
Abstract
Activation of peroxisome proliferator-activated receptor gamma (PPARγ) has been linked to induction of differentiation, cell growth inhibition and apoptosis in several types of human cancer. However, the possible effects of PPARγ agonists on human oral squamous cell carcinoma have not yet been reported. In this study, treatment with 15-deoxy-Δ12,14-PGJ2 (15-PGJ2), a natural PPARγ ligand, induced a significant reduction of oral squamous cell carcinoma cell growth, which was mainly attributed to upregulation of apoptosis. Interestingly, rosiglitazone and ciglitazone, two members of the thiazolidinedione family of PPARγ activators, did not exert a growth inhibitory effect. Given the critical role that the oncogene signal transducer and activator of transcription 3 (Stat3) plays in head and neck carcinogenesis, its potential regulation by PPARγ ligands was also examined. Treatment of oral squamous cell carcinoma cells with 15-PGJ2 induced an initial reduction and eventual elimination of both phosphorylated and unphosphorylated Stat3 protein levels. In contrast, other PPARγ did not induce similar effects. Our results provide the first evidence of significant antineoplastic effects of 15-PGJ2 on human oral squamous cell carcinoma cells, which may be related to downmodulation of Stat3 and are at least partly mediated through PPARγ-independent events.
Similar content being viewed by others

Main
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor family, which function as ligand-dependent, sequence-specific activators of transcription. The PPAR family consists of three distinct molecules, termed α, δ (β, FFAR or NUC-1), and γ, encoded by separate genes and characterised by specific tissue and developmental distribution patterns (Mangelsdorf et al, 1995; Lemberger et al, 1996; Gelman et al, 1999; Kersten et al, 2000). A variety of natural and pharmacological ligands, including prostaglandins, the fibrate class of hypolipidaemic drugs, the anti-diabetic drugs thiazolidinediones, and certain non-steroidal anti-inflammatory drugs, are able to bind to and activate PPARs (Forman et al, 1995, 1997; Yu et al, 1995; Lehmann et al, 1997). Upon activation, PPARs heterodimerise with the retinoic X receptor (RXR) and bind to peroxisome proliferator response elements (PPREs), located in the promoter region of target genes, driving their transcription (Mangelsdorf et al, 1995).
PPARs were initially described as molecular targets for compounds that induce peroxisome proliferation (Issemann and Green, 1990). However, shortly after their discovery, it became apparent that the physiologic role of PPARs extends far beyond peroxisome proliferation, involving such diverse processes as lipid homeostasis, insulin sensitisation, inflammation, and cell proliferation (Lemberger et al, 1996; Gelman et al, 1999; Kersten et al, 2000). Today, PPARs are recognised as key regulators of lipid homeostasis, playing fundamental roles in adipogenesis and fat catabolism (Lemberger et al, 1996; Kersten et al, 2000). Potential roles of PPARs in the treatment of diabetes mellitus (Vamecq and Latruffe, 1999), in inflammation control (Gelman et al, 1999), and in the regulation of atherosclerosis and thrombosis (Vamecq and Latruffe, 1999; Duez et al, 2001) have also been described.
The role of PPARγ in the acquisition of an adipocyte phenotype, through the control of the expression of genes that promote cell cycle withdrawal, drive differentiation and induce apoptosis (Shao and Lazar, 1997; Debril et al, 2001), prompted many investigators to study the potential function of PPARγ in neoplasia. Multiple lines of evidence suggest that PPARs, especially PPARγ, play an important role in modulating cell proliferation and tumour growth (Gelman et al, 1999; Kersten et al, 2000; Debril et al, 2001; Park et al, 2001; Rosen and Spiegelman, 2001). Ligand-induced PPARγ activation has been shown to promote differentiation and to induce cell growth inhibition and apoptosis in several types of human cancer, including colon cancer (Sarraf et al, 1998; Kitamura et al, 1999; Yang and Frucht, 2001), breast cancer (Elstner et al, 1998; Mueller et al, 1998), lung cancer (Chang and Szabo, 2000), prostate cancer (Kubota et al, 1998; Butler et al, 2000), gastric cancer (Sato et al, 2000), liposarcoma (Tontonoz et al, 1997; Demetri et al, 1999), and leukaemia (Sugimura et al, 1999). Histological and biochemical evidence indicate that PPARγ ligands induce tumour cell differentiation in patients with advanced liposarcoma (Demetri et al, 1999). Consistent with its possible function as a tumour suppressor gene, PPARγ has been reported to be functionally mutated in sporadic cases of colon cancer (Sarraf et al, 1999); nonetheless, the overall incidence of PPARγ mutations in human malignancies seems to be very rare (Ikezoe et al, 2001).
We recently showed that the non-steroidal anti-inflammatory drug (NSAID) sulindac induces cell growth inhibition and apoptosis in human oral squamous cell carcinoma (SCCa) cells, accompanied by upregulation of the mRNA and protein expression of PPARγ. Treatment with antisense PPARγ oligonucleotides abolished the cell growth inhibitory effect of the NSAID sulindac. These results suggest that upregulation of PPARγ expression and activation may be, at least partially, responsible for sulindac's antineoplastic effect (Nikitakis et al, 2002a). However, it is unknown whether ligand-induced PPARγ activation may alter the cell growth of oral SCCa. Here, we assessed the effects that the natural PPARγ ligand 15-deoxy-Δ12,14-PGJ2 (15-PGJ2) and the synthetic PPARγ ligands rosiglitazone and cigliatazone have on cell growth, apoptosis and cell proliferation in oral SCCa cells. Moreover, we explored the possibility that PPARγ activation may affect the expression and activation of Stat3, an oncogene that plays a critical role in head and neck carcinogenesis (Grandis et al, 1998, 2000; Bromberg et al, 1999; Bowman et al, 2000) and is downregulated by sulindac in oral SCCa cells (Nikitakis et al, 2002b).
