
Abstract
Aim:
High mobility group box protein 1 (HMGB1) and receptor for the advanced glycation end product (RAGE) play pivotal roles in vascular inflammation and atherosclerosis. The aim of this study was to determine whether the HMGB1-RAGE axis was involved in the actions of simvastatin on vascular inflammation and atherosclerosis in ApoE−/− mice.
Methods:
Five-week old ApoE−/− mice and wild-type C57BL/6 mice were fed a Western diet. At 8 weeks of age, ApoE−/− mice were administered simvastatin (50 mg·kg−1·d−1) or vehicle by gavage, and the wild-type mice were treated with vehicle. The mice were sacrificed at 11 weeks of age, and the atherosclerotic lesions in aortic sinus were assessed with Oil Red O staining. Macrophage migration was determined with scanning EM and immunohistochemistry. Human umbilical vein endothelial cells (HUVECs) were used for in vitro study. Western blots were used to quantify the protein expression of HMGB1, RAGE, vascular cell adhesion molecule-1 (VCAM-1) and monocyte chemoattractant protein-1 (MCP-1).
Results:
Vehicle-treated ApoE−/− mice exhibited significant increases in aortic inflammation and atherosclerosis as well as enhanced expression of HMGB1, RAGE, VCAM-1, and MCP-1 in aortic tissues as compared to the wild-type mice. Furthermore, serum total cholesterol, triglyceride and LDL levels were markedly increased, while serum HDL level was decreased in vehicle-treated ApoE−/− mice. Administration with simvastatin in ApoE−/− mice markedly attenuated the vascular inflammation and atherosclerotic lesion area, and decreased the aortic expression of HMGB1, RAGE, VCAM-1, and MCP-1. However, simvastatin did not affect the abnormal levels of serum total cholesterol, triglyceride, LDL and HDL in ApoE−/− mice. Exposure of HUVECs to HMGB1 (100 ng/mL) markedly increased the expression of HMGB1, RAGE and VCAM-1, whereas pretreatment of the cells with simvastatin (10 μmol/L) blocked the HMGB1-caused changes.
Conclusion:
Simvastatin inhibits vascular inflammation and atherosclerosis in ApoE−/− mice, which may be mediated through downregulation of the HMGB1-RAGE axis.
Similar content being viewed by others

Introduction
Atherosclerosis is a chronic inflammatory disease involving complex interactions among multiple cytokines, macrophages, T lymphocytes and endothelial cells1. As a proinflammatory mediator, high mobility group box protein 1 (HMGB1) upregulates adhesion molecules, chemokines and cytokines, which, in turn, stimulate macrophage infiltration and contribute to atherosclerosis2,3. Additionally, it has been reported that blockade of HMGB1 suppresses vascular inflammation and attenuates atherosclerosis in ApoE−/− mice4. Meanwhile, the receptor for advanced glycation end product (RAGE), an important receptor for HMGB1, is also involved in the progression of atherosclerosis5. Interruption of the ligand-RAGE axis by multiple approaches improves endothelial dysfunction and suppresses vascular inflammation and atherosclerosis6,7,8.
Previous studies suggest that statins inhibit atherosclerosis partly via their pleiotropic effects, including improvement of endothelial dysfunction, anti-inflammatory actions and anti-oxidative effects beyond lipid lowering9,10. Recently, studies showed that statins attenuate HMGB1 expression in atherosclerotic rats11, reduce brain HMGB1 and RAGE expression in a brain ischemia model12, and decrease lung HMGB1 and plasma HMGB1 levels in golden Syrian hamsters fed a Western diet13. In addition, statins downregulate RAGE expression in the plaques of patients with type 2 diabetes14 as well as in a diabetic model of ApoE−/− mice15.
Whether the HMGB1-RAGE axis is involved in the statin-dependent inhibition of vascular inflammation and atherosclerosis, however, has not been determined. This study was therefore performed to investigate the role of the HMGB1-RAGE axis in the effects of simvastatin on vascular inflammation and atherosclerosis in ApoE−/− mice.
