
Abstract
Insulin stimulates the transport of glucose into fat and muscle cells. Although the precise molecular mechanisms involved in this process remain uncertain, insulin initiates its actions by binding to its tyrosine kinase receptor, leading to the phosphorylation of intracellular substrates. One such substrate is the Cbl protooncogene product1. Cbl is recruited to the insulin receptor by interaction with the adapter protein CAP, through one of three adjacent SH3 domains in the carboxy terminus of CAP2. Upon phosphorylation of Cbl, the CAP–Cbl complex dissociates from the insulin receptor and moves to a caveolin-enriched, triton-insoluble membrane fraction3. Here, to identify a molecular mechanism underlying this subcellular redistribution, we screened a yeast two-hybrid library using the amino-terminal region of CAP and identified the caveolar protein flotillin. Flotillin forms a ternary complex with CAP and Cbl, directing the localization of the CAP–Cbl complex to a lipid raft subdomain of the plasma membrane. Expression of the N-terminal domain of CAP in 3T3-L1 adipocytes blocks the stimulation of glucose transport by insulin, without affecting signalling events that depend on phosphatidylinositol-3-OH kinase. Thus, localization of the Cbl–CAP complex to lipid rafts generates a pathway that is crucial in the regulation of glucose uptake.
Similar content being viewed by others

Main
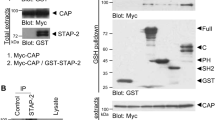
To identify proteins that may participate in the localization and function of Cbl and CAP, we screened a yeast two-hybrid library derived from 3T3-L1 adipocytes4 using as bait the N-terminal region of CAP, which contains the sorbin homology domains but lacks the SH3 domains (CAPΔSH3, Fig. 1a). Three independent clones were isolated encoding the protein flotillin5. To examine the binding specificity of flotillin and CAP, we incubated glutathione-Ssepharose-bound flotillin (GST–flotillin) with lysates from human embryonic kidney (HEK293T) cells overexpressing either Flag-epitope-tagged full-length CAP or Flag-epitope-tagged CAPΔSH3. As seen in Fig. 1b, GST–flotillin bound to both Flag–CAP and Flag–CAPΔSH3. It also precipitated endogenous Cbl from the extracts in the presence of CAP but not when CAPΔSH3 was co-expressed. These data indicate that the interaction of flotillin with Cbl requires full-length CAP to form a flotillin–CAP–Cbl ternary complex.

a, Flag-epitope-tagged expression constructs of CAP and CAPΔSH3 mutant. b, Flag-epitope-tagged CAP (left) or CAPΔSH3 (right) overexpression lysates from HEK293T cells were incubated with either glutathione agarose or glutathione-agarose-bound GST–flotillin for 1 h at 4 °C. Precipitates were subjected to SDS–polyacrylamide gel electrophoresis (PAGE), transferred to nitrocellulose and blotted with monoclonal Flag M2 antibody (Stratagene) and polyclonal c-Cbl antibody (Santa Cruz).
Triton-insoluble microdomains of the plasma membrane have been implicated in signal transduction6,7. Flotillin is an integral membrane protein that associates with the triton-insoluble fraction enriched in the caveolar protein caveolin5. Moreover, flotillin can physically associate in hetero-oligomeric structures with caveolin in detergent-solubilized cells8. The physiochemical properties of proteins that are enriched in lipid rafts make it difficult to perform standard, triton-soluble, co-immunoprecipitation assays. We therefore examined the subcellular localization of CAP, CAPΔSH3, endogenous caveolin and flotillin by confocal immunofluorescence microscopy of intact cells and by immunofluorescence following preparation of plasma membrane sheets. After transfection of 3T3-L1 adipocytes by electroporation9, both Flag–CAP and Flag–CAPΔSH3 showed diffuse staining of the cytoplasm, with a significant portion found nonuniformly distributed around the plasma membrane (see Supplementary Information Fig. 1). Immunostaining of endogenous caveolin followed by a Texas Red secondary antibody revealed caveolin in the plasma membrane, as reported6,7. Merging the two images shows that a significant portion of both CAP and CAPΔSH3 proteins are found at the plasma membrane, similar to the endogenous caveolin I.