Materials and methods
Cell lines and cell culture
Experiments were performed using established cell lines of human oral SCCa (SCC-4, -9, -15 and -25) obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA). Cells were cultured in a 1:1 mixture of Ham's F12 and Dulbecco's Modified Eagle's Medium (DMEM) containing 10% foetal bovine serum (FBS), 100 units of penicillin, 100 μg ml−1 streptomycin and 0.4 μg ml−1 hydrocortisone (Sigma Chemical Co., St. Louis, MO, USA) at 37°C in a 5% CO2 air atmosphere. Cells were subcultured by disaggregation with trypsin (0.1%)–EDTA (0.01%) in phosphate buffered saline (PBS) at pH 7.5.
Quantitative reverse transcriptase polymerase chain reaction
To estimate the mRNA levels of PPARγ, real-time quantitative reverse transcriptase polymerase chain reaction (RT–PCR) was performed using a PE Applied Biosystems protocols. Total RNA was isolated using the TRIZOL Reagent (BRL/Life Technologies) and the concentration of RNA was determined using spectrophotometry. The forward and reverse PPARγ primers were selected using Primer Express software (PE Applied Biosysystems, Foster City, CA, USA) as follows: 5′-TATCGACCAGCTGAATCCAGAG-3′ (forward) and 5′-TCGCCTTTGCTTTGGTCA-3′ (reverse). For the PCR reaction, a SYBR® Green PCR kit (PE Applied Biosystems) was used and the analyses were performed in triplicate. For each well, 5 μl of 25 nM RNA extract were added to a solution consisting of: 25 μl Master Mix solution (SYBR Green PCR Buffer, AmpErase® UNG, AmpliTaq Gold® DNA Polymerase, dATP, dCTP, dGTP, dUTP and 25 mM MgCl2), 1 μl of RNAse inhibitor, 0.25 μl of reverse transcriptase (MultiScribe), 1 μl of each primer and 16.75 μl of water. The amplification process included 30 min at 48°C, 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Thermal cycling and fluorescence detection were performed using an ABI 5700 Prism (PE Applied Biosystems). Relative quantitation of the signal of PPARγ mRNA followed. The signal of the target mRNA was normalised by comparison with the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA signal. The normalised amount of PPARγ mRNA present in each cell line was calculated by arbitrarily designating SCC25 cells as a calibrator using a comparative Ct method following PE Applied Biosystems protocols.
Immunocytochemistry
Cells were plated on chamber slides (LabTech Nalge/Nunc) at a density of 5 × 104 cells/chamber and were allowed to grow until almost confluent. The cells were then rinsed with Hanks' balanced salt solution (HBSS) and fixed with 95% ethanol for 20 min. PPARγ protein expression was ascertained by immunocytochemical analysis (Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc-1984) 1:50. The presence of antibody staining was determined after incubation of the sections with a secondary antibody (Biogenex, San Ramon, CA, USA, HK327-UG) 1:20, followed by the application of StrepABComplex/HRP (Dako, Carpinteria, CA, USA, K0377), and diaminobenzidine (DAB). The slides were counterstained with Harris's haematoxylin. The intensity of the immunostaining was classified as: 0 (negative), 1 (weakly positive), 2 (moderately positive), and 3 (strongly positive).
Immunohistochemistry
Ten cases of oral squamous cell carcinoma were randomly selected from the files of the Department of Diagnostic Sciences and Pathology. No patients were identified for these studies. The histological slides were reviewed to confirm diagnosis.
Five-micron sections of paraffin-embedded tissue were mounted on glass slides, deparaffinised and rehydrated. An antigen retrieval procedure was performed by placing the sections in Citra-solution (HK086-9K; Biogenex, San Ramon, CA, USA) inside a plastic pressure cooker, which was positioned in a microwave oven (Kenmore; Sears, Chicago, IL, USA). The specimens were treated by two cycles, 15 min each, at a high level and at level 4, respectively. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide and non-specific protein was blocked with a universal blocking reagent (HK085-5K; Biogenex). Sections were then treated with PPARγ antibody, followed by incubation with secondary antibody, application of StrepABComplex/HRP and staining with diaminobenzidine (DAB) and Harris haematoxylin, using the aforementioned reagents and dilutions.
Cell growth inhibition
Cells were plated on 24-well plates at a density of 5 × 104 cells well−1. After 24 h, the growth medium was supplemented with DMSO at a concentration of 0.1% or with either one of the following: 15-deoxy-Δ12,14-Prostaglandin J2 (15-PGJ2) (Cayman Chemical, Ann Arbor, MI, USA) at concentrations of 10 or 20 μM, rosiglitazone (Cayman Chemical) at concentrations of 25 or 50 μM, and ciglitazone (Biomol, Plymouth Meeting, PA, USA) at concentrations of 25 or 50 μM. All three compounds were dissolved in 100% DMSO, so that the final concentration of DMSO did not exceed 0.1%. Following incubation for 24, 48 or 72 h, either treated or untreated cells were removed enzymatically and counted using a Coulter Counter (Coulter Model ZI, Coulter Corporation, Miami, FL, USA). The per cent of growth was determined setting as 100% the growth of cells treated only with the vehicle (0.1% DMSO). All analyses were performed in triplicate.