Materials and methods
Animals
Animal studies were performed with the approval of the Institutional Animal Care and Use Committee of Fudan University. Male ApoE−/− mice (C57BL/6 background; provided by Beijing Vital River Laboratories Animal Technology Co Ltd, Beijing, China) and C57BL/6 mice (wild-type) were used in this study. At 5 weeks of age, the mice were placed on a Western diet (21% fat, 0.15% cholesterol). At 8 weeks of age, ApoE−/− mice were randomized to treatment with either simvastatin (50 mg·kg−1·d−1, FWD Chem, Shanghai, China; as in reference16) or vehicle (0.5% carboxymethyl cellulose sodium solution) by gavage for 3 weeks. Wild-type mice were treated with vehicle (0.5% carboxymethyl cellulose sodium solution). All mice were sacrificed at 11 weeks of age.
Quantification of atherosclerotic lesions
Mice were fasted for 4 h and then anesthetized. Hearts were removed for analysis of atherosclerotic lesions. Cryostat sections were prepared and embedded in OCT compound. Serial sections (8 μm thick) were collected from the level of the aortic valve leaflets up to approximately 480 μm above the leaflets in the aortic sinus. Every other section was retrieved and placed onto slides; 5 sections were placed onto each slide for a total of 6 slides. Sections were stained with Oil Red O and counterstained with hematoxylin and light green. Quantification of the atherosclerotic lesion area was performed by a blinded observer6,17.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated as previously described18 and cultured in ECM (ScienCell Research Laboratories, San Diego, CA, USA). HUVECs were pretreated with simvastatin (10 μmol/L, Sigma, St Louis, MO, USA), goat anti-human RAGE IgG (4 μg/mL, R&D, Minneapolis, MN, USA) or PBS for 1 h prior to exposure to HMGB1 (100 ng/mL, Sigma, St Louis, MO, USA) for 24 h. Untreated cells were used as controls.
Western blot
Aortic tissues and HUVECs were retrieved and subjected to disruption by an ultrasonic homogenizer in RIPA containing protease and phosphatase inhibitors (Pierce Biotechnology, Rockford, IL, USA). Protein concentrations were determined using an assay from Pierce (Rockford, IL, USA). Equal amounts of protein (8 or 20 μg/sample) were separated on 10% SDS-PAGE gels and transferred to PVDF membranes, which were blocked by BSA (5%) for 1 h at room temperature. Immunoblotting was performed overnight using rabbit anti-HMGB1 IgG (1:800, Abcam, Cambridge, UK), rabbit anti-RAGE IgG (1:600, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-vascular cell adhesion molecule-1 (VCAM-1) IgG (1:200, Wuhan Boster Bio-engineering Limited Company, Wuhan, China) or rabbit anti-monocyte chemoattractant protein-1 (MCP-1) IgG (1:200, Wuhan Boster Bio-engineering limited company, Wuhan, China). HRP-conjugated goat anti-rabbit antibody (1:4000, Jackson ImmunoResearch, PA, USA) was employed to identify the sites of primary antibody binding. The enhanced chemiluminescence (ECL) detection system (TIANGEN Biotech, Beijing, China) was used to visualize the immunoreactions. After completion, membranes were stripped of bound antibodies and reprobed with antibody to β-actin (Kangchen Biotechnology, Shanghai, China). Quantitative analysis of the band density was performed using Quantity One software. All bands were normalized to β-actin.
Scanning electron microscope
En face of the aortic arches were fixed in a 2.5% glutaraldehyde buffer, gold-coated with an ion sputter, and observed under a scanning electron microscope (Hitachi, Japan) with a voltage of 20 kV. According to a previous study19, monocytes can be morphologically identified by their pseudopodia around the cellular surface.