To examine the plasma membrane localization of caveolin in relation to flotillin, we prepared plasma membrane sheets from 3T3-L1 adipocytes and immunostained them with antibodies to caveolin and flotillin. Caveolin and flotillin were localized together in circular rosette structures in the plasma membrane (see Supplementary Information Fig. 2). The co-distribution of flotillin and caveolin in the plasma membrane was unaffected by insulin stimulation. These flotillin and caveolin-enriched structures probably consist of collections of caveolae or lipid rafts10.
To investigate further the dynamics of endogenous Cbl protein association with flotillin, we took a similar approach using immunofluorescence of isolated plasma membrane sheets (Fig. 2). As described above, exposure of cells to insulin for 2–60 min had no effect on the overall plasma membrane distribution of flotillin. Although Cbl was not detected in the plasma membrane sheets in the basal state, there was transient translocation of Cbl to the plasma membrane, which was apparent after 2 and 5 min of insulin stimulation. This time-dependent translocation of Cbl to the plasma membrane is similar to the time course of insulin-stimulated Cbl tyrosine phosphorylation11. Furthermore, merging of these images showed that Cbl localized with flotillin in rosette-type structures, again suggesting the insulin-dependent formation of a ternary flotillin–CAP–Cbl complex.

Representative confocal immunofluorescence images of flotillin and Cbl localized to plasma membrane sheets isolated from basal cells or those treated with insulin (Ins) for the indicated times. The colocalization of FITC-labelled flotillin and Texas Red-labelled Cbl is shown.
Finally, to assess biochemically the relationship between caveolin, flotillin and Cbl, we subjected basal and insulin-stimulated 3T3-L1 adipocytes to non-detergent homogenization and sucrose gradient fractionation (see Supplementary Information Fig. 3). Consistent with the immunofluorescence of isolated plasma membrane sheets, caveolin was found primarily in the low-density membrane regions of these gradients (fractions 3 and 4), and was not affected by insulin treatment. Similarly, flotillin was found in the fractions containing caveolin. Cbl was primarily confined to the dense regions of the gradient (fractions 8–12) in the basal state. However, following insulin stimulation, a portion of the Cbl protein was redistributed to the low-density fractions containing caveolin and flotillin. Together, these data provide strong evidence that caveolin and flotillin are colocalized in distinct subdomains of the plasma membrane, and that insulin induces the translocation of Cbl to these caveolin- and flotillin-enriched compartments through its interaction with CAP.
To assess the potential role of CAP in early signalling events, we next overexpressed CAP or CAPΔSH3 in 3T3-L1 adipocytes and evaluated insulin-stimulated tyrosine phosphorylation. Both proteins were found in the triton-soluble and insoluble fractions (Fig. 3a), and expressed at levels around fivefold higher than the endogenous CAP protein (data not shown). Expression of CAP or CAPΔSH3 had no effect on the tyrosine phosphorylation of the insulin receptor β-subunit or its predominant substrate, IRS-1 (Fig. 3b). Similarly, there was no change in the ability of insulin to stimulate the phosphorylation of two downstream targets, AKT/protein kinase B and mitogen-activated protein kinase (MAPK), that are activated as a consequence of well characterized phosphorylation cascades (Fig. 3b). As AKT activation occurs as a direct consequence of phosphatidylinositol-3-OH kinase (PI(3)K) stimulation12, these data also show that the expression of CAPΔSH3 did not affect the stimulation of PI(3)K activity by insulin.

a, A representative FLAG immunoblot of the triton-soluble (TS) fraction (top) and triton-insoluble pellet (TIP) fraction (bottom) of 3T3-L1 adipocytes expressing LacZ, CAP or CAPΔSH3. b, Lysates from basal and insulin-treated adipocytes expressing LacZ, CAP or CAPΔSH3 were immunoblotted with anti-phosphotyrosine (top), phospho-AKT (middle) and phospho-MAPK (bottom) antibodies. c, Cbl was immunoprecipitated from the triton-soluble fraction of lysates from basal and insulin-treated adipocytes expressing LacZ, CAP or CAPΔSH3. Immunoprecipitates were resolved by SDS–PAGE, immunoblotted with monoclonal phosphotyrosine antibodies and re-probed with the Cbl antibody. A representative blot is shown within the graph. Films were scanned and quantified, and the ratio of total Cbl to phospho-Cbl was calculated. d, Cbl was immunoprecipitated from the triton-insoluble pellet fraction of lysates from basal and insulin-treated adipocytes expressing LacZ, CAP or CAPΔSH3. Immunoprecipitates were resolved and analysed as described in Fig. 2c. Double asterisk: significant difference, P < 0.01, n = 4.