Cell cycle analysis
Cells were treated either with the vehicle alone (0.1% DMSO) or with 10 or 20 μM of 15-PGJ2 dissolved in 100% DMSO for 72 h. Cells were dissociated using trypsin-EDTA in PBS, pelleted and resuspended in 1 ml of PBS. While vortexing, 5 ml of 70% ethanol in distilled water were added dropwise. The cells were incubated at 4°C for 30 min and then were centrifuged at 2500 g for 5 min. The pellet was resuspended in 200 μl of two-fold propidium iodide (PI) dye and 200 μl of RNAse A (2 mg ml−1) and the tubes were incubated for 45 min in the dark at 4°C. Cells were filtered through nylon mesh prior to analysis. The DNA content of cells stained with PI and was measured with a FACScan instrument using Cell Quest software (Becton/Dickinson). To determine the proportion of cells in G0-G1, S and G2-M, cell cycle analysis of DNA histograms was performed using ModFitLTV2.0 (PMac). All analyses were performed in duplicate.
Apoptosis analysis
Apoptosis was assessed by Annexin V-FITC staining (BD Biosciences). In essence, cells were treated either with the vehicle alone (0.1% DMSO) or with 10 or 20 μM of 15-PGJ2 dissolved in 100% DMSO for 72 h. Then, cells were washed twice with cold PBS and resuspended in 1 × binding buffer at a concentration of 1 × 106 cell ml−1. One hundred microlitres of the solution were transferred to a 5 ml culture tube and 5 μl of Annexin V-FITC and 5 μl of PI were added. The mixture was gently vortexed and incubated for 15 min in the dark. Four hundred microlitres of 1 × binding buffer were added to each tube and FACScan analysis of the samples was performed within 1 h using a FACS scan equipped with Cell Quest software (Becton Dickenson). The following controls were used to set up compensation and quadrants: (1) unstained cells, (2) cells stained with Annexin V-FITC alone, and (3) cells stained with PI alone. The percentage of cells that have been induced to undergo apoptosis was determined by subtracting the percentage of apoptotic cells in the untreated population from the percentage of apoptotic cells in the treated population. All analyses were performed in duplicate.
Protein lysate preparation and Western blotting
Cells were plated on 6-well plates at a density of 5 × 104 cells well−1 and were allowed to grow to 80% confluency. Then, 15-PGJ2 at 20 μM concentration, rosiglitazone at 50 μM concentration, or ciglitazone at 50 μM concentration, was added to the medium. All three compounds were dissolved in 100% DMSO, so that the final concentration of DMSO did not exceed 0.1%. Following incubation for various time periods, the cells were washed twice with cold PBS, lysed in RIPA buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% deoxycholic acid, sodium salt, 0.1% sodium dodecyl sulphate (SDS), 100 μg ml−1 phenylmethylsulphonyl fluoride, 1 μg ml−1 aprotinin, 1 mM dithiothreitol, and 1 mM sodium orthovanadate) for 10 min, and scraped. The extracts were centrifuged at 40000 g for 15 min at 4°C. Protein concentrations were measured and equalised using Bio-Rad protein assay (Bio-Rad Laboratories, Richmond, CA, USA) according to the manufacturer's instructions.
Western blot analysis was performed using phospho-Stat3 (Tyr 705) antibody (1:500 dilution) (Cell Signaling Technology, Beverly, MA, USA) according to the manufacturer's instructions. Blots were stripped (20 mM dithiothreitol, 2% SDS, and 67.5 mM Tris-HCl (pH 6.7)) and then reprobed sequentially with Stat3 (Tyr 705) antibody (1:1000 dilution) (Cell Signaling Technology, Beverly, MA, USA) and with actin antibody (1:500 dilution) (Sigma, Saint Louis, MO, USA).
Statistical analysis
Data were submitted to an analysis of variance with repeated measures (time and dosage), using Newman–Keuls post hoc comparisons where appropriate (Statistica for Windows, StatSoft, Inc., Tulsa, OK, USA). An alpha value of P⩽0.05 was used in all models and post hoc comparisons.
Results
Expression of PPARγ in oral SCCa cell lines and tissues
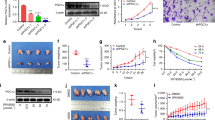
Quantitative RT–PCR analysis showed mRNA expression for PPARγ in all four oral SCCa cell lines. The lowest levels of PPARγ mRNA were expressed in SCC25 and the highest in SCC9 (Table 1). Protein expression of PPARγ was detected by means of immunocytochemistry; PPARγ immunostaining, primarily in a cytoplasmic location, was evident in all four oral SCCa cell lines (Figure 1A,B). Immunohistochemistry for PPARγ in tumour specimens of patients with oral SCCa revealed PPARγ production by the tumour cells, which was limited to the well-differentiated areas of the tumours (Figure 1C).

(A, B) Immunocytochemical detection of PPARγ protein expression in oral SCCa cells (A, SCC25; B, SCC9). (C) Immunohistochemical detection of PPARγ in specimens of oral SCCa; immunostaining was limited to the well-differentiated areas of the tumours.
