
Abstract
Some patients with idiopathic pulmonary fibrosis (IPF) have histopathologic features suggesting connective tissue disease (CTD); however, their clinical course and prognosis remain unclear. Thus, we aimed to investigate the clinical course and prognosis of these patients with histologic autoimmune features. Among 114 patients with biopsy-proven IPF, the histologic features were semi-quantitatively graded, and CTD scores (range: 0–9) were calculated as the sum of each score of plasma cell infiltration, lymphoid aggregates, and germinal centres. Patients with high CTD scores (≥ 4) were classified into the interstitial pneumonia with histologic autoimmune features (IP-hAF) group. The mean age of the patients was 60.0 years; 74.6% were men, 69.3% were ever-smokers, and 35.1% had IP-hAF. During follow-up, the IP-hAF group showed slower decline in lung function, and better prognosis (median survival, 48.7 vs. 40.4 months; p = 0.015) than the no-IP-hAF group. On multivariate Cox analysis, IP-hAF was an independent prognostic factor (hazard ratio, 0.522; p = 0.016), along with the lower diffusing capacity for carbon monoxide, higher scores of reticulation and honeycombing, and usual interstitial pneumonia pattern on high-resolution computed tomography. Patients with IPF having histologic autoimmune features show distinct clinical characteristics and better outcome than those without histologic autoimmune features.
Similar content being viewed by others


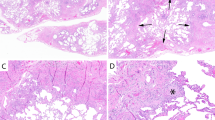
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive fibrosing interstitial pneumonia of unknown cause, characterized by a poor prognosis and median survival of 3 years1. The current guideline recommends performing a comprehensive study to exclude connective tissue diseases (CTDs) before diagnosing IPF2. However, despite extensive evaluation, some patients with IPF occasionally show clinical, serologic, and/or morphologic features suggesting CTDs, but do not fully satisfy the diagnostic criteria for defined CTDs3,4,5. Recently, the European Respiratory Society (ERS)/American Thoracic Society (ATS) task force devised a new term, interstitial pneumonia with autoimmune features (IPAF), for these disease entities6.
Some histopathologic features observed in patients with IPF were reported to suggest the presence of CTD7,8,9. A previous study on 100 patients with the usual interstitial pneumonia (UIP) pattern diagnosed via surgical lung biopsy (39 with CTD and 61 with IPF) showed that patients with CTD-UIP had higher germinal centre and total inflammation scores than those with IPF/UIP, and that a high germinal centre score (odds ratio [OR], 2.948; p = 0.001) was an independent discriminating factor for CTD-UIP7. Flaherty et al. also showed that in 108 patients with biopsy-proven UIP (99 with IPF and 9 with CTD-UIP), the mean fibroblastic focus score was a significant predicting factor (OR, 8.31; p = 0.002) for the presence of IPF-UIP rather than CTD-UIP8. In addition, Kono et al. reported that in 111 patients initially diagnosed with IPF, the presence of lymphoid aggregates with germinal centres (hazard ratio [HR], 3.367; p < 0.01) was significantly associated with the occurrence of CTD (9.0% of the study population)9. Nevertheless, the impact of histopathologic findings suggesting CTD on the clinical course and prognosis of patients with IPF remains unknown.
Therefore, we aimed to investigate the clinical significance of histopathologic features suggesting CTD in patients with IPF. Accordingly, we compared the clinical course and outcomes between patients with IPF having histopathologic autoimmune features and those without histopathologic autoimmune features.
Material and methods
Study population
Between February 2000 and November 2007, 187 patients were diagnosed with IPF via surgical lung biopsy at Asan Medical Center, Seoul, Republic of Korea. Among them, 73 patients with unavailable histopathologic slides and/or high-resolution computed tomography (HRCT) images were excluded in this study. All patients underwent thorough systemic history taking, physical examination, and serological testing for CTD at the time of the initial diagnosis, and patients with other known causes of interstitial lung disease (ILD) or those who met the definite diagnostic criteria for CTD were excluded10,11,12,13,14,15. All patients fulfilled the IPF diagnostic criteria of the ATS/ERS/Japanese Respiratory Society/Latin American Thoracic Association1, and those with histopathologic features that should raise concerns about the likelihood of not UIP pattern including prominent lymphoplasmacytic infiltration was excluded. Some of the patients in the present study had been included in a previous study7. This study was approved by the Institutional Review Board of Asan Medical Center (2014-0911). The requirement for informed consent was waived by the Institutional Review Board of Asan Medical Center, because of the retrospective nature of the study. All methods were performed in accordance with the Declaration of Helsinki and the relevant guidelines.