Immunohistochemistry
Cryostat sections were fixed in buffered formalin (10%). After 30 min of blockade with 5% nonimmune serum solution, the cryosections were incubated with rat anti-F4/80 antibody (1:100 dilution) (AbD Serotec, Oxford, UK), rabbit anti-HMGB1 IgG (1:100, Abcam, Cambridge, UK) or rabbit anti-RAGE IgG (1:50, Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4 °C and then visualized with goat anti-rat IgG conjugated with CF568 (Biotium, Hayward, CA, USA) or goat anti-rabbit IgG conjugated with Alexa Flour 546 (Invitrogen, Carlsbad, CA, USA). The atherosclerotic plaques were specially selected to quantify the relative F4/80-positive areas in the lesions using Image J software.
Enzyme-linked immunosorbent assay
A mouse HMGB1 ELISA kit (Uscn Life Science Inc, Wuhan, China) was used to measure serum HMGB1 concentrations according to the manufacturer's instructions.
Serum lipid analysis
Blood was drawn from the inferior vena cava in fasted mice. Total cholesterol, triglyceride, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) levels in serum were measured using kits from the Nanjiang Jiancheng Bioengineering Institute (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Statistical analysis
Data are expressed as mean±SD. Multiple group comparisons were performed using a one-way ANOVA followed by the post-hoc Tukey test. P<0.05 was considered statistically significant.
Results
Effect of simvastatin on atherosclerosis in ApoE−/− mice
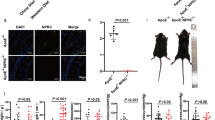
After 3 weeks of intervention, aortic sinus assays were performed to evaluate lesion formation. No atherosclerotic lesions were observed in wild-type mice (Figure 1A, 1D). Compared with ApoE−/− mice, the atherosclerotic lesion area was markedly reduced in simvastatin-treated ApoE−/− mice (≈64% reduction), measured as 159253±22423.2 μm2 vs 56854.5±11029.9 μm2, respectively; P<0.01 (Figure 1B, 1C, and 1D).

Simvastatin 50 mgkg−1·d−1 for 3 weeks impacts atherosclerosis in ApoE−/− mice. Shown are representative aortic sinus sections stained with Oil Red O (A–C; Scale bar=50 μm). (D) Mean atherosclerotic lesion area in the aortic sinus was determined. n=5–6 mice per condition. ApoE−/−/S: ApoE−/− mice treated with simvastatin. bP<0.05, cP<0.01 vs wild-type. fP<0.01 vs ApoE−/−.
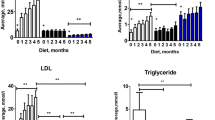
Additionally, serum lipid levels were determined. As shown in Table 1, serum total cholesterol, triglyceride, and LDL levels were markedly increased in ApoE−/− mice compared with wild-type mice; P<0.01. In contrast, serum HDL levels were decreased in ApoE−/− mice compared with wild-type mice; P<0.01. However, there were no differences in serum total cholesterol, triglyceride, LDL or HDL levels between ApoE−/− mice treated with vehicle or simvastatin. Together, these results suggest that simvastatin attenuated atherosclerosis in a lipid-independent manner.
Effect of simvastatin on vascular inflammation in ApoE−/− mice
To investigate the effect of simvastatin on vascular inflammation, we determined the degree of macrophage adhesion and infiltration. Macrophage adhesion and subendothelial migration were observed in en face aortic arches of ApoE−/− mice using scanning electron microscope, but these were not found in wild-type mice or simvastatin-treated ApoE−/− mice (Figure 2A–2D). In addition, a reduction of approximately 41% of F4/80-expressing macrophage content in atherosclerotic lesions was observed in simvastatin-treated ApoE−/− mice compared with ApoE−/− mice; P<0.05 (Figure 2F–2H).