Effect of PPARγ agonists on cell growth inhibition
Oral SCC25 cells treated with 10 or 20 μM of 15-PGJ2 exhibited reduction in cell growth (Figure 2); similar results were obtained from the other oral SCCa cell lines. Combining the results from all four cell lines, a significant main effect for time (F2,6=20.6, P⩽0.01) and interaction of time vs dosage (F4,12=17.4, P⩽0.0001) was observed, reflecting that increases in dosage and time of treatment were associated with decreases in cell growth.

(A, B) Effect of 15-deoxy-Δ12,14-PGJ2 on growth of oral SCC25 cells. The cells were treated with 0.1% DMSO, 10 μM 15-deoxy-PGJ2 or 20 μM 15-deoxy-PGJ2 for various periods of time (24, 48, or 72 h). In (A), the real number of cells is depicted, while in (B), the per cent of cell growth is shown, after setting the growth of cells treated only with the vehicle as 100%. A statistically significant reduction of cell growth was observed. Similar results were obtained from the other oral SCCa cell lines tested. (C) Effect of rosiglitazone (ROSI), ciglitazone (CIGL) and 15-deoxy-Δ12,14-PGJ2 on growth of oral SCC9 cells. The cells were treated with 0.1% DMSO, 50 μM rosiglitazone, 50 μM ciglitazone or 20 μM of 15-deoxy-PGJ2 for 72 h. Only 15-PGJ2 induced a significant cell growth inhibition. All tested oral SCCa cell lines gave similar results.
In contrast, treatment with rosiglitazone or ciglitazone did not induce significant cell growth inhibition in any of the tested cell lines (P>0.05), even when the highest dosage of treatment (i.e. 50 μM) for the longest duration (i.e. 72 h) was employed (Figure 2C).
Effect of 15-deoxy-Δ12,14-PGJ2 on apoptosis and cell proliferation
We tested next whether the cell growth inhibition induced by 15-PGJ2 was due to alterations in apoptosis or cell proliferation rates.
Treatment of oral SCCa cells with 10 or 20 μM of 15-PGJ2 did not significantly affect the percentage of cells in the S phase of the cell cycle, which remained stable or was only slightly increased; however, an increase in the percentage of cells in the G2 phase of the cell cycle was observed, accompanied by a corresponding reduction of cells in the G1 phase (Table 2). These results indicate a relative accumulation of cells in the G2 phase, which may interfere with cell cycle progression. On the other hand, significant 5–8-fold increases in the levels of apoptosis resulted following treatment with 15-deoxy-Δ12,14-PGJ2 for 72 h at concentrations ranging from 10 to 20 μM (F2,4=9.5, P⩽0.05) (Figure 3).

Effect of 15-deoxy-Δ12,14-PGJ2 on levels of apoptosis of oral SCC25 and SCC9 cells treated with 0.1% DMSO or 20 μM of the drug for 72 h. Treatment with 15-deoxy-Δ12,14-PGJ2 resulted in a statistically significant increase in apoptosis compared to DMSO-treated control cells. Similar results were obtained from the other oral SCCa cell lines tested.
Effects of PPAR agonists on Stat3 phosphorylation and expression
Because of the critical role of Stat3 in head and neck carcinogenesis (Grandis et al, 1998, 2000) and our recent demonstration of sulindac-mediated downmodulation of Stat3 in oral SCCa (Nikitakis et al, 2002b), we explored the hypothesis that ligand-mediated PPARγ activation causes changes in Stat3 expression and activation.
Forty-five minutes of treatment with 20 μM of 15-PGJ2 resulted in a significant reduction of phosphorylated Stat3 in SCC9 cells. Longer treatment, up to 9 h, did not induce further reduction in phosphorylated Stat3 levels. In contrast, phosphorylated Stat3 levels were eliminated after 24, 48 or 72 h of treatment with 20 μM of 15-deoxy-PGJ2 (Figure 4A). Stat3 protein expression levels in SCC9 cells also exhibited a small decrease after 45 min of treatment with 20 μM 15-PGJ2. Nonetheless, the decrease in protein expression of Stat3 could not account for the reduction in phosphorylated levels of this protein. Stat3 protein levels were not further reduced as a result of longer, up to 24 h, treatment. However, 48 h of treatment induced further significant reduction of Stat3 expression, which was eliminated after 72 h of treatment (Figure 4A). In accordance with the aforementioned results, phosphorylated Stat3 levels were eliminated and Stat3 protein expression levels were either eliminated or severely reduced by 72 h of 15-deoxy-PGJ2 treatment with 20 μM in all tested cell lines (Figure 4B). These effects could not be attributed to a nonspecific reduction of protein expression, as the protein expression levels of actin were not significantly affected by treatment (Figure 4A,B). On the contrary, treatments with rosiglitazone or ciglitazone at 50 μM concentrations failed to affect the phosphorylation or expression of Stat3 (Figure 4B).