Data collection
Clinical and survival data of all patients were obtained from medical records, telephonic interviews, and/or the records of the National Health Insurance Service of Korea. All clinical parameters were obtained within 1 month before surgical lung biopsy. Spirometry, diffusing capacity of the lung for carbon monoxide (DLco), and total lung capacity (TLC) determined using plethysmography were measured according to the ERS/ATS recommendations16,17.
Pathologic evaluation
The pathologic slides were reviewed by two thoracic pathologists (J.S.S. and S.J.J.) blinded to the clinico-radiologic information. The following histologic characteristics were semi-quantitatively graded according to a previously described grading system with a slight modification and presented in Supplementary Material Figs. 1 to 77: plasma cell infiltration, lymphoid aggregates, the overall extent of mononuclear cell interstitial inflammation (total inflammation), organizing pneumonia, fibroblastic foci, intra-alveolar macrophages, and stromal fibrosis (all scored between 0 and 3), as well as honeycombing and pleural changes. Honeycombing was scored according to the measured size of the largest honeycombing spaces in the biopsy specimen: score 0, none; 1, < 1 mm; 2, 1–3 mm; 3, 3–5 mm; and 4, > 5 mm. Pleural changes were scored according to the level of inflammatory change of the pleura: score 1, normal; 2, pleural fibrosis; 3, fibrinous pleuritis, and 4, fibrosis and fibrinous pleuritis. We counted the number of germinal centres on three independent low-power fields (4×) using light microscopy. Thereafter, germinal centres were scored according to the average number of germinal centres in these three areas: 0, none; 1, < 5; 2, ≥ 5 but < 10; and 3, ≥ 10. The presence of perivascular collagen was also investigated. Disagreement (≥ 2 grades) between the two pathologists was resolved via consensus.
Of these pathologic parameters, the scores of plasma cell infiltration, lymphoid aggregates, and germinal centres were considered to represent distinctive histologic features of CTD-ILD7,18, and their sum was defined as the CTD score.
Radiologic evaluation
The HRCT images acquired at the time of diagnosis were reviewed by two thoracic radiologists (S.Y.L. and E.J.C.) in a blinded manner. The extent of reticular abnormality, emphysema, ground-glass opacity, traction bronchiectasis, consolidation, and honeycombing were semi-quantitatively scored on a scale of 25% for all lobes (0- to 4-point scales). Radiologic features associated with non-specific interstitial pneumonia (NSIP) or CTD, such as the presence of peribronchovascular distribution, subpleural sparing, pleural or pericardial effusion, and oesophageal dilatation, were also evaluated19,20. Overall, the HRCT pattern was categorised as UIP or non-UIP1. The UIP pattern was defined as a subpleural, basal predominance of reticular abnormalities, honeycombing with or without traction bronchiectasis, and the absence of findings inconsistent with a UIP pattern including extensive ground-glass opacity, micronodules, discrete cysts, mosaic attenuation, or segmental/lobar consolidation1. Disagreement between the two readers was resolved via consensus.
Statistical analysis
All values were presented as mean ± standard deviation for continuous variables or as percentages for categorical variables. Student’s t-test or Mann–Whitney U test was used for continuous data, and Pearson’s chi-square test or Fisher’s exact test was used for categorical data. Receiver operating characteristic (ROC) curve analysis was performed to confirm the optimal cut-off value of CTD scores for predicting mortality in the study population. Changes in lung function were evaluated using a paired t-test for intragroup comparison and Student’s t-test for intergroup comparison. Survival was assessed using Kaplan–Meier survival curves, and the differences between groups were evaluated using the log-rank test. To identify variables that predicted histologic autoimmune features or survival, we used logistic or Cox regression analysis with backward elimination; variables with p < 0.2 in the univariate analysis were entered into the multivariate models. All statistical analyses were performed using IBM SPSS Statistics for Windows/Macintosh, Version 23.0 (IBM Corp., Armonk, USA). A value of p < 0.05 was considered significant (two-tailed).