The phosphorylation of Cbl by insulin requires the recruitment of the protein to the insulin receptor by CAP2. As CAPΔSH3 does not bind either Cbl or the insulin receptor, overexpression of CAP or CAPΔSH3 had no effect on insulin-stimulated Cbl phosphorylation in the triton-soluble fraction (Fig. 3c). However, CAPΔSH3 overexpression significantly reduced the appearance of tyrosine phosphorylated Cbl in the triton-insoluble fraction (Fig. 3d). These data indicate that the expression of the N-terminal flotillin-binding domain of CAP may be able to block the interaction of the endogenous CAP–Cbl complexes with flotillin, thus preventing the appearance of phosphorylated Cbl in the triton-insoluble plasma membrane subdomain.
To investigate the role of the association of the CAP–Cbl complex with flotillin in insulin action, we evaluated the effect of CAPΔSH3 expression on insulin-stimulated [14C]2-deoxyglucose uptake in 3T3-L1 adipocytes (Fig. 4a). Although wild-type CAP had no effect, expression of CAPΔSH3 led to a reduction of about 50% in insulin-stimulated glucose uptake, without affecting basal activity. Similar experiments were done to evaluate the effect of LacZ, CAP or CAPΔSH3 on glycogen and lipid synthesis (Fig. 4b, c). As observed for glucose uptake, the expression of CAPΔSH3 inhibited the insulin-stimulated incorporation of [14C]glucose into glycogen and lipid, whereas expression of CAP had no effect. Considering that the average efficiency of transfection by electroporation in these cells is about 50% (ref. 9), the observed reduction in glucose utilization may indicate that the expression of the CAPΔSH3 mutant almost completely blocked the metabolic actions of insulin.

3T3-L1 adipocytes were transfected by electroporation and the stimulation of [14C]2-deoxyglucose uptake (a), [14C]-glucose incorporation into glycogen (b) and [14C]-glucose incorporation into lipid (c) by insulin were measured. Results are the mean ± standard error of three separate experiments, each performed in triplicate. Double asterisk: significant difference, P < 0.01, n = 3.
The stimulation of glucose uptake and utilization by insulin is dependent upon the translocation of the insulin-responsive glucose transporter GLUT4 from intracellular storage sites to the plasma membrane. To investigate how CAPΔSH3 blocks glucose transport, the translocation of GLUT4 was directly examined. We electroporated 3T3-L1 adipocytes with pcDNA3, CAP or CAPΔSH3 together with a construct encoding an enhanced green fluorescent protein fusion of GLUT4 (GLUT4–EGFP; Fig. 5a). As expected, insulin stimulated the translocation of GLUT4–EGFP to the plasma membrane in cells transfected with pcDNA3 or CAP, as detected by the presence of a continuous rim of cell-surface EGFP fluorescence. In contrast, expression of CAPΔSH3 markedly inhibited GLUT4–EGFP translocation to the cell surface in response to insulin. Expression of CAPΔSH3 not only decreased the number of cells displaying GLUT4–EGFP at the cell surface, but also decreased the plasma membrane intensity in those cells that still had observable rim fluorescence (arrows). Quantification of the number of transfected cells displaying any visual rim fluorescence indicated that following pcDNA3 or wild-type CAP transfection, about 65% of the cells displayed insulin-stimulated translocation, which was reduced to less than 30% in cells transfected with CAPΔSH3.

a, Differentiated 3T3-L1 adipocytes were transfected with GLUT4–EGFP plus vector, Flag–CAP or Flag–CAPΔSH3, and allowed to recover for 18 h. The cells were treated without or with 100 nM insulin for 30 min. Cells were fixed and fluorescence visualized by confocal microscopy. This is a representative collection of 6–9 cell images per field. b, Number of GLUT4–EGFP transfected cells displaying visually detectable plasma membrabe (PM) rim fluorescence. c, d, Number of EGFP–IRAP- and CI/MPR–EGFP-transfected cells displaying visually detectable rim fluorescence. These data were obtained by blind counting of more than 75 cells from three independent experiments. Double asterisk: significant difference, P < 0.01, n = 3.