(A) 15-deoxy-PGJ2 inhibits phosphorylation and production of Stat3 in oral SCCa cells. Oral SCC9 cells were treated with normal medium (NM) or 20 μM 15-deoxy-PGJ2 for 45 min, or 1.5, 3, 4.5, 9, 24, 48, 72 h, as indicated. Cells were lysed, blotted with antibody to phosphorylated Stat3 (phospho Stat3), and then sequentially stripped and reprobed with antibodies to Stat3 and to actin. (B) 15-deoxy-PGJ2, but not rosiglitazone or ciglitazone, inhibits phosphorylation and production of Stat3 in oral SCCa cells. Oral SCCa cells, derived from cell lines SCC4, 15, or 25, were treated for 48 h with normal medium (NM), 50 μM ciglitazone (CI), 50 μM rosiglitazone (RO), or 20 μM 15-deoxy-PGJ2 (PG), as indicated. Cells were lysed, blotted with antibody to phosphorylated Stat3 (phospho Stat3), and then sequentially stripped and reprobed with antibodies to Stat3 and to actin.
Notably, no changes in PPARγ protein expression were induced by either 15-deoxy-PGJ2 or thiazolidinediones (Data not shown).
Discussion
The capacity of PPARγ to promote differentiation, cell cycle withdrawal and apoptosis has encouraged extensive investigation of its potential anticancer activity in multiple types of human cancer (see Introduction). We recently showed that NSAID sulindac upregulates PPARγ expression and activity and relies on PPARγ availability for its antineoplastic activities on oral SCCa (Nikitakis et al, 2002a). These observations prompted us to investigate the possibility that oral SCCa cells are also responsive to treatment with direct PPARγ ligands.
15-PGJ2, a natural PPARγ ligand, exerted a statistically significant growth inhibitory effect on oral SCCa cells. Significant increases in the levels of apoptosis were observed, suggesting that 15-PGJ2 cell growth inhibitory effect is primarily mediated through induction of apoptosis. Nonetheless, accumulation of cells in the G2 phase of the cell cycle was also noted, supporting the concept that interference of 15-PGJ2 with cell cycle progression may also contribute to its activity. In contrast, the well-characterised PPARγ ligands rosiglitazone and ciglitazone did not inhibit the cell growth of oral SCCa cells, implying that PPARγ activation is not sufficient for inducing antineoplastic effects on oral SCCa cells. These results raise the possibility that the antineoplastic properties of 15-PGJ2 are, at least partially, mediated through PPARγ-independent mechanisms.
PPARγ-independent effects of 15-PGJ2 have been previously reported in other systems, including chondrocytes, myofibroblasts, mesangial cells, inflammatory cells and cells of the nervous system (Petrova et al, 1999; Castrillo et al, 2000; Rossi et al, 2000; Straus et al, 2000; Boyault et al, 2001; Li et al, 2001; Janabi, 2002; Ward et al, 2002). Several mechanisms have been implicated as responsible for the effects of 15-PGJ2 on these cells and may also partly explain its antineoplastic properties. Negative regulation of the NF-κB pathway through inhibition of IκB kinase (IKK) and abrogation of the DNA binding ability of NF-κB, has emerged as a major pathway of PPARγ-independent 15-PGJ2 activity (Petrova et al, 1999; Castrillo et al, 2000; Rossi et al, 2000; Straus et al, 2000; Boyault et al, 2001; Janabi, 2002;). 15-PGJ2-mediated NF-κB inhibition has been linked to downregulation of inducible nitric oxide synthase (iNOS) and abrogation of cyclooxygenase-2 (COX-2) transactivation (Petrova et al, 1999; Castrillo et al, 2000; Rossi et al, 2000; Straus et al, 2000; Boyault et al, 2001; Janabi, 2002). Similarly, Ward et al (2002) recently showed that 15-PGJ2 exploits PPARγ-independent inhibition of NF-κB activation to induce caspase-dependent apoptosis in granulocytes. Other mediators that have been implicated in PPARγ-independent properties of 15-PGJ2 and can potentially have a similar function in cancer cells include AP-1 (Boyault et al, 2001), MAP kinase (Harris et al, 2002; Lennon et al, 2002), and reactive oxygen species (Li et al, 2001; Lennon et al, 2002). The latter has been shown to act as intermediates for the induction of apoptosis caused by 15-PGJ2 in human myofibroblasts (Li et al, 2001). Identification of the molecular pathways that mediate the PPARγ-independent antineoplastic effects of 15-PGJ2 should be thoroughly addressed in future studies. In this respect, the recent observations of Clay et al (2001) that early de novo gene expression is necessary for 15-PGJ2-induced apoptosis in breast cancer cells may be of particular relevance.
Although our data strongly support the existence of PPARγ-independent effects of 15-PGJ2 on oral SCCa cells, recruitment of PPARγ-mediated pathways cannot be ruled out. However, the precise molecular mechanisms that are responsible for the antineoplastic properties of PPARγ are not well understood. An association has been suggested between PPARγ and COX-2, which has also been implicated in various human cancers, including head and neck SCCa (Chan et al, 1999; Dannenberg et al, 2001). PPARγ activators may inhibit COX-2 expression, possibly through negative interference with NF-κB and/or AP-1 activation (Inoue et al, 2000; Subbaramaiah et al, 2001; Yang and Frucht, 2001). There is also evidence that supports the function of PPARγ ligands as potent inhibitors of angiogenesis in vivo and in vitro, providing an additional mechanism that may partially account for the anticancer properties of PPARγ (Bishop-Bailey and Hla, 1999; Xin et al, 1999). Finally, cross-talk between PPARγ and other signalling molecules, such as NF-κB, AP-1 and STAT (Ricote et al, 1998; Zhou and Waxman, 1999a, b), may contribute significantly to the effects of PPARγ on tumour growth. The possible contribution, if any, of PPARγ activation to growth inhibition induced by 15-PGJ2 treatment warrants further exploration.