Results
Study population
The median follow-up period was 40 months. The mean age of the patients was 60.0 years; 74.6% were men, and 69.3% were ever-smokers (Table 1). During follow-up, 80 (70.2%) patients died. and no significant difference was observed in treatment between survivors and non-survivors. Non-survivors had lower lung function (forced vital capacity [FVC] and TLC) than survivors (Table 1). Non-survivors also had higher scores of reticulation and honeycombing; lesser frequent subpleural sparing; more frequent UIP pattern on HRCT; and lower lymphoid aggregate, and CTD scores on surgical lung biopsy than survivors (Tables 2, 3).
Prognostic factors in IPF
On univariate Cox analysis, lower lung function (FVC, DLco, and TLC), poorer exercise capacity (distance and the lowest oxygen saturation during the 6MWT), higher scores of reticulation and honeycombing, and a UIP pattern on HRCT were significant predictors of mortality (Supplementary Material Table 1). In addition, higher fibroblastic focus scores and lower germinal centre scores were significant prognostic factors. On multivariate Cox analysis, lower CTD scores were independently associated with an increased risk of mortality (HR, 0.837; 95% confidence interval [CI], 0.724–0.969; p = 0.017) in patients with IPF, along with lower DLco, higher scores of reticulation and honeycombing, and a UIP pattern on HRCT (Table 4). On ROC analysis, the optimal cut-off level of CTD scores for predicting mortality in patients with IPF was 3.5 (sensitivity, 72.5%; specificity, 52.9%; p = 0.024), and patients with high CTD scores (≥ 4) were classified into the interstitial pneumonia with histologic autoimmune features (IP-hAF) group.
Baseline characteristics
Histopathologic findings of typical IP-hAF patients were shown in Supplementary Material Fig. 8. The IP-hAF group included 35.1% of patients, and comprised more women and never-smokers than the no-IP-hAF group (Table 5). Additionally, the IP-hAF group showed a tendency of lower DLco than the no-IP-hAF group. However, no significant differences were observed in radiologic parameters between the groups except for higher consolidation scores (0.3 vs. 0.1; p = 0.079) and more frequent presence of peribronchovascular distribution (47.5% vs. 30.1%; p = 0.066) in the IP-hAF group than in the no-IP-hAF group (Supplementary Material Table 2). The IP-hAF group had higher scores of lymphoid aggregates (2.3 vs. 1.2; p < 0.001), plasma cell infiltration (1.8 vs. 0.5; p < 0.001), germinal centres (1.1 vs. 0.1; p < 0.001), total inflammation (2.2 vs. 1.3; p < 0.001), pleural changes (2.8 vs. 2.2; p = 0.001), perivascular collagen (0.2 vs. 0.1; p = 0.014), and CTD score (5.1 vs. 1.8; p < 0.001) than the no-IP-hAF group (Supplementary Material Table 3). The IP-hAF group also had lower scores of honeycombing (2.2 vs. 2.7; p = 0.016) and stromal fibrosis (1.5 vs. 1.9; p = 0.012) than the no-IP-hAF group.
On univariate logistic analysis, female sex and never-smoking were significantly associated with the IP-hAF group (Supplementary Material Table 4). However, on multivariate logistic analysis, female sex (OR, 6.624; 95% CI, 2.548–17.222; p < 0.001) and higher scores of consolidation (OR, 4.072; 95% CI, 1.293–12.823; p = 0.016) were independently associated with the IP-hAF group.
Changes in lung function
Among all patients (n = 114), 96 (84.2%) were treated with steroids and/or immunosuppressants, and the proportion of treated patients did not differ between the IP-hAF and no-IP-hAF groups (Table 5). During follow-up, the IP-hAF group did not show any decline in lung function (FVC, DLco, and TLC); however, the no-IP-hAF group showed a significant decline for 6 or 12 months after diagnosis (Fig. 1, Supplementary Material Table 5). A comparison of the decline in lung function revealed that the no-IP-hAF group had a greater decline in DLco after 6 months and in FVC, DLco, and TLC after 12 months than the IP-hAF group (Supplementary Material Table 5).