The insulin-responsive aminopeptidase (IRAP) is localized in the same compartments as GLUT4 in adipocytes, and undergoes an identical pattern of insulin-stimulated translocation13,14. As expected, expression of CAPΔSH3 blocked insulin-stimulated translocation of EGFP–IRAP, in a manner identical to that observed with EGFP–GLUT4 (Fig. 5c). Insulin also stimulates the net exocytosis of the cation-independent mannose-6-phosphate/IGF-2 receptor (CI/MPR). This protein is located in compartments distinct from the GLUT4/IRAP-containing vesicles, and travels between the trans-Golgi network and the late endosomes in the absence of insulin15,16,17. Expression of CAPΔSH3 had no significant effect on the insulin-stimulated translocation of a CI/MPR–EGFP fusion protein (Fig. 5d ), in contrast to its effects on GLUT4 and IRAP.
As the determination of translocation in these intact cell assays relies on the qualitative strength of plasma membrane rim fluorescence from overexpressed EGFP-fusion proteins, we also examined the translocation of the endogenous GLUT4 and GLUT1 proteins in isolated plasma membrane sheets from microinjected 3T3-L1 adipocytes (Fig. 6). To identify the plasma membrane sheets derived from the microinjected cells, the wild-type CAP and CAPΔSH3 complementary DNAs were microinjected into the cells with the maltose-binding protein (MBP)-Ras cDNA18. In the absence of insulin, microinjection of MBP-Ras and CAPΔSH3 resulted in the identification of four MBP-Ras positive sheets in the fields shown in Fig. 6 (arrows).

Differentiated 3T3-L1 adipocyte nuclei were microinjected with 0.2 mg ml-1 of MBP-Ras plus either the CAP or CAPΔSH3 cDNA. After 24 h recovery, cells were treated without or with 100 nM insulin for 30 min. Plasma membrane sheets were prepared and MBP-Ras was detected with a FITC-labelled anti-MBP antibody and GLUT4 with a Cy5-labelled anti-GLUT4 antibody. These are representative images obtained from five independent experiments and visualization of 60–91 individual plasma membrane sheets.
In the absence of insulin, GLUT4 remained intracellular, with low levels of plasma membrane GLUT4 in both the microinjected and surrounding non-microinjected cells. Insulin stimulation resulted in the appearance of GLUT4 at the plasma membrane in the non-microinjected cells, but not in the cells microinjected with CAPΔSH3. In contrast, microinjection of cells with the wild-type CAP cDNA had no significant effect on GLUT4 translocation, compared with the surrounding non-microinjected cells. Quantification of these data showed that insulin stimulated the translocation of GLUT4 in 87 ± 6% of the cells microinjected with wild-type CAP. In contrast, expression of CAPΔSH3 resulted in a marked reduction, with only 28 ± 10% of the plasma membrane sheets displaying any detectable GLUT4 translocation.
Like CI/MPR, the GLUT1 glucose transporter resides in vesicle compartments distinct from GLUT4, and undergoes modest insulin-stimulated plasma membrane translocation19. To assess the specificity of CAPΔSH3, we examined the translocation of the endogenous GLUT1 protein in microinjected 3T3-L1 adipocytes (see Supplementary Information Fig. 4). Insulin modestly stimulated the translocation of GLUT1. However, this effect of insulin was not impaired by the microinjection of either wild-type CAP or CAPΔSH3 cDNAs. Under these conditions, 94 ± 2% and 87 ± 6% of the cells microinjected with wild-type CAP and CAPΔSH3, respectively, displayed insulin-stimulated GLUT1 translocation. Together, these data show that the CAPΔSH3 mutant has a dominant-interfering effect on insulin-stimulated translocation of GLUT4/IRAP-containing vesicles to the plasma membrane, without affecting general exocytosis or other insulin-regulated vesicle trafficking processes.