Constitutive activation of Stat3 plays an important role in the tumorigenesis of various types of human cancer (Catlett-Falcone et al, 1999; Fernandes et al, 1999), and abrogation of Stat3 signalling has been correlated with stimulation of cell proliferation, prevention of apoptosis and tumour formation (Bromberg et al, 1999; Bowman et al, 2000). Based on accumulating evidence that Stat3 is upregulated in head and neck SCCa (Grandis et al, 1998, 2000), we investigated whether treatment with PPARγ ligands exerts an effect on Stat3 protein expression and Stat3 tyrosine phosphorylation. Treatment of oral SCCa cells with 15-PGJ2 at concentrations that cause significant reduction of cell growth resulted in downregulation of both Stat3 expression and phosphorylation. Given that targeting of Stat3 in head and neck SCCa has been linked to significant growth inhibition and induction of apoptosis both in vitro and in vivo (Grandis et al, 1998, 2000), our results suggest that the ability of 15-PGJ2 to downregulate Stat3 may be aetiologically related to its growth inhibitory and apoptotic effects. Interestingly, the mode of Stat3 downregulation (i.e. reduction of Stat3 phosphorylated levels after 45 min and elimination of both Stat3 phosphorylated and unphosphorylated levels after 72 h) was very similar to that induced by sulindac sulphide in the same cell lines (Nikitakis et al, 2002b). Although sulindac's growth inhibitory effect was dependent on PPARγ availability, its ability to induce Stat3 downmodulation was independent of its ability to act as a PPARγ ligand. Similarly, the inability of PPARγ activation trough rosiglitizone and ciglitazone stimulation to affect the phosphorylation and expression levels of Stat3 entails that activation of PPARγ-independent mechanisms is necessary for 15-PGJ2-mediated Stat3 downmodulation.
Both PPARγ-dependent and PPARγ-independent mechanisms may come into play during Stat3 downregulation by 15-PGJ2. Direct protein–protein interactions or indirect mechanisms, such as competition for common co-activators or modulation of inhibitors of transcriptional activity, have been suggested as possible mediators of STAT-PPAR cross-talk and could account for the Stat3 inhibitory effect of PPARγ activation (Zhou and Waxman, 1999a, b). Cytokine stimulation results in phosphorylation of STATs through the mediation of the Janus kinase (JAK) family of protein tyrosine kinases (Darnell et al, 1994). Accordingly, the recently proposed association between PPARγ and cytokine expression may represent another possible connection between Stat3 and PPARγ. Indeed, PPARγ agonists have been shown to suppress monocyte elaboration of inflammatory cytokines (Jiang et al, 1998) and to inhibit IL-1β-induced expression of IL-8 in colon cancer cell lines (Su et al, 1999). 15-PGJ2-mediated inhibition of cytokine production and function may also ensue through PPARγ-independent pathways; for example, 15-PGJ2 has been shown to modulate IL-1β effects in human chondrocytes (Boyault et al, 2001) and to inhibit TNF-α and IL-6 production in human macrophages without PPARγ mediation. Inhibition of cytokine expression by 15-PGJ2 would conceivably result in decreased activation of STAT molecules that mediate the transduction of the cytokine signal from the cell surface to the nucleus. Head and neck cancer cells have been shown to express a variety of pro-inflammatory and pro-angiogenic cytokines (Chen et al, 1999; Ondrey et al, 1999) and to respond to IL-6 stimulation with upregulation of Stat3 phosphorylation and promotion of cell growth; reversal of this process could participate in the observed effects of 15-PGJ2 on oral SCCa cells. In that aberrant tumour growth factor-α/epidermal growth factor receptor signalling has been demonstrated to play a major role in Stat3 constitutive activation of head and neck SCCa cells, possible interference of 15-PGJ2 with this pathway, at the level of the ligand or the receptor, constitutes another distinct possibility.
In summary, we demonstrated that 15-PGJ2, a specific natural PPARγ ligand, inhibits growth of oral SCCa cells, which may be related to its capacity to downregulate the oncogene Stat3. The ineffectiveness of rosiglitazone and ciglitazone to cause similar effects strongly suggests that 15-PGJ2 effects are at least partly mediated through PPARγ-independent mechanisms. Delineation of these mechanisms, as well as determination of the potential contribution of PPARγ activation, not only will provide an explanation for 15-PGJ2 anticancer qualities but it will also enhance our understanding of critical signalling pathways for oral SCCa carcinogenesis.
