 degradation in cancer stem cells23. In response to cisplatin or ionizing radiation, activated ATM (Ataxia-telangiectasia) increases PD-L1 protein stability by inhibiting its proteasome-dependent degradation in MDA-MB-231 cells resulting in reduced T cell activity24, whereas the mechanism of PD-L1 degradation is unclear. This finding suggests that chemotherapy or radiation could decrease the response rates of PD-L1/PD-1 blockade by increasing PD-L1 expression in cancer cells. Taken together, PD-L1 undergoes ubiquitination and degradation, while cancer cell exhibits the ability to inhibit this process by multiple pathways resulting in tumor immunosuppression (Fig. 1).
The pathways of PD-L1 degradation by autophagy
Autophagy induces degradation of cytoplasmic materials and organelles in lysosomes, which plays an important role in maintaining cellular homeostasis25,26. In addition to the proteasome-dependent degradation discussed above, PD-L1 undergoes autophagic degradation by HIP1R and PKCα/GSK3β/MITF pathways15,16. HIP1R contains a lysosomal targeted signal and binds to PD-L1, which subsequently delivers PD-L1 into lysosomes for autophagic degradation and enhances T cell killing of cancer cells15. In addition to the directly regulatory mechanism of HIP1R-mediated PD-L1 autophagic degradation, SA-49 activates PKCα/GSK3β/MITF pathway-mediated lysosome biogenesis, leading to PD-L1 autophagic degradation; consequently this enhances T cell activity and inhibits tumor growth27. Since autophagy is usually non-selective degradation of substrates25, why does increased lysosome biogenesis degrade only PD-L1 protein rather than other intracellular proteins? This needs to be further addressed. Romero et al.16 reported that the region (225–240 aa) of PD-L1 was the potential surface metalloproteases (ADAM10/17) cleavage site in triple-negative breast cancer, which subsequently generated N-terminal (~24 kDa) fragments that were released outside and C-terminal (~13 kDa) fragments that were degraded by lysosomes, and the activators of ADAM10/17 (ionomycin/PMA) enhanced this event, whereas the mechanism of PD-L1 degradation by lysosomes is still unclear.
Although HIP1R induces PD-L1 autophagic degradation15, cancer cells have exhibited the ability to inhibit PD-L1 autophagic degradation by binding to CMTM6 or palmitoylation modification by DHHC3 (palmitoyltransferase ZDHHC3)28,29. The binding of CMTM6 to plasma membrane PD-L1 and recycling endosomes, leading to inhibition of endocytosed PD-L1 degradation, subsequently enhances PD-L1 protein stability and promotes tumor immune escape28, whereas H1A (PD-L1 antibody) abolishes the binding of PD-L1 to CMTM6, resulting in PD-L1 degradation by lysosomes30. PD-L1 modification by glycosylation and palmitoylation results in inhibition of its endosomal sorting-mediated autophagic degradation29,31. In response to EGF, active EGFR induces N-glycosyltransferase B3GNT3 expression, leading to B3GNT3-mediated glycosylation of PD-L1, which subsequently inhibits PD-L1 degradation resulting in immunosuppression in a breast xenograft tumor model31. Palmitoyltransferase DHHC3 induces PD-L1 palmitoylation at cystine-272, inhibits its ubiquitination and endosomal sorting-mediated autophagic degradation, and subsequently enhances PD-L1 protein stability and immune suppression in a colon tumor model29. On the other hand, Sigma 1 mainly binds to glycosylated PD-L1 and maintains PD-L1 protein stability. In contrast, Sigma 1 inhibitor IPAG induces PD-L1 autophagic degradation in breast and prostate cancer cells, thereby leading to enhanced T cell activity32. Collectively, PD-L1 undergoes autophagic degradation, whereas cancer cells exhibit the ability to maintain its protein stability, leading to tumor immunosuppression (Fig. 2).