The stimulation of glucose transport by insulin is a complex, multistep process involving the movement of intracellularly sequestered GLUT4-containing vesicles to the cell surface, followed by docking and fusion with the plasma membrane19. The activation of PI(3)K and its downstream kinases AKT and/or protein kinase Cζ/λ (PKCζ/λ) are required for this process, as inhibitors and dominant-negative mutants of PI(3)K, AKT and PKCζ/λ can completely block insulin-stimulated glucose transport20. There is compelling evidence, however, that activation of PI(3)K-dependent pathways is not sufficient to induce glucose transport. Other growth factors and adhesion molecules that can activate PI(3)K and its downstream kinases have no effect on glucose transport or GLUT4 translocation21,22,23. Furthermore, cell-permeable derivatives of phosphatidylinositol 3,4,5-trisphosphate can stimulate GLUT4 translocation only when cells are pre-treated with insulin24. These data suggest that insulin must generate at least two independent signals to stimulate glucose transport, one dependent on and another independent of PI(3)K.
What is the second signal needed for insulin-stimulated glucose transport? Our data indicate that this pathway is probably initiated by the phosphorylation of Cbl and its subsequent segregation into lipid raft subdomains of the plasma membrane. This process is independent of wortmannin (data not shown) and requires the adapter protein CAP2. CAP expression and Cbl phosphorylation correlate well with insulin sensitivity25. The appearance of phosphorylated Cbl in the triton-insoluble fraction, its association with the src kinase fyn, and the subsequent phosphorylation of the caveolar protein caveolin are specifically stimulated by insulin. Insulin-stimulated Cbl tyrosine phosphorylation is observed only in insulin-sensitive cells and tissues in which CAP is also expressed1,3,11. Although inhibition of Cbl phosphorylation in the triton-insoluble complexes with the CAPΔSH3 mutant does not interfere with PI(3)K-dependent and ras-dependent signalling, disruption of this association markedly attenuates insulin-stimulated GLUT4 translocation and glucose uptake. Whether the GLUT4 protein itself resides in the triton-insoluble microdomains of the plasma membrane remains uncertain26,27,28. However, it is possible that the association of the CAP–Cbl complex with other proteins in lipid rafts may lead to the segregated generation of a specific signal, perhaps directing fusion of the GLUT4 vesicle to the plasma membrane in a domain near the lipid raft.
Methods
Cell culture and transfection of HEK293Tcells and 3T3-L1 adipocytes
HEK293T cells were maintained in Dulbecco's Eagle Medium (DMEM) containing 10% fetal bovine serum at 37 °C and 5% CO2. HEK293T cells were transfected with a mammalian CaPO4 transfection kit as described by the manufacturer (Stratagene). 3T3-L1 preadipocytes were cultured in DMEM containing 25 mM glucose, 10% calf serum at 37 °C and 5% CO2. Confluent cultures were induced to differentiate into adipocytes as described29. 3T3-L1 adipocytes were transiently transfected by electroporation as described9. The cells were then allowed to adhere to collagen-coated tissue culture dishes for 30–48 h before serum starvation and insulin treatment. The average efficiency of transfection was 50% as evaluated by β-galactosidase staining.
In vitro protein associations
The Flag–CAP expression construct was made as described2. We constructed Flag-epitope-tagged CAPΔSH3 by restriction digestion, removal of the SH3 domains of the CAP cDNA and religation of the CAP expression construct. CAPΔSH3 does not associate with Cbl or the insulin receptor. We lysed HEK293T cells overexpressing CAP or CAPΔSH3 in HNTG buffer containing protease inhibitors and sodium orthovanadate29. Lysates from HEK293T cells were incubated with GST alone or GST–flotillin immobilized on glutathione agarose beads for 1 h at 4 °C. Beads were then washed extensively with HNTG and retained proteins were eluted in 4 × Laemmli sample buffer, heated for 5 min at 100 °C, separated by SDS–PAGE and immunoblotted with either a Flag antibody (Stratagene) or a polyclonal c-Cbl antibody (Santa Cruz).
GLUT4-EGFP, IRAP-EGFP and CI/M6PR-EGFP translocation assays
We constructed all EGFP fusion proteins and performed translocation assays as described9. Briefly, 3T3-L1 adipocytes were electroporated with 50 µg of the EGFP-tagged plasmid and 200 µg of empty vector, CAP or CAPΔSH3 constructs. Adipocytes were replated, allowed to recover overnight and stimulated with or without 100 nM insulin for 30 min. The cells were then fixed and EGFP fluorescence was analysed by confocal immunofluorescence microscopy.