Comparison of changes in lung function between the interstitial pneumonia with histologic autoimmune features (IP-hAF) group and no-IP-hAF group among patients with idiopathic pulmonary fibrosis (IPF). (a) Changes in FVC. (b) Changes in DLco. (c) Changes in TLC. Each symbol with error bars represents the mean and standard deviation. * means p < 0.05. FVC, forced vital capacity; DLco, diffusing capacity of the lung for carbon monoxide; TLC, total lung capacity.
When stratified according to treatment, the IP-hAF group did not show any decline in lung function regardless of treatment (Supplementary Material Fig. 9a–c); however, in the no-IP-hAF group, the treatment group showed a significant decline in FVC, DLco, and TLC for 6 or 12 months after diagnosis, and the no-treatment group showed only a significant decline in FVC for 12 months after diagnosis (Supplementary Material Fig. 9d–f). A comparison of the decline in lung function revealed no significant difference between the treatment and non-treatment groups (Supplementary Material Table 6).
Clinical course and outcome
During follow-up, CTDs occurred in 2 (5%) patients (1 rheumatoid arthritis and 1 Sjogren syndrome) in the IP-hAF group; however, none occurred in the no-IP-hAF group (p = 0.121). The IP-hAF group showed better survival (median survival, 48.7 vs. 40.4 months; p = 0.015) than the no-IP-hAF group (Fig. 2). Likewise CTD score, IP-hAF was also an independent prognostic factor (HR, 0.522; 95% CI, 0.307–0.886; p = 0.016) in patients with IPF, along with lower DLco, higher scores of reticulation and honeycombing, and a UIP pattern on HRCT, as seen on multivariate Cox analysis (Table 6, Supplementary Material Table 7).

Comparison of survival curves between the interstitial pneumonia with histologic autoimmune features (IP-hAF) group and no-IP-hAF group among patients with idiopathic pulmonary fibrosis (IPF).
Discussion
This study revealed significant histologic autoimmune features in one-third of those in the IPF cohort. The IP-hAF group included patients who were more frequently women and never-smokers, had more stable lung function and frequent CTD development, and had better survival than the no-IP-hAF group. IP-hAF was an independent risk factor for mortality in patients with IPF.
Some histologic features, such as lymphoplasmacytic infiltration and lymphoid aggregates with or without germinal centres, have been reported to suggest CTD7,21. A previous study on 100 patients with a UIP pattern diagnosed via surgical lung biopsy (39 with CTD and 61 with IPF) reported that patients with CTD-UIP had higher total inflammation and germinal centre scores than those with IPF/UIP7. Ozasa et al. studied 105 patients with idiopathic interstitial pneumonia (IIP; 79 with UIP and 26 with NSIP) and 49 with CTD-ILD and showed that plasma cell infiltration, lymphoid follicle with germinal centres, and airspace fibrin were predictive of CTD-ILD21. On the basis of these results, lymphoplasmacytic infiltration and interstitial lymphoid aggregates with germinal centres were suggested as histopathologic patterns of the morphologic domain in IPAF6. In our study, one-third of patients with IPF showed these findings. Our findings are supported by a previous study by Omote et al.22 on 44 patients with IIP and autoantibodies (25 with UIP, 13 with NSIP, and 6 with other histologic patterns). They reported observing two or more characteristic histologic features suggesting CTD (e.g. prominent plasmacytic infiltration and lymphoid aggregates with germinal centres) in 60% of patients with histologic UIP22.