References
Bishop-Bailey D, Hla T (1999) Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta12, 14-prostaglandin J2. J Biol Chem 274: 17042–17048
Boyault S, Simonin MA, Bianchi A, Compe E, Liagre B, Mainard D, Becuwe P, Dauca M, Netter P, Terlain B, Bordji K (2001) 15-Deoxy-delta12,14-PGJ2, but not troglitazone, modulates IL-1beta effects in human chondrocytes by inhibiting NF-kappaB and AP-1 activation pathways. FEBS Lett 501: 24–30
Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19: 2474–2488
Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell Jr JE (1999) Stat3 as an oncogene. Cell 98: 295–303
Butler R, Mitchell SH, Tindall DJ, Young CY (2000) Nonapoptotic cell death associated with S-phase arrest of prostate cancer cells via the peroxisome proliferator-activated receptor gamma ligand, 15-deoxy-delta12,14-prostaglandin J2. Cell Growth Differ 11: 49–61
Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L (2000) Inhibition of IkappaB kinase and IkappaB phosphorylation by 15-deoxy-Delta(12,14)-prostaglandin J(2) in activated murine macrophages. Mol Cell Biol 20: 1692–1698
Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS, Jove R (1999) Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10: 105–115
Chan G, Boyle JO, Yang EK, Zhang F, Sacks PG, Shah JP, Edelstein D, Soslow RA, Koki AT, Woerner BM, Masferrer JL, Dannenberg AJ (1999) Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res 59: 991–994
Chang TH, Szabo E (2000) Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor gamma in non-small cell lung cancer. Cancer Res 60: 1129–1138
Chen Z, Malhotra PS, Thomas GR, Ondrey FG, Duffey DC, Smith CW, Enamorado I, Yeh NT, Kroog GS, Rudy S, McCullagh L, Mousa S, Quezado M, Herscher LL, Van Waes C (1999) Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res 5: 1369–1379
Clay CE, Atsumi GI, High KP, Chilton FH (2001) Early de novo gene expression is required for 15-deoxy-Delta 12,14-prostaglandin J2-induced apoptosis in breast cancer cells. J Biol Chem 276: 47131–47135
Dannenberg AJ, Altorki NK, Boyle JO, Lin DT, Subbaramaiah K (2001) Inhibition of cyclooxygenase-2: an approach to preventing cancer of the upper aerodigestive tract. Ann N Y Acad Sci 952: 109–115
Darnell Jr JE, Kerr IM, Stark GR (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264: 1415–1421
Debril MB, Renaud JP, Fajas L, Auwerx J (2001) The pleiotropic functions of peroxisome proliferator-activated receptor gamma. J Mol Med 79: 30–47
Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, Spiegelman BM, Singer S (1999) Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci USA 96: 3951–3956
Duez H, Fruchart JC, Staels B (2001) PPARS in inflammation, atherosclerosis and thrombosis. J Cardiovasc Risk 8: 187–194
Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, Shintaku P, Said JW, Heber D, Koeffler HP (1998) Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci USA 95: 8806–8811
Fernandes A, Hamburger AW, Gerwin BI (1999) ErbB-2 kinase is required for constitutive stat 3 activation in malignant human lung epithelial cells. Int J Cancer 83: 564–570
Forman BM, Chen J, Evans RM (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA 94: 4312–4317
Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM (1995) 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 83: 803–812
Gelman L, Fruchart JC, Auwerx J (1999) An update on the mechanisms of action of the peroxisome proliferator-activated receptors (PPARs) and their roles in inflammation and cancer. Cell Mol Life Sci 55: 932–943
Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, Tweardy DJ (1998) Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest 102: 1385–1392
Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, Johnson DE, Huang L, He Y, Kim JD (2000) Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci USA 97: 4227–4232
Harris SG, Smith RS, Phipps RP (2002) 15-deoxy-Delta 12,14-PGJ2 induces IL-8 production in human T cells by a mitogen-activated protein kinase pathway. J Immunol 168: 1372–1379
Ikezoe T, Miller CW, Kawano S, Heaney A, Williamson EA, Hisatake J, Green E, Hofmann W, Taguchi H, Koeffler HP (2001) Mutational analysis of the peroxisome proliferator-activated receptor gamma gene in human malignancies. Cancer Res 61: 5307–5310
Inoue H, Tanabe T, Umesono K (2000) Feedback control of cyclooxygenase-2 expression through PPARgamma. J Biol Chem 275: 28028–28032
Issemann I, Green S (1990) Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347: 645–650
Janabi N (2002) Selective inhibition of cyclooxygenase-2 expression by 15-deoxy-Delta(12,14)(12,14)-prostaglandin J(2) in activated human astrocytes, but not in human brain macrophages. J Immunol 168: 4747–4755
Jiang C, Ting AT, Seed B (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391: 82–86
Kersten S, Desvergne B, Wahli W (2000) Roles of PPARs in health and disease. Nature 405: 421–424
Kitamura S, Miyazaki Y, Shinomura Y, Kondo S, Kanayama S, Matsuzawa Y (1999) Peroxisome proliferator-activated receptor gamma induces growth arrest and differentiation markers of human colon cancer cells. Jpn J Cancer Res 90: 75–80
Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, Miyoshi I, Koeffler HP (1998) Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res 58: 3344–3352
Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA (1997) Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem 272: 3406–3410
Lemberger T, Desvergne B, Wahli W (1996) Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol 12: 335–363
Lennon AM, Ramauge M, Dessouroux A, Pierre M (2002) MAP kinase cascades are activated in astrocytes and preadipocytes by 15-deoxy-delta 12-14-prostaglandin J2 and the thiazolidinedione ciglitazone through peroxisome proliferator activator receptor gamma-independent mechanisms involving reactive oxygenated species. J Biol Chem 277: 29681–29685
Li L, Tao J, Davaille J, Feral C, Mallat A, Rieusset J, Vidal H, Lotersztajn S (2001) 15-deoxy-Delta 12,14-prostaglandin J2 induces apoptosis of human hepatic myofibroblasts. A pathway involving oxidative stress independently of peroxisome-proliferator-activated receptors. J Biol Chem 276: 38152–38158