Single cell microinjection and plasma membrane sheet isolation
We grew 3T3-L1 adipocytes on 35-mm tissue culture dishes. Before microinjection, the medium was changed to Lebovitz's L-15 medium containing 0.1% bovine serum albumin and microinjection was carried out as described30. The cells were allowed to recover for 24 h and plasma membrane sheets were prepared as described18. The bound plasma membranes sheets were then subjected to confocal fluorescent microscopy.
Metabolic assays
We determined 2-deoxyglucose uptake, glycogen synthesis and lipogenesis as described29.
Immunoprecipitations and immunoblotting
After insulin treatment, we washed electroporated adipocytes with PBS and lysed them in MBS buffer containing protease inhibitors and sodium orthovanadate. Lysed cells were kept at 4 °C to prevent degradation of caveolar complexes. The triton-insoluble pellet fraction was generated as described11. This triton-insoluble pellet fraction was solubilized in SOL buffer containing protease inhibitors and sodium orthovanadate as described11. Lysates and immunoprecipitations were resolved by SDS–PAGE and transferred to nitrocellulose for immunoblot analysis.
Immunoprecipitations were performed using a polyclonal c-Cbl antisera (Santa Cruz). We used monoclonal 4G10 anti-phosphotyrosine antibodies (Upstate Biotechnology) to blot for IR, IRS-1 and Cbl tyrosine phosphorylation. Polyclonal phospho-AKT and phospho-MAPK antibodies (New England Biolabs) were used to probe for activated AKT and MAP kinase. Monoclonal caveolin I antibodies (Transduction Labs) were used to evaluate protein content of the resolved TIP fractions. We used horseradish peroxidase (HRP)-conjugated secondary antibodies and ECL chemiluminesence for immunoblot detection.
References
Ribon, V. & Saltiel, A. R. Insulin stimulates tyrosine phosphorylation of the proto-oncogene product of c-Cbl in 3T3-L1 adipocytes. Biochem. J. 324, 839–845 ( 1997).
Ribon, V., Printen, J. A., Hoffman, N. G., Kay, B. K. & Saltiel, A. R. A novel, multifuntional c-Cbl binding protein in insulin receptor signaling in 3T3-L1 adipocytes. Mol. Cell. Biol. 18, 872–879 (1998).
Mastick, C. C., Brady, M. J. & Saltiel, A. R. Insulin stimulates the tyrosine phosphorylation of caveolin. J. Cell Biol. 129, 1523– 1531 (1995).
Printen, J. A., Brady, M. J. & Saltiel, A. R. PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science 275, 1475–1478 (1997).
Bickel, P. E. et al. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J. Biol. Chem. 272, 13793–13802 ( 1997).
Anderson, R. G. The caveolae membrane system. Annu. Rev. Biochem. 67 , 199–225 (1998).
Harder, T. & Simons, K. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr. Opin. Cell. Biol. 9, 534–542 ( 1997).
Volonte, D. et al. Flotillins/cavatellins are differentially expressed in cells and tissues and form a hetero-oligomeric complex with caveolins in vivo. Characterization and epitope-mapping of a novel flotillin-1 monoclonal antibody probe. J. Biol. Chem. 274, 12702–12709 (1999).
Thurmond, D. C. et al. Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J. Biol. Chem. 273, 33876–33883 (1998).
Gustavsson, J. et al. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 13, 1961– 1971 (1999).
Mastick, C. C. & Saltiel, A. R. Insulin-stimulated tyrosine phosphorylation of caveolin is specific for the differentiated adipocyte phenotype in 3T3-L1 cells. J. Biol. Chem. 272, 20706–20714 (1997).
Corvera, S. & Czech, M. P. Direct targets of phosphoinositide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol. 8, 442–446 (1998).
Keller, S. R., Scott, H. M., Mastick, C. C., Aebersold, R. & Lienhard, G. E. Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J. Biol. Chem. 270, 23612– 23618 (1995); erratum, ibid 270, 30236 (1995)..
Kandror, K. V. & Pilch, P. F. gp160, a tissue-specific marker for insulin-activated glucose transport. Proc. Natl Acad. Sci. USA 91, 8017–8021 ( 1994).
Oka, Y., Mottola, C., Oppenheimer, C. L. & Czech, M. P. Insulin activates the appearance of insulin-like growth factor II receptors on the adipocyte cell surface. Proc. Natl Acad. Sci. USA 81, 4028–4032 (1984).