PD-L1 degradation and antitumor activity
Cancer cells exhibit the ability to inhibit PD-L1 degradation and maintain its protein stability by deubiquitination or glycosylation of PD-L113,22,23, while PD-L1 induces proteasome-dependent degradation by the GSK3β pathway in response to osimertinib or MTI-31 in EGFR mutant non-small cell lung cancer (NSCLC) cells33,34, and MTI-31 induces PD-L1 degradation and increases T-cell proliferation, which is associated with inhibition of tumor growth in a lung cancer tumor model34. Moreover, the ATR kinase inhibitor VE822 induces proteasomal degradation of PD-L1, leading to increased T cell killing of breast cancer cells24. In addition to proteasomal degradation, SA-49-induced PD-L1 autophagic degradation by the PKCα/GSK3β/MITF pathway results in enhanced T cell killing of cancer cells27. Similarly, Sigma 1 inhibitor IPAG induces PD-L1 autophagic degradation in breast and prostate cancer cells, leading to increased T cell activity32. Pharmacological inhibition of palmitoyltransferase DHHC3 by 2-bromopalmitate (2-BP) promotes PD-L1 autophagic degradation and enhances antitumor activity in a colon tumor model29. In addition, the chimeric PD-LYSO peptide with PD-L1 binding and lysosomal sorting sequences of HIP1R effectively targets PD-L1 for autophagic degradation and increases T cell killing of colon cancer cells15. These findings suggest that PD-L1 degradation by treatment with drugs effectively enhances tumor immunotherapy (Table 1).
Combination therapy
Since PD-L1 protein undergoes degradation in cancer cells in response to the drugs gefitinib13, curcumin22, metformin21, and etoposide23 when combined with anti-PD-1, anti-CTLA4, or anti-Tim3 antibody, we observe that combination therapy effectively improves tumor immunotherapy (Table 2). The specific anti-glycosylated PD-L1 (gPD-L1) antibody could target glycosylated PD-L1, resulting in PD-L1 degradation; thus, the conjugated STM108 (anti-gPD-L1) with MMAE (monomethyl auristatin E) effectively enhances antitumor activity in a breast tumor model31. In addition, the combination of H1A, a specific anti-PD-L1 antibody for PD-L1 autophagic degradation, with cisplatin significantly increases antitumor activity30. Either CDK4/6 or mTOR inhibitors increase PD-L1 protein levels by disruption of CDK4/6/cullin3SPOP or mTORC1/p70S6K/β-TrCP pathway-mediated PD-L1 ubiquitination and degradation11,12, while the combination of CDK4/6 inhibitors with PD-L1/PD-1 blockade effectively enhances tumor immunotherapy11. These findings suggest that the effect of antitumor drugs could be counteracted by increasing PD-L1 expression, leading to cancer cell immune escape. Still, the combination of inhibitors with PD-1/PD-L1 blockade may provide a strategy for cancer therapy. Taken together, the rational combination therapy could effectively enhance antitumor activity (Table 2).
Conclusion
Increasing evidence suggests that PD-L1 protein degradation effectively promotes cancer immunotherapy (Table 1) and the combination therapy significantly enhances this event (Table 2), which provides a potential strategy to increase the response rates of PD-1/PD-L1 blockade in cancer immunotherapy. Although PD-L1 antibody (H1A, STM108) could induce PD-L1 degradation in lysosomes30,31, it is still unclear whether the FDA-approved agents that target PD-L1 (atezolizumab, etc.) or PD-1 (nivolumab, etc.) could induce PD-L1 degradation. The mechanism of PD-L1 degradation is elusive in some studies such as the interaction of CMTM6 with PD-L1 leading to inhibition of PD-L1 degradation by both ubiquitination10 and autophagy30, and hence it is needed to further determine the correlation of these two pathways. In addition, inhibition of the mTOR pathway reduces PD-L1 protein levels in NSCLC cell lines34,35, but the other reports are opposite in the same type of cancer cells12. These contradictory findings may be derived from different PD-L1 antibodies or inhibitors. GSK3β/β-TrCP or AMPK/ERAD pathway induces PD-L1 ubiquitination and degradation. As a secreted trans-membrane protein, although PD-L1 protein is synthesised in the cytoplasm, it will be targeted to the endoplasmic reticulum (ER) by its signal peptide and enter into the ER. How does GSK3β or AMPK induce the extracellular fragment of PD-L1 phosphorylation? Moreover, it remains unclear how membrane PD-L1 protein can be translocated into the cytoplasm and degraded. Is there any other E3 ligase or autophagy receptor for PD-L1 degradation by proteasomes or lysosomes? Furthermore, does the cleaved cytoplasm fragment of PD-L1 by ADAM10/1716 have an additional intracellular function? These issues need to be further clarified, which may contribute to the understanding of cancer immunosuppression by PD-L1/PD-1 blockade for cancer patients.
References
Balkwill, F. & Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 357, 539–545 (2001).
Galon, J. et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 232, 199–209 (2014).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 24, 207–212 (2012).
Yokosuka, T. et al. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 209, 1201–1217 (2012).