Our results showed that female sex and consolidation on HRCT were independent predicting factors of IP-hAF. Previous studies also reported female predominance in IPAF cohorts (52.1–71.4%)23,24,25,26. Alhamad et al. studied 118 patients (61 with IPF, 29 with CTD-UIP, and 28 with lung-dominant CTD-UIP) and reported that patients with CTD-UIP and lung-dominant CTD-UIP were more likely to be female than patients with IPF (72.4% with CTD-UIP, 67.9% with lung-dominant CTD-UIP, and 47.5% with IPF; p = 0.042)27. The radiologic organizing pneumonia pattern, characterized by consolidation, is considered a morphologic domain in the IPAF criteria6. Yoshimura et al. studied 194 patients with chronic fibrosing interstitial pneumonia (163 with IPF and 31 with NSIP) and reported more frequent NSIP with a combined organizing pneumonia pattern (12.5 vs. 4.3%) on HRCT in the IPAF group than in the non-IPAF group28.
In our study, lung function declines over 12 months were slower in the IP-hAF group than in the no-IP-hAF group. Our findings were consistent with those of previous reports22,29,30. Omote et al. studied 23 patients with histologic UIP pattern and CTD features and reported no significant deterioration in lung function during 12 months (baseline FVC, 80.6%; 1-year FVC, 78.4%; p = 0.48)22. Kinder et al. studied 59 patients (30 with IPF and 29 with undifferentiated CTD [UCTD]) and showed that UCTD, rather than IPF, was associated with a substantial improvement in FVC (odds ratio, 8.23; 95% CI, 1.27–53.2; p = 0.03) during follow-up29. Likewise, Collins et al. studied 124 patients with well-defined ILD and reported that the mean change in DLco over 12 months significantly differed between patients with IPAF and those with IPF (6.3 vs. − 2.9% predicted; p < 0.001); moreover, a subgroup analysis among those with UIP in each group showed similar results30. In other aspects, the anti-inflammatory treatment group showed a faster decline in lung function than the no-treatment group in the no-IP-hAF group in our study. This finding might be attributed to the harmful effects of anti-inflammatory treatment in patients with IPF31.
The IP-hAF group had better survival than the no-IP-hAF group in our study. Previous studies support our results32,33. Lim et al. studied 305 patients with ILD (54 with IPAF, 175 with IPF, and 76 with CTD-ILD) and reported that the IPAF group had better survival (mean survival, 73.3 vs. 50.7 months; p < 0.001) than the IPF group32. Another study on 44 patients with UCTD-UIP and 499 with IPF reported that the UCTD-UIP group showed better survival (median survival, 31 vs. 26 months; p = 0.042) than the IPF group33. However, others reported contradicting findings23,34. Oldham et al. studied 422 patients with IIP or UCTD and showed that patients with IPAF and a UIP pattern (n = 98) had similar survival to those with IPF (n = 268; p = 0.51)23. Strand et al. studied 321 patients with IPF and 19 with UCTD-UIP and showed no statistical difference in survival between the patients with UCTD-UIP and IPF (median survival, 3.8 vs. 4.4 years; p = 0.95)34. These contradictory results might be attributed to the difference in the definition of autoimmune features; we used histopathologic findings suggesting CTD for classifying patients with IPF and significant autoimmune features. Our results also suggest that the morphologic domain may further influence the prognosis.
Our study has some limitations. First, a selection bias might exist because we restricted patients to those who underwent surgical lung biopsy at a single centre. However, the clinical features of our patients were comparable to those in previous studies22,29. Second, CTD was possibly missed in some of our patients with IIP because minor symptoms and signs could be ignored. However, we used a standardised survey form to identify suspected symptoms and signs of CTD and consulted rheumatologists to determine whether early CTD could be excluded when suspected symptoms and signs were observed. Third, around 80% of patients received steroid therapy. Patients enrolled in this study were diagnosed between February 2000 and November 2007, at which time, steroid therapy was considered as standardized therapy. Although anti-inflammatory treatment has been shown to be harmful in IPF patients, our results suggest that these effects might be different in the IP-hAF group; among the IP-hAF group, changes in lung function were stabilized in the treatment group. Fourth, we excluded those with histopathologic features that should raise concerns about the likelihood of not UIP pattern including prominent lymphoplasmacytic infiltration. Therefore, the whole picture of IP-hAF of UIP pattern is not clear in our study. Finally, we defined the IP-hAF group by using an optimal cut-off level of CTD scores for predicting mortality, instead of the development of CTD, because of the small number of events. However, previous reports suggest that autoimmune features may portend a better prognosis32,33. Despite these limitations, our study is potentially the first to investigate the clinical implications of histologic autoimmune features in patients with IPF.