Wardzala, L. J., Simpson, I. A., Rechler, M. M. & Cushman, S. W. Potential mechanism of the stimulatory action of insulin on insulin- like growth factor II binding to the isolated rat adipose cell. Apparent redistribution of receptors cycling between a large intracellular pool and the plasma membrane. J. Biol. Chem. 259, 8378– 8383 (1984).
Malide, D. & Cushman, S. W. Morphological effects of wortmannin on the endosomal system and GLUT4-containing compartments in rat adipose cells. J. Cell Sci. 110, 2795– 2806 (1997).
Min, J. et al. Synip: a novel insulin-regulated syntaxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol. Cell 3, 751–760 (1999).
Pessin, J. E., Thurmond, D. C., Elmendorf, J. S., Coker, K. J. & Okada, S. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J. Biol. Chem. 274, 2593–2596 ( 1999).
Czech, M. P. & Corvera, S. Signaling mechanisms that regulate glucose transport. J. Biol. Chem. 274, 1865 –1868 (1999).
Wiese, R. J., Mastick, C. C., Lazar, D. F. & Saltiel, A. R. Activation of mitogen-activated protein kinase and phosphatidylinositol 3′-kinase is not sufficient for the hormonal stimulation of glucose uptake, lipogenesis, or glycogen synthesis in 3T3-L1 adipocytes. J. Biol. Chem. 270, 3442–3446 (1995).
Isakoff, S. J. et al. The inability of phosphatidylinositol 3-kinase activation to stimulate GLUT4 translocation indicates additional signaling pathways are required for insulin-stimulated glucose uptake. Proc. Natl Acad. Sci. USA 92, 10247–10251 (1995).
Guilherme, A. & Czech, M. P. Stimulation of IRS-1-associated phosphatidylinositol 3-kinase and Akt/protein kinase B but not glucose transport by beta1-integrin signaling in rat adipocytes. J. Biol. Chem. 273, 33119–33122 (1998).
Jiang, T. et al. Membrane-permeant esters of phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 11017– 11024 (1998).
Ribon, V., Johnson, J. H., Camp, H. S. & Saltiel, A. R. Thiazolidinediones and insulin resistance: peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc. Natl Acad. Sci. USA 95, 14751– 14756 (1998).
Kandror, K. V., Stephens, J. M. & Pilch, P. F. Expression and compartmentalization of caveolin in adipose cells: coordinate regulation with and structural segregation from GLUT4. J. Cell Biol. 129, 999– 1006 (1995).
Scherer, P. E. et al. Induction of caveolin during adipogenesis and association of GLUT4 with caveolin-rich vesicles. J. Cell Biol. 127, 1233–1243 (1994).
Gustavsson, J., Parpal, S. & Stralfors, P. Insulin-stimulated glucose uptake involves the transition of glucose transporters to a caveolae-rich fraction within the plasma membrane: implications for type II diabetes. Mol. Med. 2, 367–372 (1996).
Lazar, D. F. et al. Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. J. Biol. Chem. 270, 20801–20807 ( 1995).
Elmendorf, J. S., Chen, D. & Pessin, J. E. Guanosine 5′-O-(3-thiotriphosphate) (GTPγS) stimulation of GLUT4 translocation is tyrosine kinase-dependent. J. Biol. Chem. 273, 13289–13296 (1998).
Acknowledgements
J.E.P. is supported by the NIH. P.E.B. is supported by the American Diabetes Association and awards from the Washington University Clinical Nutrition Research Unit and Diabetes Research Training Center.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article
Cite this article
Baumann, C., Ribon, V., Kanzaki, M. et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature 407, 202–207 (2000). https://doi.org/10.1038/35025089
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/35025089
This article is cited by
-
Tirzepatide prevents neurodegeneration through multiple molecular pathways
Journal of Translational Medicine (2024)
-
KSRP improves pancreatic beta cell function and survival
Scientific Reports (2024)
-
Phosphoproteomics reveals rewiring of the insulin signaling network and multi-nodal defects in insulin resistance
Nature Communications (2023)
-
FLOT1 knockdown inhibits growth of AML cells through triggering apoptosis and pyroptosis
Annals of Hematology (2023)
-
Complementary omics strategies to dissect p53 signaling networks under nutrient stress
Cellular and Molecular Life Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}