Simvastatin impacts macrophage adhesion and infiltration in ApoE−/− mice. Scanning electron microscope analysis of macrophage adhesion and subendothelial migration in the aortic arch (A–D). At higher magnification (insert), many pseudopodia were apparent around the macrophages (C). The red arrows indicate macrophages adhering to endothelial cells, and the white arrows indicate subendothelial macrophages. Macrophage accumulation in atherosclerotic lesions was examined by immunohistochemical staining of F4/80 (E–G). The special extent marked by white lines shown in F and G was used to detect the F4/80 positive area, and the relative number of macrophages in the lesion was quantified (H); n=3 per group. All scale bars=100 μm. ApoE−/−/S: ApoE−/− mice treated with simvastatin. cP<0.01 vs wild-type. eP<0.05 vs ApoE−/−.
Effects of simvastatin on the HMGB1-RAGE axis in ApoE−/− mice
We measured vascular HMGB1 and RAGE levels to gain insight into the mechanisms underlying simvastatin-suppressed vascular inflammation and atherosclerosis. Compared with wild-type mice, vascular HMGB1 and RAGE expression were significantly increased in ApoE−/− mice by Western blot; P<0.05 (Figure 3H and 3I, respectively). However, this was reversed by simvastatin. Furthermore, immunohistochemistry showed a similar phenomenon in atherosclerotic lesions (Figure 3B–3G). Considering that there were similar regions in adjacent sections of aortic sinus13, we found that HMGB1 staining was observed primarily in the cytoplasm of macrophages of atherosclerotic lesions (Figure 2F and 3C). We also found that serum HMGB1 levels were increased in ApoE−/− mice compared with wild-type mice; however, the difference was not significant (40.1±12.2 ng/mL vs 54.5±4.3 ng/mL, P=0.12). In addition, no significant differences in serum HMHB1 levels were found between ApoE−/− mice treated with vehicle or those treated with simvastatin (54.5±4.3 ng/mL vs 50.4±9.2 ng/mL, P=0.81). These data suggested that attenuated HMGB1 levels in atherosclerotic lesions, but not in serum, were associated with suppression of vascular inflammation and atherosclerosis by simvastatin.

Simvastatin 50 mgkg−1·d−1 impacts the HMGB1-RAGE axis in ApoE−/− mice. Serum HMGB1 levels were detected by ELISA (A). Aortic tissues were subjected to Western blot to detect HMGB1 (H), RAGE (I), VCAM-1 (J) and MCP-1 (K) expression normalized to levels of β-actin; n=3 per group. Immunohistochemistry for HMGB1 (B–D) and RAGE (E–G) in atherosclerotic lesions; all scale bars represent 100 μm. At higher magnification (insert of C), HMGB1 (red) was generally present in the cytoplasm but not the nuclei (blue). ApoE−/−/S: ApoE−/− mice treated with simvastatin. bP<0.05, cP<0.01 vs wild-type. eP<0.05, fP<0.01 vs ApoE−/−.
Subsequently, we determined the levels of HMGB1-RAGE downstream inflammatory molecules, VCAM-1 and MCP-1, which contribute to macrophage migration and atherosclerosis20. The expression of VCAM-1 and MCP-1 was increased in ApoE−/− mice compared with wild-type mice, while there were no significant differences between ApoE−/− mice treated with simvastatin or those treated with vehicle; P<0.05 (Figure 3J and 3K, respectively). These findings indicate that the HMGB1-RAGE axis may play a critical role in the reduction of vascular inflammation and atherosclerosis by simvastatin.
Effects of simvastatin on the HMGB1-RAGE axis in HUVECs treated with HMGB1
To further elucidate the roles of simvastatin in the HMGB1-RAGE axis, we assessed the effect of simvastatin on the expression of HMGB1, RAGE, and VCAM-1 in HUVECs. HMGB1 markedly raised expression of VCAM-1, HMGB1, and RAGE in HUVECs; P<0.01 (Figure 4A–4D). In the presence of simvastatin, however, levels of VCAM-1, HMGB1, and RAGE were significantly decreased compared with HUVECs incubated with HMGB1; P<0.01 (Figure 4A–4D). Similarly, the expression of VCAM-1 and RAGE was also reduced by anti-RAGE antibodies; P<0.01 (Figure 4A–4D). Meanwhile, there were no significant differences in the levels of VCAM-1, HMGB1, and RAGE in HUVECs pretreated with simvastatin compared with those treated with anti-RAGE antibodies. These results suggest that simvastatin reduced VCAM-1 expression in HMGB1-treated HUVECs, in part via the HMGB1-RAGE axis.