In conclusion, patients with IPF and significant histologic autoimmune features were more frequently women and non-smokers and had more frequent CTD development, more stable lung function, and better prognosis than those without these features. These findings suggest that this group may be classified as a specific subgroup of patients with IPF.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Raghu, G. et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824. https://doi.org/10.1164/rccm.2009-040GL (2011).
Raghu, G. et al. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 198, e44–e68. https://doi.org/10.1164/rccm.201807-1255ST (2018).
Mosca, M., Tani, C., Neri, C., Baldini, C. & Bombardieri, S. Undifferentiated connective tissue diseases (UCTD). Autoimmun. Rev. 6, 1–4. https://doi.org/10.1016/j.autrev.2006.03.004 (2006).
Mosca, M., Tavoni, A., Neri, R., Bencivelli, W. & Bombardieri, S. Undifferentiated connective tissue diseases: the clinical and serological profiles of 91 patients followed for at least 1 year. Lupus 7, 95–100. https://doi.org/10.1191/096120398678919787 (1998).
Bodolay, E. et al. Five-year follow-up of 665 Hungarian patients with undifferentiated connective tissue disease (UCTD). Clin. Exp. Rheumatol. 21, 313–320 (2003).
Fischer, A. et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur. Respir. J. 46, 976–987. https://doi.org/10.1183/13993003.00150-2015 (2015).
Song, J. W. et al. Pathologic and radiologic differences between idiopathic and collagen vascular disease-related usual interstitial pneumonia. Chest 136, 23–30. https://doi.org/10.1378/chest.08-2572 (2009).
Flaherty, K. R. et al. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am. J. Respir. Crit. Care Med. 167, 1410–1415. https://doi.org/10.1164/rccm.200204-373OC (2003).
Kono, M. et al. Usual interstitial pneumonia preceding collagen vascular disease: a retrospective case control study of patients initially diagnosed with idiopathic pulmonary fibrosis. PLoS ONE 9, e94775. https://doi.org/10.1371/journal.pone.0094775 (2014).
Bohan, A. & Peter, J. B. Polymyositis and dermatomyositis (second of two parts). N. Engl. J. Med. 292, 403–407. https://doi.org/10.1056/NEJM197502202920807 (1975).
Tan, E. M. et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 25, 1271–1277. https://doi.org/10.1002/art.1780251101 (1982).
Vitali, C. et al. Preliminary criteria for the classification of Sjogren’s syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum. 36, 340–347. https://doi.org/10.1002/art.1780360309 (1993).
Smolen, J. S. & Steiner, G. Mixed connective tissue disease: to be or not to be?. Arthritis Rheum. 41, 768–777 (1998).
Aletaha, D. et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 69, 1580–1588. https://doi.org/10.1136/ard.2010.138461 (2010).
van den Hoogen, F. et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 65(2737–2747), 2013. https://doi.org/10.1002/art.38098 (2013).
Macintyre, N. et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur. Respir. J. 26, 720–735. https://doi.org/10.1183/09031936.05.00034905 (2005).
Wanger, J. et al. Standardisation of the measurement of lung volumes. Eur. Respir. J. 26, 511–522. https://doi.org/10.1183/09031936.05.00035005 (2005).
Fischer, A., West, S. G., Swigris, J. J., Brown, K. K. & du Bois, R. M. Connective tissue disease-associated interstitial lung disease: a call for clarification. Chest 138, 251–256. https://doi.org/10.1378/chest.10-0194 (2010).
Capobianco, J. et al. Thoracic manifestations of collagen vascular diseases. Radiographics 32, 33–50. https://doi.org/10.1148/rg.321105058 (2012).
Bryson, T., Sundaram, B., Khanna, D. & Kazerooni, E. A. Connective tissue disease-associated interstitial pneumonia and idiopathic interstitial pneumonia: similarity and difference. Semin. Ultrasound CT MR 35, 29–38. https://doi.org/10.1053/j.sult.2013.10.010 (2014).