Iwai, Y., Hamanishi, J., Chamoto, K. & Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 24, 26 (2017).
Yarchoan, M., Hopkins, A. & Jaffee, E. M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 377, 2500–2501 (2017).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015).
Ritprajak, P. & Azuma, M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol. 51, 221–228 (2015).
Casey, S. C. et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016).
Mezzadra, R. et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110 (2017).
Zhang, J. et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95 (2018).
Deng, L. et al. Inhibition of mTOR complex 1/p70 S6 kinase signaling elevates PD-L1 levels in human cancer cells through enhancing protein stabilization accompanied with enhanced beta-TrCP degradation. Oncogene 38, 6270–6282 (2019).
Li, C. W. et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 7, 12632 (2016).
Zhou, S. et al. Neddylation inhibition upregulates PD-L1 expression and enhances the efficacy of immune checkpoint blockade in glioblastoma. Int. J. Cancer 145, 763–774 (2019).
Wang, H. et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat. Chem. Biol. 15, 42–50 (2019).
Romero, Y., Wise, R. & Zolkiewska, A. Proteolytic processing of PD-L1 by ADAM proteases in breast cancer cells. Cancer Immunol. Immunother. 69, 43–55 (2019).
Ciechanover, A., Heller, H., Elias, S., Haas, A. L. & Hershko, A. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc. Natl. Acad. Sci. USA 77, 1365–1368 (1980).
Finley, D., Ciechanover, A. & Varshavsky, A. Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. Cell 37, 43–55 (1984).
Hershko, A., Heller, H., Elias, S. & Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258, 8206–8214 (1983).
Xu, C., Kim, N. G. & Gumbiner, B. M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 8, 4032–4039 (2009).
Cha, J. H. et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 71, 606–620. e607 (2018).
Lim, S. O. et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939 (2016).
Hsu, J. M. et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 9, 1908 (2018).
Sun, L. L. et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 8, 1307–1316 (2018).
Kaur, J. & Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472 (2015).
Gou, Q. et al. PPARdelta is a regulator of autophagy by its phosphorylation. Oncogene 39, 4844–4853 (2020).
Zhang, N. et al. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1. EBioMedicine 40, 151–162 (2019).
Burr, M. L. et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105 (2017).
Yao, H. et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 3, 306–317 (2019).
Tu, X. et al. PD-L1 (B7-H1) Competes with the RNA exosome to regulate the DNA damage response and can be targeted to sensitize to radiation or chemotherapy. Mol. Cell 74, 1215–1226. e1214 (2019).
Li, C. W. et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell 33, 187–201. e110 (2018).
Maher, C. M. et al. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 16, 243–255 (2018).
Jiang, X. M. et al. Osimertinib (AZD9291) decreases programmed death ligand-1 in EGFR-mutated non-small cell lung cancer cells. Acta Pharm. Sin. 38, 1512–1520 (2017).
Zhang, Q. et al. A novel mTORC1/2 inhibitor (MTI-31) inhibits tumor growth, epithelial-mesenchymal transition, metastases, and iImproves antitumor immunity in preclinical models of lung cancer. Clin. Cancer Res. 25, 3630–3642 (2019).
Lastwika, K. J. et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 76, 227–238 (2016).
Acknowledgements
This work was supported by National Natural Science Foundation of China (81972618). Supported by Changzhou Science and Technology Program. Grant No. CJ20200004.
Author information
Authors and Affiliations
Contributions
Qian Gou and Yongzhong Hou wrote the manuscript. Chen Dong, huihui Xu, Bibimaryam Khan, Jianhua Jin, Qian Liu, and Juanjuan Shi corrected the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by H.-U. Simon
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gou, Q., Dong, C., Xu, H. et al. PD-L1 degradation pathway and immunotherapy for cancer. Cell Death Dis 11, 955 (2020). https://doi.org/10.1038/s41419-020-03140-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03140-2
This article is cited by
-
Prognostic value and immunological role of PD-L1 gene in pan-cancer
BMC Cancer (2024)
-
Exercise accelerates recruitment of CD8+ T cell to promotes anti-tumor immunity in lung cancer via epinephrine
BMC Cancer (2024)
-
Pharmaceutical targeting of OTUB2 sensitizes tumors to cytotoxic T cells via degradation of PD-L1
Nature Communications (2024)
-
High MAL2 expression predicts shorter survival in women with triple-negative breast cancer
Clinical and Translational Oncology (2024)
-
Potential role of immune cell therapy in gynecological cancer and future promises: a comprehensive review
Medical Oncology (2024)