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P (1995) The nuclear receptor superfamily: the second decade. Cell 83: 835–839
Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM (1998) Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1: 465–470
Nikitakis NG, Hebert C, Lopes MA, Reynolds MA, Sauk JJ (2002a) PPARγ-mediated antineoplastic effect of NSAID sulindac on human oral squamous carcinoma cells. Int J Cancer 98: 817–823
Nikitakis NG, Hamburger AV, Sauk JJ (2002b) The nonsteroidal anti-inflammatory drug sulindac causes downregulation of signal transducer and activator of transcription 3 (Stat3) in human oral squamous cell carcinoma cells. Cancer Res 62: 1004–1007
Ondrey FG, Dong G, Sunwoo J, Chen Z, Wolf JS, Crowl-Bancroft CV, Mukaida N, Van Waes C (1999) Constitutive activation of transcription factors NF-(kappa)B, AP-1, and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express pro-inflammatory and pro-angiogenic cytokines. Mol Carcinog 26: 119–129
Park BH, Breyer B, He TC (2001) Peroxisome proliferator-activated receptors: roles in tumorigenesis and chemoprevention in human cancer. Curr Opin Oncol 13: 78–83
Petrova TV, Akama KT, Van Eldik LJ (1999) Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc Natl Acad Sci USA 96: 4668–4673
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391: 79–82
Rosen ED, Spiegelman BM (2001) Ppargamma:a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem 276: 37731–37734
Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG (2000) Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 403: 103–108
Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher C, Spiegelman BM (1998) Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med 4: 1046–1052
Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, de la Chapelle A, Spiegelman BM, Eng C (1999) Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell 3: 799–804
Sato H, Ishihara S, Kawashima K, Moriyama N, Suetsugu H, Kazumori H, Okuyama T, Rumi MA, Fukuda R, Nagasue N, Kinoshita Y (2000) Expression of peroxisome proliferator-activated receptor (PPAR)gamma in gastric cancer and inhibitory effects of PPARgamma agonists. Br J Cancer 83: 1394–1400
Shao D, Lazar MA (1997) Peroxisome proliferator activated receptor gamma, CCAAT/enhancer-binding protein alpha, and cell cycle status regulate the commitment to adipocyte differentiation. J Biol Chem 272: 21473–21478
Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK (2000) 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci USA 97: 4844–4849
Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD (1999) A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest 104: 383–389
Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ (2001) Peroxisome proliferator-activated receptor gamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J Biol Chem 276: 12440–12448
Sugimura A, Kiriyama Y, Nochi H, Tsuchiya H, Tamoto K, Sakurada Y, Ui M, Tokumitsu Y (1999) Troglitazone suppresses cell growth of myeloid leukemia cell lines by induction of p21WAF1/CIP1 cyclin-dependent kinase inhibitor. Biochem Biophys Res Commun 261: 833–837
Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, Brun RP, Mueller E, Altiok S, Oppenheim H, Evans RM, Spiegelman BM (1997) Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci USA 94: 237–241
Vamecq J, Latruffe N (1999) Medical significance of peroxisome proliferator-activated receptors. Lancet 354: 141–148
Ward C, Dransfield I, Murray J, Farrow SN, Haslett C, Rossi AG (2002) Prostaglandin D2 and its metabolites induce caspase-dependent granulocyte apoptosis that is mediated via inhibition of I kappa B alpha degradation using a peroxisome proliferator-activated receptor-gamma-independent mechanism. J Immunol 168: 6232–6243
Xin X, Yang S, Kowalski J, Gerritsen ME (1999) Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J Biol Chem 274: 9116–9121
Yang WL, Frucht H (2001) Activation of the PPAR pathway induces apoptosis and COX-2 inhibition in HT-29 human colon cancer cells. Carcinogenesis 22: 1379–1383
Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, Brown M, Lazar MA (1995) Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem 270: 23975–23983
Zhou YC, Waxman DJ (1999a) Cross-talk between janus kinase-signal transducer and activator of transcription (JAK-STAT) and peroxisome proliferator-activated receptor-alpha (PPARalpha) signaling pathways. Growth hormone inhibition of pparalpha transcriptional activity mediated by stat5b. J Biol Chem 274: 2672–2681
Zhou YC, Waxman DJ (1999b) STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain. J Biol Chem 274: 29874–29882
Acknowledgements
This work was supported in part by PHS grants DE 12606 and DE 13118.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Nikitakis, N., Siavash, H., Hebert, C. et al. 15-PGJ2, but not thiazolidinediones, inhibits cell growth, induces apoptosis, and causes downregulation of Stat3 in human oral SCCa cells. Br J Cancer 87, 1396–1403 (2002). https://doi.org/10.1038/sj.bjc.6600618
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6600618
Keywords
This article is cited by
-
The multifaceted role of STAT3 pathway and its implication as a potential therapeutic target in oral cancer
Archives of Pharmacal Research (2022)
-
Treatment of primary CLL cells with bezafibrate and medroxyprogesterone acetate induces apoptosis and represses the pro-proliferative signal of CD40-ligand, in part through increased 15dΔ12,14,PGJ2
Leukemia (2009)
-
PPARγ-independent induction of growth arrest and apoptosis in prostate and bladder carcinoma
BMC Cancer (2006)
-
The Biological Functions of the Versatile Transcription Factors STAT3 and STAT5 and New Strategies for their Targeted Inhibition
Journal of Mammary Gland Biology and Neoplasia (2006)
-
15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) Induces Cell Death Through Caspase-independent Mechanism in A172 Human Glioma Cells
Neurochemical Research (2006)