Ozasa, M. et al. Proposed method of histological separation between connective tissue disease-associated interstitial pneumonia and idiopathic interstitial pneumonias. PLoS ONE 13, e0206186. https://doi.org/10.1371/journal.pone.0206186 (2018).
Omote, N. et al. Lung-dominant connective tissue disease: clinical, radiologic, and histologic features. Chest 148, 1438–1446. https://doi.org/10.1378/chest.14-3174 (2015).
Oldham, J. M. et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur. Respir. J. 47, 1767–1775. https://doi.org/10.1183/13993003.01565-2015 (2016).
Dai, J. et al. Clinical features, risk factors, and outcomes of patients with interstitial pneumonia with autoimmune features: a population-based study. Clin. Rheumatol. 37, 2125–2132. https://doi.org/10.1007/s10067-018-4111-5 (2018).
Ito, Y. et al. Serological and morphological prognostic factors in patients with interstitial pneumonia with autoimmune features. BMC Pulm. Med. 17, 111. https://doi.org/10.1186/s12890-017-0453-z (2017).
Chartrand, S. et al. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir. Med. 119, 150–154. https://doi.org/10.1016/j.rmed.2016.09.002 (2016).
Alhamad, E. H. et al. Comparison of three groups of patients with usual interstitial pneumonia. Respir. Med. 106, 1575–1585. https://doi.org/10.1016/j.rmed.2012.07.009 (2012).
Yoshimura, K. et al. Distinctive characteristics and prognostic significance of interstitial pneumonia with autoimmune features in patients with chronic fibrosing interstitial pneumonia. Respir. Med. 137, 167–175. https://doi.org/10.1016/j.rmed.2018.02.024 (2018).
Kinder, B. W. et al. Undifferentiated connective tissue disease-associated interstitial lung disease: changes in lung function. Lung 188, 143–149. https://doi.org/10.1007/s00408-009-9226-7 (2010).
Collins, B. F. et al. Idiopathic interstitial pneumonia associated with autoantibodies: a large case series followed over 1 year. Chest 152, 103–112. https://doi.org/10.1016/j.chest.2017.03.004 (2017).
Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 366, 1968–1977. https://doi.org/10.1056/NEJMoa1113354 (2012).
Lim, J. U. et al. Interstitial pneumonia with autoimmune features show better survival and less exacerbations compared to idiopathic pulmonary fibrosis. BMC Pulm. Med. 19, 120. https://doi.org/10.1186/s12890-019-0868-9 (2019).
Kim, H. C. et al. Interstitial pneumonia related to undifferentiated connective tissue disease: pathologic pattern and prognosis. Chest 147, 165–172. https://doi.org/10.1378/chest.14-0272 (2015).
Strand, M. J. et al. Pulmonary function and survival in idiopathic vs secondary usual interstitial pneumonia. Chest 146, 775–785. https://doi.org/10.1378/chest.13-2388 (2014).
Acknowledgements
We express our gratitude to the Minkyu Han in Department of Clinical Epidemiology and Biostatistics, Asan Medical Center, for valuable advice in the statistical analysis.
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and Technology (NRF-2019R1A2C2008541) and a Grant (2020IP0083) from the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea.
Author information
Authors and Affiliations
Contributions
J.W.S. is the guarantor of the paper and takes responsibility for the integrity of the work as a whole. H.C.K., S.P., H.Y.Y., and J.W.S. made substantial contributions to acquisition, analysis, and interpretation of data. J.S.S. and S.J.J. reviewed pathologic specimens. E.J.C. and S.Y.L. evaluated the HRCT images. H.C.K., S.P., H.Y.Y., and J.W.S. performed statistical analysis and interpreted the data. H.C.K., S.P., and J.W.S. drafted the initial manuscript. All authors discussed the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, H.C., Song, J.S., Park, S. et al. Histologic features suggesting connective tissue disease in idiopathic pulmonary fibrosis. Sci Rep 10, 21137 (2020). https://doi.org/10.1038/s41598-020-78140-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78140-5
This article is cited by
-
Lung fibrosis in autoimmune diseases and hypersensitivity: how to separate these from idiopathic pulmonary fibrosis
Rheumatology International (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