Simvastatin 50 mgkg−1·d−1 impacts the HMGB1-RAGE axis in HMGB1-treated HUVECs. Proteins extracted from HUVECs were subjected to Western blot to detect HMGB1 (B), RAGE (C), and VCAM-1 (D) expression normalized to levels of β-actin; n=4 per group. Representative bands are shown (A). From left to right: lane 1, control; lane 2, HUVECs stimulated with HMGB1; lane 3, HUVECs stimulated with anti-RAGE antibodies and HMGB1; and lane 4, HUVECs stimulated with simvastatin and HMGB1. cP<0.01 vs HUVECs without HMGB1. fP<0.01 vs HUVECs stimulated with HMGB1.
Discussion
Evidence suggests that HMGB1 and RAGE play pivotal roles in inflammation and atherosclerosis3,5 and that they are the key target for the suppression of vascular inflammation and atherosclerosis4,6,7,8. Meanwhile, recent studies have shown that statins suppress HMGB1 and/or RAGE levels in atherosclerotic rats, rats with brain ischemia, diabetic mice and human subjects11,12,13,14,15. However, it is unknown whether the HMGB1-RAGE axis is involved in the effects of simvastatin on vascular inflammation and atherosclerosis. In the present study, we showed that ApoE−/− mice displayed markedly increased vascular inflammation and atherosclerosis, as well as upregulation of the HMGB1-RAGE axis, compared with wild-type mice. We also found that simvastatin inhibited the HMGB1-RAGE axis, vascular inflammation and atherosclerosis in ApoE−/− mice. Finally, we demonstrated that HMGB1 upregulated VCAM-1 in HUVECs partly via the RAGE pathway—an effect that was negated by simvastatin.
HMGB1, a 30-kDa nuclear protein, can be released by inflammatory cells3 or necrotic cells21 to trigger inflammation. Kalinina et al3 found that increased HMGB1 in atherosclerotic plaques, primarily associated with macrophages, induced endothelial cells to upregulate the expression of VCAM-1 and MCP-1 via the RAGE signaling pathway2. As critical mediators for macrophage migration, VCAM-1 and MCP-1 aggravate macrophage infiltration into atherosclerotic lesions where HMGB1 is over-expressed by macrophages, and then further promote atherosclerotic progression20,22. Consistent with these findings, ApoE−/− mice displayed significantly enhanced aortic expression of HMGB1, RAGE, VCAM-1, and MCP-1 as well as increased macrophage infiltration and atherosclerosis compared with wild-type mice. In addition, HMGB1 expression was mostly associated with macrophages in lesions, suggesting that macrophages may be the main source of HMGB1 in plaque. These observations suggest that HMGB1 enforces macrophage infiltration and promotes atherosclerotic progression through the RAGE signaling pathway. Moreover, the blockade of HMGB1 or RAGE improved endothelial dysfunction and suppressed vascular inflammation and atherosclerosis4,6,7,8. Overall, these data clearly indicate that the HMGB1-RAGE axis is a potential therapeutic target for treating inflammation and atherosclerosis.
Statins, as 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, have been reported to suppress atherosclerosis partly by their pleiotropic effects, including improvement of endothelial dysfunction, anti-inflammatory actions and anti-oxidative effects beyond lipid lowering9,10. Recently, increasing evidence demonstrated that statins attenuate HMGB1 and/or RAGE expression in atherosclerotic rats, rats with brain ischemia, diabetic mice and human subjects11,12,13,14,15. However, the role of the HMGB1-RAGE axis in the effects of statins on vascular inflammation and atherosclerosis in ApoE−/− mice is poorly understood. The results of this study revealed that simvastatin not only decreased aortic expression of HMGB1, RAGE, VCAM-1, and MCP-1 but also markedly attenuated macrophage accumulation and atherosclerosis in ApoE−/− mice. Moreover, HMGB1 in lesions but not in serum, which was decreased by simvastatin, was closely associated and consistent with the reduced macrophage infiltration. Similar to our results, Kanellakis et al4 reported that inhibition of HMGB1 in plaques attenuated macrophage accumulation and deterioration of atherosclerosis lesions by reduction of VCAM-1 and MCP-1 expression via the RAGE signaling pathway. However, we did not find a significant difference in lipid profiles between ApoE−/− mice treated with vehicle or those treated with simvastatin. Because the cholesterol-lowering effect of statins results mainly from enhanced hepatic uptake of LDL by increased LDL receptor (LDLr) expression and reduced cholesterol synthesis16, statins may not lower serum lipids in ApoE−/− mice, as they are deficient in a critical ligand (ApoE) for LDLr and LDLr-related proteins. These findings suggest that simvastatin's inhibition of the HMGB1-RAGE axis in lesions beyond lipid lowering may establish a negative inhibitory loop in the inflammatory response and consequently suppress vascular inflammation and atherosclerotic progression.
VCAM-1 is a pivotal mediator for macrophage migration and atherosclerosis8,20; therefore, we further determined the effects of simvastatin on the HMGB1-RAGE axis including HMGB1, RAGE, and VCAM-1 in HUVECs. In line with a previous study2, our data revealed that HMGB1 upregulated VCAM-1 expression through the RAGE pathway. Furthermore, simvastatin normalized VCAM-1, HMGB1, and RAGE expression in HMGB1-incubated HUVECs, which produced similar effects as the inhibition of the RAGE pathway by an anti-RAGE antibody. These results indicate that the HMGB1-RAGE interaction may induce upregulation of VCAM-1, which then contributes to macrophage migration and further promotes atherosclerosis. Although not significant, the expression of HMGB1 and VCAM-1 was lower in HUVECs pretreated with simvastatin than those pretreated with the anti-RAGE antibody, thus suggesting that receptors beyond RAGE, such as TLR423, may be involved in the amelioration of HMGB1-induced endothelial activation by simvastatin. Therefore, simvastatin may alleviate inflammation and consequently inhibit atherosclerotic progression partly through downregulation of the HMGB1-RAGE axis.
In conclusion, our data demonstrate that simvastatin suppresses vascular inflammation and atherosclerosis in ApoE−/− mice, presumably as a result of the downregulation of the HMGB1-RAGE axis in atherosclerotic plaques. Together with previous studies, these findings indicate that the HMGB1-RAGE axis may be involved in the pleiotropic effects of statins in suppressing inflammation and atherosclerosis, although the underlying mechanisms need to be further elucidated.
Author contribution
Hong JIANG and Yun-zeng ZOU designed the research; Ming LIU, Ying YU, Peng YU, and Jian-guo JIA performed the research; Lei ZHANG and Pei-pei ZHANG analyzed data; Ming LIU and Ying YU wrote the paper; Hong JIANG, Rui-zhen CHEN, and Jun-bo GE revised the manuscript.
References
Ross R . Atherosclerosis — an inflammatory disease. N Engl J Med 1999; 340: 115–26.
Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003; 101: 2652–60.
Kalinina N, Agrotis A, Antropova Y, DiVitto G, Kanellakis P, Kostolias G, et al. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterioscler Thromb Vasc Biol 2004; 24: 2320–5.
Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, et al. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2011; 31: 313–9.
Cipollone F, Iezzi A, Fazia M, Zucchelli M, Pini B, Cuccurullo C, et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation 2003; 108: 1070–7.
Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr, Chow WS, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 1998; 4: 1025–31.
Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002; 106: 2827–35.
Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest 2008; 118: 183–94.
Sorrentino S, Landmesser U . Nonlipid-lowering effects of statins. Curr Treat Options Cardiovasc Med 2005; 7: 459–66.
Zhou Q, Liao JK . Pleiotropic effects of statins: basic research and clinical perspectives. Circ J 2010; 74: 818–26.
Yin YX, Yao YM, Liu RM, Zhai HX, Li L, Zhang JJ, et al. The effect of simvastatin on the expression of high mobility group box-1 protein in atherosclerotic rats. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 2010; 22: 306–8.
Wang L, Zhang X, Liu L, Yang R, Cui L, Li M . Atorvastatin protects rat brains against permanent focal ischemia and downregulates HMGB1, HMGB1 receptors (RAGE and TLR4), NF-kappaB expression. Neurosci Lett 2010; 471: 152–6.
Haraba R, Suica VI, Uyy E, Ivan L, Antohe F . Hyperlipidemia stimulates the extracellular release of the nuclear high mobility group box 1 protein. Cell Tissue Res 2011; 346: 361–8.
Cuccurullo C, Iezzi A, Fazia ML, De Cesare D, Di Francesco A, Muraro R, et al. Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler Thromb Vasc Biol 2006; 26: 2716–23.
Calkin AC, Giunti S, Sheehy KJ, Chew C, Boolell V, Rajaram YS, et al. The HMG-CoA reductase inhibitor rosuvastatin and the angiotensin receptor antagonist candesartan attenuate atherosclerosis in an apolipoprotein E-deficient mouse model of diabetes via effects on advanced glycation, oxidative stress and inflammation. Diabetologia 2008; 51: 1731–40.
Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, et al. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol 2007; 27: 1706–21.
Baglione J, Smith JD . Quantitative assay for mouse atherosclerosis in the aortic root. Methods Mol Med 2006; 129: 83–95.
Baudin B, Bruneel A, Bosselut N, Vaubourdolle M . A protocol for isolation and culture of human umbilical vein endothelial cells. Nat Protoc 2007; 2: 481–5.
Nathan L, Pervin S, Singh R, Rosenfeld M, Chaudhuri G . Estradiol inhibits leukocyte adhesion and transendothelial migration in rabbits in vivo: possible mechanisms for gender differences in atherosclerosis. Circ Res 1999; 85: 377–85.
Libby P . Inflammation in atherosclerosis. Nature 2002; 420: 868–74.
Scaffidi P, Mistell T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–5.
Li W, Sama AE, Wang H . Role of HMGB1 in cardiovascular diseases. Curr Opin Pharmacol 2006; 6: 130–5.
Yang J, Huang C, Yang J, Jiang H, Ding J . Statins attenuate high mobility group box-1 protein induced vascular endothelial activation: a key role for TLR4/NF-kappaB signaling pathway. Mol Cell Biochem 2010; 345: 189–95.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No 81170221) and the Janssen Research Council China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, M., Yu, Y., Jiang, H. et al. Simvastatin suppresses vascular inflammation and atherosclerosis in ApoE−/− mice by downregulating the HMGB1-RAGE axis. Acta Pharmacol Sin 34, 830–836 (2013). https://doi.org/10.1038/aps.2013.8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.8
Keywords
This article is cited by
-
Control of the post-infarct immune microenvironment through biotherapeutic and biomaterial-based approaches
Drug Delivery and Translational Research (2023)
-
Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19?
Molecular Medicine (2020)
-
Cardiomyocyte-restricted high-mobility group box 1 (HMGB1) deletion leads to small heart and glycolipid metabolic disorder through GR/PGC-1α signalling
Cell Death Discovery (2020)
-
TLRs and RAGE are elevated in carotid plaques from patients with moderate-to-severe obstructive sleep apnea syndrome
Sleep and Breathing (2020)
-
Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction
Scientific Reports (2016)
